

Abstract
Background & Aims
Chronic injury results in regeneration of normal pancreatic tissue and formation of a metaplasia of ductal phenotype. Metaplastic ductal lesions are seen in pancreatitis as well as in specimens of pancreatic cancer, and are thought to represent a condition with increased risk of neoplasia. Acinar-to-ductal transdifferentiation is thought to be the source of this metaplasia. This has been suggested for flat duct-like lesions called tubular complexes and for lesions exhibiting a mucinous metaplasia. However, available studies are based on interpretation of static data rather than on direct evidence. Transdifferentiation from acinar to ductal cells has never been confirmed in the adult pancreas.
Methods
Here we use Cre-loxP-based genetic lineage tracing in vivo to investigate whether transdifferentiation of acinar cells contributes to regeneration and metaplasia in pancreatitis.
Results
The results show that transdifferentiation does not play a role in regeneration of normal tissue. Acinar cells are regenerated by preexisting acinar cells and not from other cell types. Three different types of metaplastic ductal lesions are observed and analyzed. Whereas the majority of metaplastic lesions are not of acinar origin, acinar-to-ductal transdifferentiation is identified in a minority of mucinous metaplastic lesions.
Conclusions
Here we provide direct evidence that acinar-to-ductal transdifferentiation occurs in the adult pancreas in vivo. However, it accounts for only a minority of metaplastic lesions.
Introduction
In many organs chronic injury results in regeneration of normal tissue and in metaplasia. Metaplasia can occur through a variety of mechanisms, including selective proliferation or loss of a cell type, differentiation of stem cells, or transdifferentiation.1 In the normal pancreas the predominant cells are acinar. In the diseased pancreas acinar tissue is replaced by metaplastic ductal lesions (MDL). MDL are seen in pancreatitis as well as in specimens of pancreatic cancer, and are thought to represent a condition with increased risk of neoplasia.2,3
Two definitions focusing on different morphologic features have been used to describe MDL: (I) tubular complexes (TC) and (II) mucinous metaplastic lesions (MML), or Pancreatic Intraepithelial Neoplasia (PanIN). TC are defined as cylindrical tubes with an often wide lumen lined by a monolayer of flattened duct-like cells.4,5 TC have been observed in human pancreatitis4,6 and pancreatic cancer, in animal models of chemical5 and surgical7 pancreatic injury and of chemical carcinogenesis,8,9 and in transgenic mouse models of pancreatic disease.10 The significance of TC is controversial. It has been proposed that they give rise to new acinar tissue5,7 or represent early cancer precursors.8,10 In recent years, the misexpression of mucins in pancreatic cancer has shifted the focus from these flat epithelial lesions to MML. In the human and mouse pancreas, MML are classified in the PanIN system according to their morphologic atypia.3,11 High-grade lesions are thought to represent direct cancer precursors.
In spite of the ductal phenotype of MDL, both the mechanisms of their formation and their cellular origin are controversial. Almost all pancreatic cell lineages, including ductal, centroacinar, acinar, and islet cells, have been proposed as the origin of MDL and neoplasia.8,12 Based on morphologic observations and interpretation of the momentary expression of lineage markers, most of these studies come to the conclusion that the flat epithelial lesions called TC are derived from acinar cells by degeneration and atrophy4,6 or transdifferentiation.7,10 Other authors propose that TC originate from ductal proliferation, or merely represent pre-existing ducts, which are less sensitive to injury than acinar cells and persist during acinar loss.13 The most prominent hypothesis remains that metaplastic lesions, including TC and more advanced lesions including neoplastic changes, are formed from acinar cells that take on a ductal phenotype, a mechanism addressed as acinar-to-ductal metaplasia.14-17
It has recently been demonstrated by genetic lineage tracing that transdifferentiation of acinar cells into duct-like cells can be observed in vitro.17 However, in vitro studies on transdifferentiation have been questioned because of the possibility of artifacts in culture, fusion and clonality.18 Even if transdifferentiation can be demonstrated in vitro, its role in living tissue remains unclear.
The aim of the present study was to use genetic lineage tracing in vivo to evaluate whether acinar-to-ductal transdifferentiation does occur in the adult pancreas and whether it contributes to regeneration or metaplasia. Elastase-CreER™ mice19 were crossed to a Z/AP reporter strain.20 Tamoxifen injection of bigenic animals results in a heritable tag, human placental alkaline phosphatase (HPAP), specifically in acinar cells or their progeny regardless of their fate. Tamoxifen-injected Ela-CreER™;Z/AP animals were subjected to pancreatitis and the resulting MDL were evaluated for HPAP expression as the indicator of acinar origin.
Materials and Methods
Transgenic in Vivo Lineage Tracing
The Elastase-CreER™ construct was generated by fusing a 200 bp enhancer of the Elastase gene to a minimal hsp68 promoter and placing the chimeric promoter upstream of a CreER™ coding sequence.19 Elastase-CreER™ mice were crossed to the Z/AP reporter strain, genotyped by PCR and tagged as previously described.19-21 Pancreatitis was induced 2.5 weeks after the last tamoxifen injection.
Animal Experiments
All experiments were approved by the institutional Subcommittee on Research Animal Care. Acute and chronic pancreatitis were induced using the cerulein model as described previously.21 CP was induced for 3 (3wCP), 6 (6wCP), or 10 weeks (10wCP).
Pancreaticoduodenectomy specimens were harvested 48 hours (AP) and 72 hours (CP) after the last cerulein injection. For assessment of proliferation by BrdU incorporation, animals were exposed to BrdU 72 hours (0.8 mg/ml in the drinking water) or 2 hours (1mg i.p.) prior to sacrifice. Specimens were fixed in 4% paraformaldehyde at 4°C for 4-5 hours, dehydrated and embedded in paraffin.
Histology, Histochemistry and Immunohistochemistry
HPAP and LacZ expression were identified as previously described.20,22 Mucins were identified by periodic acid-Schiffs (PAS) and Alcian blue (pH 2.8) staining using standard protocols. Expression of HPAP, amylase, cytokeratins, gastric mucins, Shh, Hes-1, caspase-3, and Ki-67, and BrdU incorporation were identified as specified in the Supplementary Methods section.
Quantitative Analysis
Tagging
For each animal the percentage of tagged acini was determined by counting acini in five 100x high power fields (HPF).
Morphologic analysis
TC were counted in AP and 3wCP in three 100x HPF per cut, three cuts per animal. MML were identified in 6wCP and 10wCP by positive staining for PAS and Alcian blue in the entirety of three HPAP/mucin double stains and serial HPAP and Alcian blue stains per animal. For immunofluorescence MML were identified by expression of gastric mucins.
Results
Tagging in the Elastase-CreER™;Z/AP Animal
To determine efficiency and specificity of acinar cell tagging in the Elastase-CreER™ mouse, expression of HPAP was assessed in tamoxifen-injected healthy Elastase-CreER™;Z/AP mice. The HPAP tag was identified at the luminal membrane of acinar cells (Fig.1) with a tagging efficiency of about 40% (Fig.2A,C), consistent with the expression pattern described in previous studies for this strain and other Elastase-CreER strains.17,19 Tagged acini were distributed throughout the pancreas. HPAP was also identified within islets with a low frequency of ~ 3.6% of islet cells (Fig.1A,B). HPAP expression was not observed in ductal and centroacinar cells (Fig.1A-D). Within the exocrine pancreas, the tagging system is highly specific for acinar cells.
Figure 1.

Tagging in the ElastaseCreER™;Z/AP animal. Identification of HPAP by BCIP/NBT (blue) (A,C) and immunofluorescence (B,D). (A, B) HPAP is identified on the luminal membrane of acinar cells (arrowheads) and infrequently in islets (arrows) but not in the ductal epithelium (stars), including terminal ducts (arrowheads) and centroacinar cells (arrows) (C,D).
Figure 2.

New acinar cells are formed from preexisting acinar cells. (A) Stable percentage of tagged acinar cells independent of the duration of pancreatitis. (B) BrdU incorporation (brown). (C,D) Identification of HPAP activity, blue (C) and immunofluorescence (D). In CP acinar cell density is decreased but the percentage of HPAP+ acinar cells remains stable. HPAP is not identified in ducts (arrows in C,D). (E) BrdU incorporation, 2 hrs (red). In the normal pancreas (left panel), proliferative activity is identified in single differentiated acinar cells (arrowhead) expressing amylase (green). In response to inflammation (right panel), proliferation is frequently identified in acinar cells (arrowheads).
New Acinar Cells Are Generated from Pre-existing Acinar Cells
The cerulein model results in apoptosis of more than 10% of acinar cells after a single episode of pancreatitis, followed by nearly complete regeneration within 96 hours.23 The mechanisms of this regeneration are unclear. It is still controversial whether in pancreatitis acinar regeneration occurs from differentiated acinar cells,24,25 or from other cell types.5,7,24
Our first aim was to determine whether exocrine regeneration occurs through transdifferentiation between acinar, centroacinar and ductal lineages, or by division of differentiated cells. Similar to the model used by Dor et al. for β-cell renewal,22 regeneration of acinar cells through division of terminally differentiated acinar cells would result in a stable percentage of tagged cells. Regeneration of acinar cells from any other (non-labeled) source would result in a progressive drop in the percentage of tagged acinar cells. Conversely, a contribution of transdifferentiated acinar cells to centroacinar and ductal renewal would result in HPAP expression in these lineages.
In Elastase-CreER™;Z/AP mice no drop in the percentage of tagged acinar cells in response to pancreatitis was observed regardless of the experiment length (Fig.2A,C,D). Furthermore, there was no drop in HPAP expression over time in pancreata of healthy control animals harvested two or eight weeks after tamoxifen exposure (data not shown). HPAP expression was not detected in ductal or centroacinar cells (Fig.2C,D).
The stable percentage of tagged cells suggests that newly formed acinar cells originate from pre-existing terminally differentiated acinar cells. Whereas cerulein pancreatitis mainly induces acinar injury, both ductal and centroacinar cells also display enhanced proliferation (Suppl.Fig.1). The absence of HPAP in these lineages in pancreatitis demonstrates that these compartments are not renewed by transdifferentiation of acinar cells.
To estimate whether enough new acinar cells are formed in this model to see a drop in tagging efficiency in the case of renewal from non-labeled sources, we exposed animals to BrdU. Sensitivity of this model was assessed by BrdU exposure, which revealed incorporation in 35% of acinar cells within 72 hours (Fig.2B). Extrapolation suggests that the acinar cell pool is renewed several times in the course of 6w and 10wCP, indicating that a significant contribution of cells other than acinar-to-acinar regeneration would have been detected. BrdU incorporation during a 2-hour exposure was used to assess proliferation. The normal pancreas has a low cell turnover of acinar cells. Proliferation is significantly increased in pancreatitis. BrdU is frequently incorporated in differentiated acinar cells (Fig.2E), centroacinar and, to a lesser extent, ductal cells (data not shown). Expression of Ki-67 mirrored these observations (Suppl.Fig.1). Thus acinar cells can enter the cell cycle in their differentiated state.
These data suggest in pancreatitis new acinar cells originate from terminally differentiated acinar cells without significant contribution of unlabeled sources.
Lineage Tracing of Metaplastic Ductal Lesions
Presently, there is no standard classification comprising all types of MDL. The terms TC, mucinous metaplasia, PanIN, and other descriptors have been used without uniformity in previous publications. The cerulein model results in three distinct types of MDL, mirroring human disease21: Type 1 tubular complexes (TC1) are lesions with a wide lumen lined by a monolayer of a few (typically 4-5) very flat, wide cells (Suppl.Fig.2A,B). Type 2 tubular complexes (TC2) are lined by many small cells and arranged in groups with branchings and anastomoses (Suppl.Fig.2C,D). Mucinous metaplastic lesions (MML) are lined by cells of variable size and height that express PAS and Alcian blue-positive mucins (Suppl.Fig.2E-H). MML with abundant supranuclear mucin have flat, basally located nuclei, features of early mPanIN.11 MML can be divided into lesions with a pure ductal phenotype (Suppl.Fig.2E,F), and mixed lesions including cells of acinar morphology (Suppl.Fig.2G,H). These mixed lesions can be found in human and mouse and contribute to the heterogeneity of PanIN.26 Thus mixed MML appear of special interest in regard to acinar-to-ductal transdifferentiation. These different types of MDL were analyzed separately.
TC1 Are Acinar Cell-Derived, but Do Not Acquire a Ductal Fate
TC1 were consistently found and analyzed in AP and 3wCP animals. Cells within TC1, which histologically resembled acinar cells, were HPAP-labeled with a frequency of 45%, a percentage expected for acinar progeny (Fig.3A,B). TC1 with HPAP expression can also contain cells that do not express HPAP (Fig.3B). This observation suggests that TC1 are derived in part from differentiated acinar cells and in part from non-labeled cells.
Figure 3.
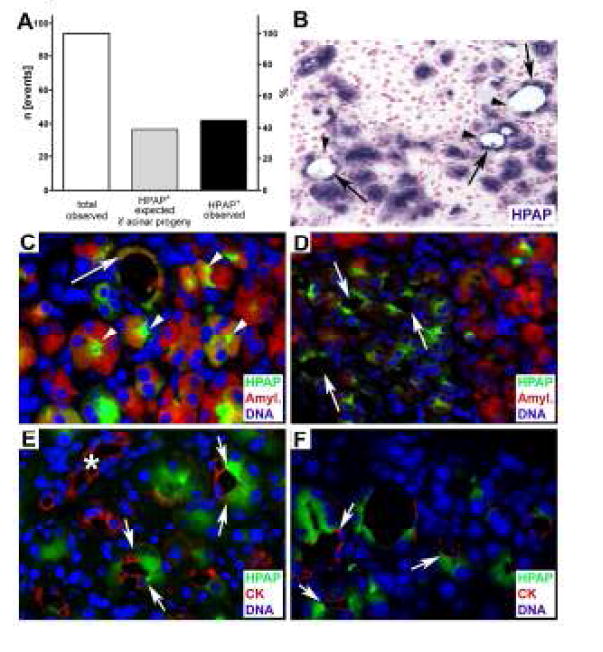
TC1 are acinar cell-derived. (A) Quantitative analysis of lineage tracing. Expected and observed numbers of HPAP+ events. (B) Normal acinar cells and TC1 (arrows) are labeled with HPAP (blue). Unlabeled duct-like cells in TC1 (arrowheads). (C,D) TC1 express both HPAP (green) and amylase (red) (arrow in C) like acini (arrowheads). With loss of cytoplasm, amylase expression is downregulated but HPAP persists (arrows in D). (E,F) Double staining for HPAP (green) and CK (red) reveals that TC1 contain cells that are, like terminal ducts (star in E), strongly CK+ but never tagged with HPAP. A clear border (arrows) between CK+ cells and acinar-derived cells is maintained both in lesions with beginning luminar dilation (E) and with wide lumen (F).
In previous studies, loss of acinar enzymes and identification of ductal markers in these lesions has been interpreted as a sign of transdifferentiation.7,24 Cytokeratins (CK) are expressed in the entire ductal system of the pancreas, whereas acinar cells are usually CK-negative. To determine whether TC1 may represent acinar cells undergoing transdifferentiation, double immunofluorescence for HPAP and amylase or CK was performed. Amylase is co-expressed in the majority of HPAP+ cells (Fig.3C). Flat HPAP+ cells that have lost amylase expression can occasionally be identified (Fig.3D). However, HPAP is not expressed in CK+ cells of ductal morphology. Instead, a clear border between CK+ and HPAP+ cells can be detected in lesions with incipient widening of the lumen (Fig.3E), and is maintained in lesions with dilated lumen (Fig.3F).
Thus, TC1 are derived from acini. The CK+ duct-like cells within TC1 are not derived from acinar cells and most likely represent adjacent cells of the terminal ductal compartment. Depending on the plane of section, TC1 may be composed entirely of acinar-derived cells or may contain cells of the terminal ductal compartment, which may result in co-expression of acinar and ductal markers within TC1 without transdifferentiation.
TC2 Are Not of Acinar Origin
The lesions defined as TC2 were found occasionally in AP and frequently in animals with 3wCP. Lineage tracing revealed no HPAP+ duct-like cells in fields of TC2, suggesting that TC2 are not a result of acinar-to-ductal transdifferentiation (Fig.4A,B). If these lesions were derived from acinar cells, given a tagging efficiency of 40% we would expect about 395.1 of the total of 1013 observed lesions to be tagged with HPAP+. In contrast to cells of ductal morphology, HPAP expression was detected in acinar cells within fields of TC2 (Fig.4B). This HPAP expression disappeared with progressive loss of acinar cells. These data suggest a specific loss of acinar cells and persistence of ducts in the formation of TC2.
Figure 4.
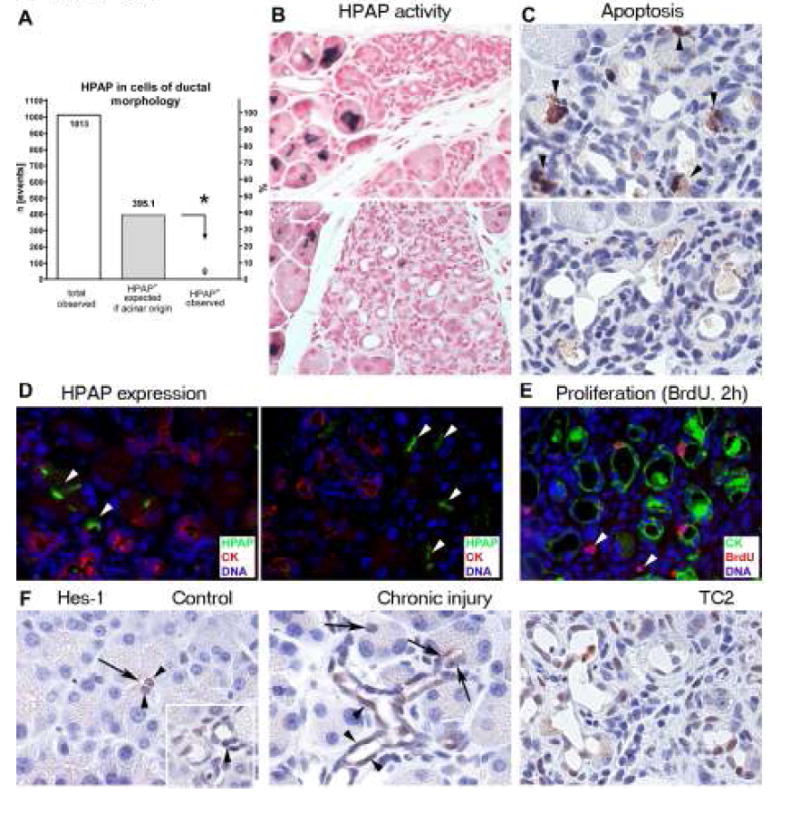
TC2 are not of acinar origin. (A) Quantitative analysis of lineage tracing. Comparison (*) of the number of expected HPAP+ events assuming acinar origin and the number of actually observed HPAP+ cells of ductal morphology. (B) HPAP activity (blue) is detected in acinar but not in duct-like cells within fields of TC and disappears with progressive loss of acinar cells. (C) The apoptosis marker cleaved caspase-3 (brown) is identified in acinar cells (arrowheads), but not in duct-like cells within fields of TC2. Fields without acinar cells are devoid of cleaved caspase-3 expression. (D) The epithelial lining of TC2 is not tagged with HPAP (green), but expresses the ductal marker CK (red). Note tagging of adjacent acinar cells (arrowheads). (E) Proliferation (BrdU-red) is frequently identified in CK+ (green) duct-like cells and occasionally in the stroma (arrowheads). (F) Hes-1 expression (brown). In controls, Hes-1 is identified in centroacinar cells (arrowheads), located near the acinar lumen (arrow) and in ductal cells (arrowhead, inset). In chronic injury, Hes-1 is almost always identified in centroacinar cells (arrows). Terminal ductal cells frequently express Hes-1 (arrowheads). TC2 mirror this expression pattern. Acini remain consistently negative for Hes-1.
To test this hypothesis, apoptosis was assessed by detection of cleaved caspase-3. Within fields of TC2, apoptosis was selectively identified in acinar, but not ductal cells (Fig.4C). Fields without acinar cells were devoid of apoptotic activity.
Immunofluorescence for HPAP, CK, and BrdU was used to better characterize the existing duct-like cells. In contrast to adjacent acini, ductal cells within TC2 were not tagged with HPAP but expressed the ductal marker CK (Fig.4D). Double staining for BrdU (2 hours exposure) and CK demonstrates that proliferation is a frequent event in the epithelial lining of TC2 (Fig.4E). These observations indicate that TC2 represent duct cells that persist and proliferate after a selective loss of acinar cells, and perhaps represent terminal ductal and/or centroacinar cells.
To better define the possible contribution of terminal ductal and centroacinar cells to TC2, expression of Hes-1 was assessed (Fig.4F). Hes-1, a transcription factor in the Notch pathway, has been described as a marker for centroacinar cells and is thought to play a role in metaplasia.16,19 Consistent with previous reports, Hes-1 was regularly identified in centroacinar cells, but also in ductal cells in the normal pancreas.19 In animals with CP, Hes-1 was regularly found in centroacinar cells and had a mosaic pattern in ductal cells. Hes-1 was neither identified in acinar cells nor acinar-derived cells within TC1. It is expressed in most but not all duct-like cells within TC2 and mirrors the expression pattern of terminal ducts plus centroacinar cells. Although not conclusive, this expression pattern supports a terminal ductal and/or centroacinar origin of TC2.
Thus, TC2 are not of acinar origin, but appear to be formed by the persistence and proliferation of preexisting terminal ductal and/or centroacinar cells after acinar cell loss.
Acinar-to-Ductal Transdifferentiation Does Occur in Vivo but Accounts for Only a Minority of MML
A large number of the MML observed in this study contained acinar cells. This morphology makes these lesions a target of special interest in a study on the contribution of acinar-to-ductal transdifferentiation to metaplasia.
HPAP expression was analyzed separately for mixed MML containing acinar cells and lesions of pure ductal morphology in animals with 6wCP and 10wCP. Of 271 MML observed, 190 (70.1%) were of mixed morphology. In mixed lesions, HPAP expression was identified at a frequency similar to that expected for acinar origin (Fig.5A,B). In the majority of these lesions HPAP expression was found in cells exhibiting acinar morphology but not in Alcian-blue+ duct-like cells (Fig.5B). Double immunofluorescence revealed that the majority of HPAP+ cells colocalized with amylase but not CK or gastric mucins. The majority of cells of ductal morphology which express CK and gastric mucins were not tagged, indicating that mixed MML contain acinar cells but that the majority of the MML are not of acinar origin (Fig.5B; Suppl.Fig.3A-F). However, 11 (5.8%) mixed lesions contain cells of ductal morphology labeled with HPAP (Fig.5A,C). Serial sections were evaluated by double immunofluorescence to confirm acinar-to-ductal transdifferentiation (Fig.5C, Suppl.Fig.3G-L): These lesions were composed of acinar cells expressing amylase and HPAP, and of HPAP+ cells which had lost amylase expression and exhibited expression of CK, gastric mucins, or both markers, indicating transdifferentiation.
Figure 5.

Acinar-to-ductal transdifferentiation accounts for a minority of MML. Separate analysis for lesions of mixed (A-C) and pure ductal phenotype (D-F). In most lesions mucinous metaplastic cells are not tagged with HPAP (B,E); In a minority of mixed and pure lesions HPAP+ ductal cells can be identified (C,F). Mixed phenotype: (A) Quantitative analysis reveals that HPAP is identified in cells of acinar morphology with a frequency similar to that expected for acinar origin. A minority (5.8%) of mixed lesions additionally contain HPAP+ mucinous cells of ductal morphology. (B) Mixed lesions, in which mucinous duct-like cells are not of acinar origin. HPAP/Alcian blue stains show that only acinar cells are tagged (blue), whereas mucinous metaplastic cells (turquoise) are untagged. Note that the lesion has formed in the terminal ductal to acinar region, whereas the draining duct (arrows) exhibits normal morphology. Serial double immunofluorescence identifies HPAP (green) in amylase+ cells (red, upper panel), but not in mucinous metaplastic cells, which are positive for both CK (red, mid panel) and gastric mucins (red, lower panel). Secreted amylase can be identified at the luminar membrane of the metaplastic ductal lining (arrowheads). (C) Mixed lesions with evidence of acinar-to-ductal transdifferentiation. Serial stains show expression of both HPAP (blue) and Alcian blue-positive mucins (turquoise) in cells of ductal morphology. Serial double immunofluorescence shows co-expression of HPAP (green) and amylase (red) in acinar cells (arrowhead), and confirm HPAP expression in duct-like cells without amylase expression (arrows, upper panel). These HPAP+ cells instead express both CK (red, middle panel) and gastric mucins (red, lower panel), suggesting transdifferentiation. Pure ductal phenotype: (D) In lesions of pure ductal phenotype HPAP+ duct-like cells are observed with a frequency (4.9%) similar to mixed lesions. (E) Most lesions (turquoise) are HPAP-. Double immunofluorescence confirms expression of HPAP (green, upper panel) in acini, but absence of HPAP in metaplastic lesions, which instead express gastric mucins (red, middle panel). There is no overlap between HPAP and mucin expression (lower panel). (F) Pure ductal lesions with evidence of acinar-to-ductal transdifferentiation: Serial stains show expression of both HPAP (blue) and Alcian blue-positive mucins (turquoise) in cells of ductal morphology. Double immunofluorescence of the same lesion confirms co-expression of HPAP (green) and the ductal marker CK (red).
Pure ductal morphology was observed in 81 lesions. Whereas the vast majority of these lesions were not tagged (Fig.5D,E) HPAP expression was identified in 4 (4.9%) lesions with pure ductal phenotype (Fig.5D,F). These lesions expressed both HPAP and ductal markers (Fig.5F), indicating that they were the result of true acinar-to-ductal transdifferentiation.
However, the lineage tracing results also indicate that the majority of MML are not of acinar origin. The consistency of observations in mixed and pure ductal lesions suggests that they represent one and the same type of lesion and appear as mixed or pure phenotype depending on the plane of section.
Of all metaplastic lesions observed, only MML exhibit morphologic similarities to PanIN.11 These lesions express Alcian blue+ mucins, including gastric mucins (Fig.5B,C,E,F), characteristics of early cancer precursors. MML were further assessed for expression of Hes-1 and Sonic Hedgehog (Shh), developmental genes that are thought to play a role early in carcinogenesis.16,27 Whereas Hes-1 expression was identified with a similar pattern in both MML (Fig.6A) and TC2, Shh was regularly and selectively identified in MML (Fig.6B). Proliferation and apoptosis were occasionally identified in these lesions (Fig.6C-E). However, progression to lesions with higher atypia according to the PanIN system, or to invasive neoplasia, was not observed. Acinar-derived and non-acinar-derived MML show no differences in morphology or in any of the molecular features tested.
Figure 6.

(A) MML exhibit a mosaic expression pattern of Hes-1 (brown). (B) Expression of Shh (brown) is frequently identified in MML. (C) Ki-67 expression (brown) and (D) incorporation of BrdU (green) demonstrate proliferative activity. (E) Apoptosis-cleaved caspase-3 (brown) is infrequently identified in MML.
Clearly, the majority of MML that form in response to chronic inflammation are not derived from acinar cells. However, acinar-to-ductal transdifferentiation infrequently occurs and contributes to a minority of MML in chronic injury.
Controls for False Negative and False Positive Lineage Tracing Results
To exclude the possibility of false negative results from epigenetic inactivation of the Z/AP reporter, additional bigenic animals with 6wCP were stained for LacZ activity. HPAP- cells with an intact reporter are expected to be LacZ+. In these animals TC2 (Suppl.Fig.4A) and MML (Suppl.Fig.4B) were LacZ+, indicating a functional reporter in these lesions. Although these results do not exclude inactivation of the Z/AP reporter in single lesions, they rule out reporter dysfunction as a mechanism that systematically produces false negative lineage tracing results.
Second, aberrant expression of HPAP or endogenous AP could result in false positive results. (i) To investigate whether aberrantly expressed endogenous AP may result in false positive, we assessed AP activity in pancreata of non-transgenic littermates with 6wCP. In these animals AP activity was never identified, ruling out endogenous AP as a source of false positive results (data not shown). (ii) To determine whether inflammation may result in HPAP activation, 6wCP was induced in Elastase-CreER™;Z/AP mice that had not been exposed to tamoxifen (n=4). In these animals, HPAP activity could be detected infrequently (0.7%) in acini (Suppl.Fig.4C,D). In contrast, HPAP was not identified in centroacinar cells, islets, or any of the observed ductal structures (n=1366). None of the 43 MML observed in these animals exhibited HPAP expression (Suppl.Fig.4C,D). These observations indicate that in response to inflammation the HPAP transgene appears to be infrequently activated in acinar cells, but not in other cell types. The frequency of this inflammation-mediated HPAP activation in acini (0.7%) is much lower than the observed frequency of HPAP expression in MML in tamoxifen-injected animals (6%). Moreover, inflammation-mediated HPAP activation was never observed in ducts or metaplastic lesions. Thus HPAP expression due to inflammation cannot account for the observed HPAP+ events that we thought indicated acinar-to-ductal transdifferentiation.
Discussion
Here we investigate by in vivo genetic lineage tracing the contribution of acinar cells to exocrine regeneration and to the formation of ductal metaplasia. Lineage tracing reveals that acinar-to-ductal metaplasia does not contribute to regeneration of normal ductal tissue, and that new acinar cells are generated from preexisting acinar cells. Three morphologically distinct types of MDL are observed. TC1 are derived from acini, but do not acquire a ductal fate. TC2 are composed of duct-like cells that are not derived from acinar cells. The majority of MML are not derived from acinar cells. However, acinar-to-ductal transdifferentiation was observed in a minority of MML. This study definitively provides direct evidence of acinar-to-ductal transdifferentiation in vivo.
Transdifferentiation in Exocrine Regeneration
The adult pancreas maintains a considerable regenerative capacity. However, the source of regeneration in different models of pancreatic injury is still unclear. Suggested mechanisms include precursor cells, transdifferentiation and a sequence of acinar de-differentiation, reactivation of developmental programs, and re-differentiation.25 It was recently shown by lineage tracing that acinar cells are renewed from preexisting acinar cells during regeneration following partial pancreatectomy.28 However, the authors did not explore acinar cell regeneration in response to inflammatory injury. Our observation of a stable percentage of acinar tagging, even after weeks of inflammatory injury, provides evidence that, similar to partial pancreatectomy,28 new acinar cells are generated from preexisting acinar cells rather than other lineages in the setting of inflammation. Both Ki-67 expression and BrdU incorporation in acinar cells expressing amylase suggests that acinar cells can enter the cell cycle in a differentiated state. Unlabeled cells, including centroacinar, ductal, hypothetical progenitor cells, and TC2, are ruled out as a significant source of acinar renewal. This tagging method may not be sensitive enough to exclude a low frequency of (trans-) differentiation in acinar regeneration. However, the assay is highly sensitive in detecting transdifferentiation of acinar cells into other lineages that undergo injury (Supplemental Data). The absence of HPAP expression in the other exocrine lineages shows that acinar-to-ductal or acinar-to-centroacinar transdifferentiation does not contribute significantly to exocrine regeneration.
Acinar-to-Ductal Transdifferentiation in the Formation of Metaplasia
Our study provides clear evidence that the majority of MDL that form in response to chronic pancreatic injury, including MML, are not of acinar origin. Scattered acinar cells found in MML are not a sign of acinar-to-ductal transdifferentiation. In contrast, lineage tracing provides clear evidence that acinar cells in the adult pancreas have the potential to transdifferentiate into metaplastic ductal cells: a minority of MML observed originated from acinar-to-ductal transdifferentiation. These events had a low frequency; approximately 5-6% of MML were tagged as acinar progeny. Using a rough and simplified model in which a lesion originates from a single cell, the tagging efficiency of 40% would suggest that about 12% of lesions were of acinar origin. Acinar-to-ductal transdifferentiation does occur in the formation of MML, but is clearly not the predominant mechanism in the cerulein model of chronic pancreatic injury. Guerra et al. recently reported that targeted expression of endogenous K-RASG12V in acinar/centroacinar cells results in inflammation-promoted PanIN and cancer, suggesting an acinar/centroacinar origin.29 The present demonstration of acinar-to-ductal transdifferentiation in cerulein pancreatitis may have important implications for such studies targeting mutations specifically to acinar cells,10,29-31 where it may occur more frequently and represent a main source of metaplasia. However, the ultimate question remains: What is the origin of metaplastic lesions in human disease? Compared to transgenic models, which target single mutations to specific cell lineages, the cerulein model results in a broader insult to the exocrine pancreas and may more closely recapitulate early events in human disease.
Centroacinar or Ductal Origin?
For TC2 and the majority of MML, the cell lineage tracing results exclude acinar cells as the cells of origin. Instead, our results suggest that both of these lesions may be derived from centroacinar and/or ductal cells. It has recently been proposed that centroacinar cells may play the central role in the formation of ductal metaplasia and neoplasia.19 In our study Hes-1, which is thought to play an important role in metaplasia and tumorigenesis,16 was identified consistently in centroacinar cells, had a mosaic pattern in terminal ductal cells, but was never detected in acinar cells. Both TC2 and MML exhibit a mosaic expression of Hes-1. One possible interpretation is that both centroacinar and ductal cells contribute to the formation of these lesions.
Implications for Neoplasia
It has been suggested that flat lesions consistent with TC2 progress to more atypical lesions and to cancer.8,10 Over the past decade the focus has been shifting from such flat epithelial lesions to MML (PanIN) as cancer precursors.3 In this study MML developed independently from TC2, in part through acinar-to-ductal transdifferentiation. MML exhibit several morphologic and molecular characteristics of mPanIN, murine cancer precursors recently defined in a consensus conference.11 However, one of the criteria of mPanIN was that lesions are “confined to native pancreatic ducts.”11 Since we show that part of the MML originate through acinar-to-ductal transdifferentiation, we refrained from calling them mPanIN and have used the descriptive term “MML” instead. In this study a progression of MML to lesions consistent with high-grade PanIN or to invasive cancer was not observed. The model used here closely recapitulates early events in chronic injury and results in metaplastic lesions similar to early cancer precursors. The contribution of acinar-to-ductal transdifferentiation to invasive cancer, however, remains to be determined.
Supplementary Material
Acknowledgments
This work was supported by the German Research Foundation and the Surgery Foundation Heidelberg, Lautenschläger Scholarship (to O.S.), the Juvenile Diabetes Research Foundation (2-2005-171), the Israel Science Foundation, the National Institutes of Health Beta Cell Biology Consortium, and the Barbara S. Goodman Career Development Award from the Israel Cancer Research Fund (to Y.D.), and NIH grant DK071329, the American College of Surgeons Clowes Award, and the Lustgarten Foundation (to S.P.T.).
The authors thank Douglas Melton for the Ela-CreER™ and Z/AP mouse strains, Samuel Ho for the anti-mucin antibody, and Tetsuo Sudo for the anti-Hes-1 antibody.
Abbreviations
- MDL
Metaplastic ductal lesions
- MML
Mucinous metaplastic lesions
- TC
Tubular complexes
- PanIN
Pancreatic Intraepithelial Neoplasia
- AP
Acute pancreatitis
- CP
Chronic pancreatitis
Footnotes
For all authors, there is no conflict of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tosh D, Slack JM. How cells change their phenotype. Nat Rev Mol Cell Biol. 2002;3:187–194. doi: 10.1038/nrm761. [DOI] [PubMed] [Google Scholar]
- 2.Parsa I, Longnecker DS, Scarpelli DG, Pour P, Reddy JK, Lefkowitz M. Ductal metaplasia of human exocrine pancreas and its association with carcinoma. Cancer Res. 1985;45:1285–1290. [PubMed] [Google Scholar]
- 3.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Kloppel G, Longnecker DS, Luttges J, Offerhaus GJ. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Willemer S, Adler G. Histochemical and ultrastructural characteristics of tubular complexes in human acute pancreatitis. Dig Dis Sci. 1989;34:46–55. doi: 10.1007/BF01536153. [DOI] [PubMed] [Google Scholar]
- 5.Lechene dIP, Iovanna J, Odaira C, Choux R, Sarles H, Berger Z. Involvement of tubular complexes in pancreatic regeneration after acute necrohemorrhagic pancreatitis. Pancreas. 1991;6:298–306. doi: 10.1097/00006676-199105000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Bockman DE, Boydston WR, Anderson MC. Origin of tubular complexes in human chronic pancreatitis. Am J Surg. 1982;144:243–249. doi: 10.1016/0002-9610(82)90518-9. [DOI] [PubMed] [Google Scholar]
- 7.Tokoro T, Tezel E, Nagasaka T, Kaneko T, Nakao A. Differentiation of acinar cells into acinoductular cells in regenerating rat pancreas. Pancreatology. 2003;3:487–496. doi: 10.1159/000075580. [DOI] [PubMed] [Google Scholar]
- 8.Bockman DE, Guo J, Buchler P, Muller MW, Bergmann F, Friess H. Origin and development of the precursor lesions in experimental pancreatic cancer in rats. Lab Invest. 2003;83:853–859. doi: 10.1097/01.lab.0000074918.31303.5a. [DOI] [PubMed] [Google Scholar]
- 9.Longnecker DS, Curphey TJ, Kuhlmann ET, Schaeffer BK. Experimental induction of pancreatic carcinomas in the hamster with N delta-(N-methyl-N-nitrosocarbamoyl)-L-ornithine. J Natl Cancer Inst. 1983;71:1327–1336. [PubMed] [Google Scholar]
- 10.Wagner M, Luhrs H, Kloppel G, Adler G, Schmid RM. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology. 1998;115:1254–1262. doi: 10.1016/s0016-5085(98)70098-8. [DOI] [PubMed] [Google Scholar]
- 11.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein A, Klimstra DS, Kloppel G, Lauwers GY, Longnecker DS, Luttges J, Maitra A, Offerhaus GJ, Perez-Gallego L, Redston M, Tuveson DA. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106. doi: 10.1158/0008-5472.CAN-05-2168. [DOI] [PubMed] [Google Scholar]
- 12.Pour PM, Weide L, Liu G, Kazakoff K, Scheetz M, Toshkov I, Ikematsu Y, Fienhold MA, Sanger W. Experimental evidence for the origin of ductal-type adenocarcinoma from the islets of Langerhans. Am J Pathol. 1997;150:2167–2180. [PMC free article] [PubMed] [Google Scholar]
- 13.Reid LE, Walker NI. Acinar cell apoptosis and the origin of tubular complexes in caerulein-induced pancreatitis. Int J Exp Pathol. 1999;80:205–215. doi: 10.1046/j.1365-2613.1999.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J Clin Invest. 2002;109:1437–1444. doi: 10.1172/JCI15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmid RM. Acinar-to-ductal metaplasia in pancreatic cancer development. J Clin Invest. 2002;109:1403–1404. doi: 10.1172/JCI15889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, Hruban RH, Ball DW, Schmid RM, Leach SD. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 17.Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ, Jr, Wright CV, Stoffers DA, Leach SD. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development. 2005;132:3767–3776. doi: 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 18.Wells WA. Is transdifferentiation in trouble? J Cell Biol. 2002;157:15–18. doi: 10.1083/jcb.200203037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanger BZ, Stiles B, Lauwers GY, Bardeesy N, Mendoza M, Wang Y, Greenwood A, Cheng KH, McLaughlin M, Brown D, Depinho RA, Wu H, Melton DA, Dor Y. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8:185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- 21.Strobel O, Dor Y, Stirman A, Trainor A, Fernandez-del Castillo C, Warshaw AL, Thayer SP. Beta cell transdifferentiation does not contribute to preneoplastic/metaplastic ductal lesions of the pancreas by genetic lineage tracing in vivo. Proc Natl Acad Sci U S A. 2007;104:4419–4424. doi: 10.1073/pnas.0605248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 23.Gomez G, Lee HM, He Q, Englander EW, Uchida T, Greeley GH., Jr Acute pancreatitis signals activation of apoptosis-associated and survival genes in mice. Exp Biol Med (Maywood) 2001;226:692–700. doi: 10.1177/153537020222600716. [DOI] [PubMed] [Google Scholar]
- 24.De Lisle RC, Grendell JH, Williams JA. Growing pancreatic acinar cells (postpancreatitis and fetal) express a ductal antigen. Pancreas. 1990;5:381–388. doi: 10.1097/00006676-199007000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Jensen JN, Cameron E, Garay MV, Starkey TW, Gianani R, Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171:263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007;117:971–977. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Sandgren EP, Quaife CJ, Paulovich AG, Palmiter RD, Brinster RL. Pancreatic tumor pathogenesis reflects the causative genetic lesion. Proc Natl Acad Sci U S A. 1991;88:93–97. doi: 10.1073/pnas.88.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–2019. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.