

Abstract
Peroxynitrite is a potent oxidant and nitrating species proposed as a direct effector of myocardial damage in a wide range of cardiac diseases. Whether peroxynitrite also acts indirectly, by modulating cell signal transduction pathways in the myocardium, has not been investigated. Here, we examined the ability of peroxynitrite to activate extracellular signal-related kinase (ERK), a MAP kinase which has been linked with hypertrophic and anti-apoptotic responses in the heart, in cultured H9C2 cardiomyocytes. Peroxynitrite elicited a concentration-and time-dependent activation of ERK, secondary to the upstream activation of MEK 1 (ERK kinase). Activation of MEK-ERK by peroxynitrite was related to the upstream activation of Raf-1 kinase, as ERK and MEK phosphorylation were prevented by the Raf-1 inhibitor BAY43–9006. These effects of peroxynitrite were not associated with the activation of p21Ras, known as a common signaling target of cellular oxidative stress. In contrast to ERK activation mediated by the epidermal growth factor (EGF), ERK activation by peroxynitrite was not prevented by AG1478 (EGF receptor inhibitor). Peroxynitrite acted through oxidative, but not nitrative chemistry, as ERK remained activated while nitration was prevented by the flavanol epicatechin. In addition to ERK, peroxynitrite also potently activated two additional members of the MAP kinase family of signaling proteins, JNK and p38. Thus, peroxynitrite activates ERK in cardiomyocytes through an unusual signaling cascade involving Raf-1 and MEK 1, independently from EGFR and P21Ras, and also acts as a potent activator of JNK and p38. These results provide the novel concept that peroxynitrite may represent a previously unrecognized signaling molecule in various cardiac pathologies.
Keywords: Peroxynitrite, Extracellular signal-regulated kinase, Cardiomyocyte, Cell signaling, MAP kinase, Nitration, Oxidation, C-Jun NH2-terminal kinase (JNK), P38 MAP kinase
1. Introduction
Mitogen-activated protein kinases (MAPKs) are a family of signaling proteins linking extracellular stimuli to various cellular responses. Three major classes of MAPKs have been described, including p38 MAP kinase, c-Jun NH2-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK), which exists as two major isoforms, ERK 1 and ERK 2 (p44 and p42 MAP kinase) [1]. While p38 and JNK are activated by environmental stress, ERK is involved in the signaling pathways triggered by growth factors, such as the epidermal growth factor (EGF), via the activation of EGF receptor (EGFR), Raf-1 kinase and MEK 1 [2]. ERK activation has recently emerged as an important mechanism involved in various cardiac pathologies, including ischemia–reperfusion, anthracycline cardiotoxicity and severe heart failure [3], where ERK has been shown to promote cardiac hypertrophy and to reduce cardiomyocyte apoptosis [4,5]. The stimuli responsible for myocardial ERK activation in these conditions are only poorly defined, and the signaling pathway leading to ERK activation in the myocardium is far from being elucidated [6]. Further studies are therefore needed to define potential mechanisms of ERK activation in the heart [3].
Peroxynitrite (ONOO−), a strong oxidant and nitrating species formed by the near-diffusion-limited reaction between superoxide (O2•−) and nitric oxide (NO•) [7], has been recently found to be a key effector of myocardial damage and dysfunction in several conditions, most notably myocardial infarction [8], anthracycline cardiomyopathy [9] and chronic heart failure [10]. ONOO− is generally considered to act as a directly biotoxic molecule, but indirect effects, related to the modulation of redox sensitive cell signaling pathways might also account for some of its biological actions in the heart. Since no association between ONOO− and ERK activation in the myocardium has been reported so far, we conducted the present study to detect a possible link between ONOO− and the ERK signaling pathway in H9C2 rat cardiomyocytes. H9C2 cells were used, as they are morphologically similar to embryonic cardiocytes and are recognized as a well suited model for the study of cardiomyocyte biology [11]. Due to potential limitations associated with the use of a clonal cell line, the effects of ONOO− on the ERK pathway were also addressed in primary ventricular cardiomyocytes isolated from mouse heart. In addition, we evaluated whether ONOO−might activate other members of the MAP kinase family, including p38 and JNK, which have been associated with pro-apoptotic responses in cardiomyocytes [6]. Our findings indicate that ONOO− acts via an unusual signaling pathway to induce ERK phosphorylation and activation, and also strongly activates the phosphorylation of p38 and JNK. These observations reveal a previously unrecognized role of ONOO− as a signaling molecule in cardiomyocytes.
2. Material and methods
2.1. Cell culture
H9C2 cells, a clonal line derived from rat heart (ATCC, Manassas, VA) were grown (5% CO2, 37 °C) in Dulbecco’s modified Eagles medium (Gibco BRL, Invitrogen, Basle, Switzerland) supplemented with 10% fetal calf serum. Cells were cultured to 80% confluence in 10 cm culture dishes or 6-well plates and were serum-starved 24 h before the experiments. Some experiments were also performed using primary ventricular mouse cardiomyocytes. Briefly, 8–10 weeks old C57BL/6 male mice were anesthetized with pentobarbital (80 mg kg−1). Hearts were removed and perfused (at 37 °C) for 5 min with modified Krebs–Henseleit bicarbonate (KHB) buffer (mM: NaCl 35, KCl 4.7, KH2PO4 1.2, Na2HPO4 16, NaHCO3 25, HEPES 10, glucose 10, sucrose 134, pH 7.4). The heart was then perfused for 12 min with KHB containing 0.1% (w/v) collagenase type II (Sigma). After perfusion, the left ventricle was minced and the cells further digested with collagenase before being filtered through a nylon mesh (300 μm). The cardiomyocytes were pelleted by 2 successive centrifugations (220 rpm, 1 min), and were used for the experiments described below immediately after isolation.
2.2. Stimulation with ONOO−
ONOO− was obtained as a 50 mM stock in 0.3 M NaOH (Upstate Biotechnology, Inc., Lake Placid, NY). All dilutions of the stock solution were made in the same concentration of NaOH. Prior to experimentation, ONOO− was quantitated spectrophotometrically (extinction coefficient at 302 nm = 1670 M−1 cm−1 [12]). For stimulation with ONOO−, cells were washed once and placed in phosphate-buffered saline (PBSgluc) (50 mM sodium phosphate, 90 mM NaCl , 5 mM KCl and 5 mM glucose, pH 7.4), to avoid reactions of ONOO− with media constituents [2,13,14]. ONOO− was delivered as a single bolus at a 1:100 dilution against one side of the dish, while rapidly swirling the medium to ensure optimal exposure of the cells to ONOO−. ONOO− was used at concentrations ranging from 50 to 500 μM, which agrees with previous studies evaluating the effects of ONOO− on various cell signaling pathways in vitro [2,13,15,16]. Such concentrations of ONOO− are physiologically relevant, as it has been estimated that the rate of peroxynitrite generation may reach up to 1 mM min−1 in an inflamed organ (lung) in vivo. [17]. Control experiments were made using a solution of decomposed ONOO−, made by leaving the solution of ONOO− overnight at room temperature [18], which contains similar amounts of nitrite, nitrate and NaOH as the ONOO− solution, but no ONOO−.
2.3. Treatment of cells
To address the role of the various signaling intermediates possibly involved in ERK activation by ONOO−, cells were treated with the following inhibitors before ONOO− stimulation: AG1478, a specific inhibitor of EGFR tyrosine kinase (100 nM, Calbiochem, San Diego, CA), BAY 43–9006, a Raf-1 kinase inhibitor (5 μM, Calbiochem) [19], and PD98059, a specific MEK 1 inhibitor (50 μM, Calbiochem). All inhibitors were added to the cells 1 h before ONOO− stimulation and remained in the medium until the end of the experiments. In additional experiments, cells were pretreated with the farnesyl transferase inhibitor FPTII (Calbiochem, 20 μM, 24 h pretreatment), a potent and selective inhibitor of Ras GTPase [20]. Also, to evaluate the potential role of tyrosine nitration in the effects of ONOO−, cells were pretreated for 1 h with the flavanol (−)-epicatechin (Sigma), a specific inhibitor of tyrosine nitration [15], at concentrations ranging from 50 to 250 μM.
2.4. Western blot analysis
After ONOO− stimulation, cells were scraped in lysis buffer (Tris–HCl 10 mM, NP40 0.5%, NaCl 0.15 M, Na3VO4 1 mM, NaF 10 mM, PMSF 1 mM, EDTA 1 mM, aprotinin 10 μg ml−1, leupeptin 10 μg ml−1 and pepstatin 1 μg ml−1). Proteins (10–20 μg) were separated by SDS-PAGE, transferred to nitrocellulose membrane, and blocked for 1 h at room temperature with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20. The membrane was then incubated overnight at 4 °C with an appropriate dilution of anti-phospho-ERK 1/2 (p44 and p42), anti-phospho-MEK 1/2, anti-phospho JNK1/2 (p54 and p46), anti-phospho-p38, anti-ERK 1/2, anti-MEK 1/2, anti-JNK1, anti-p38 (all from Cell Signaling, Beverly, MA) or anti-nitrotyrosine (Upstate Biotechnology) primary antibody, followed by incubation for 1 h with a 1:5000 dilution of the appropriate horseradish peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA). The immunoblot signal was visualized using enhanced chemiluminescence (ECL,Amersham Biosciences, Otelfingen, Switzerland). Densitometric analysis of ECL autoradiographs were performed using a Personal Densitometer and TotalLab Software.
2.5. Immunoprecipitation and ERK 1/2 kinase assay
ERK 1/2 activity was measured using a non-radioactive p42/p44 MAP Kinase assay kit (Cell Signaling, Beverly, MA), according to the manufacturer’s instructions. Briefly, phospho-ERK 1/2 was immuno-precipitated from 200 μg of protein with an immobilized anti-phospho-ERK1/2 mAb at 4 °C overnight. The immunoprecipitate was resuspended in kinase buffer containing 25 mM Tris (pH 7.5), 5 mM β-glycerophosphate, 10 mM MgCl2, 0.1 mM Na3VO4, and 2 mM DTT. The kinase reaction was performed using 2 μg Elk-1 fusion protein (a specific substrate of phospho-ERK 1/2) and 0.2 mM ATP at 30 °C for 30 min. The phosphorylation of Elk-1 was then determined following immunoblotting, by using a primary polyclonal anti-phospho-Elk-1 antibody (1:1000) and chemiluminescence detection. Densitometric analysis was performed as above.
2.6. Ras activation assay
Ras activation was determined using a non-radioactive assay kit (Upstate, Lake Placid, NY, USA), as previously described [21]. Briefly, activated P21Ras from H9C2 lysates was precipitated (45 min, 4 °C) using a GST fusion protein corresponding to the Ras binding domain of Raf-1 (residues 1–149) conjugated to agarose beads. After extensive washing, the beads were boiled in 2× Laemmli buffer, and the eluted proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes, at 4 °C overnight, and visualized by using a goat anti-mouse secondary antibody conjugated to horseradish peroxidase (Bio-Rad) and a chemiluminescence detection system. Total p21 ras immunoblotting was used as an internal control. Densitometric analysis was performed as above. Positive and negative controls were obtained by loading cell extracts with either GTPcS (a non-hydrolyzable analog of GTP, positive control) or GDP (negative control).
2.7. Presentation of data and statistical analysis
Results of the densitometric analyses are shown as mean ± S.E.M. In the experiments comparing only two conditions, statistical analysis was performed using Student’s t-test, and a P value < 0.05 was considered significant. In all the other experiments, statistical analysis was performed using analysis of variance for repeated measurements, after logarithmic transformation of the densitometric data to stabilize variances. When the F value was significant, further pairwise comparisons were carried out with an adjustment for multiple comparison made using the Bonferroni procedure, as modified by Hochberg and Benjamini [22]. The alpha level of all tests was set at 0.05.
3. Results
3.1. Activation of ERK1/2 by ONOO−
Incubation of cells with 50 μM or more of ONOO− for 15 min resulted in a concentration-dependent increased phosphorylation of ERK1/2 (p44/p42, Fig. 1A), with no change in the level of non-phosphorylated ERK 1/2. At 500 μM, ONOO− (Fig. 1B) elicited a time-dependent ERK phosphorylation, starting at 2 min and reaching a maximum between 15 and 30 min. The phosphorylation of ERK by ONOO− was associated with a stimulated activity of ERK, as evidenced by an increased phosphorylation of the transcription factor Elk-1 in the ERK kinase assay experiment (Fig. 1c). ERK phosphorylation and activation were not observed in cells stimulated with decomposed ONOO− (DC). As illustrated in Fig. 2, stimulation with 500 μM ONOO− for 15 min also induced a significant phosphorylation of both p44 and p42 MAP kinase in primary mouse ventricular cardiomyocytes.
Fig. 1.
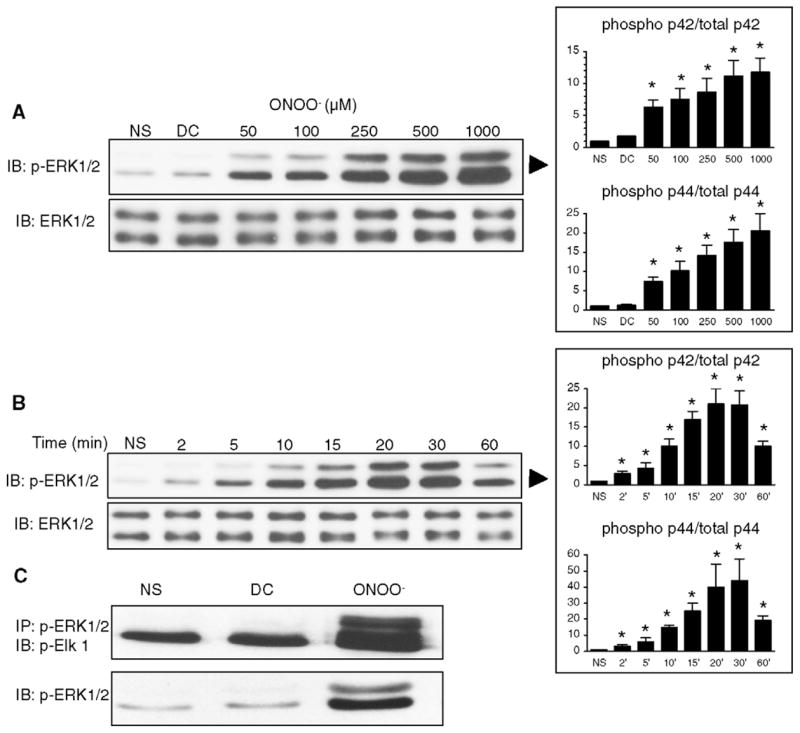
ONOO− induces the phosphorylation and the activity of ERK 1/2 in H9C2 cardiomyocytes. A. Cells were treated with increasing concentrations of ONOO− for 15 min. Phosphorylation of ERK 1/2 was induced at concentrations of ONOO− of 50 μM and above. Decomposed ONOO− (DC) did not phosphorylate ERK. Total ERK was comparable in all the conditions of stimulation. B. Cells were exposed to 500 μM ONOO− for 2–60 min. ERK phosphorylation was maximally induced at 20 min of ONOO− exposure. C. Cells were either not stimulated (NS) or activated with ONOO− (500 μM) or decomposed ONOO− (DC) for 15 min. ERK activity was assessed by Elk-1 phosphorylation after immunoprecipitation of phosphorylated ERK 1/2. As expected, ERK activity correlated with ERK phosphorylation. Densitometric analyses are shown as mean ± S.E.M. of at least four independent experiments. * P < 0.05 vs. NS.
Fig. 2.
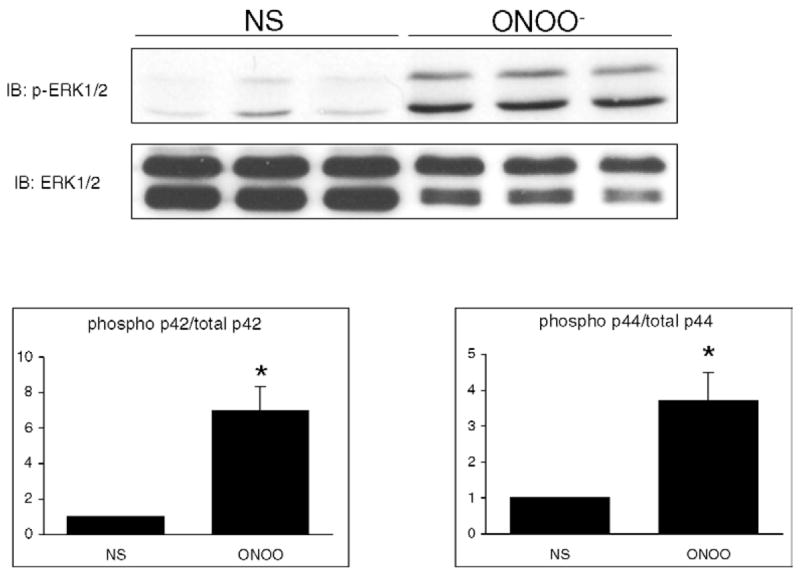
ONOO− induces the phosphorylation of ERK 1/2 in primary mouse ventricular cardiomyocytes. Primary cardiomyocytes were either NS or treated with 500 μM ONOO− for 15 min. ONOO− induced a significant phosphorylation of both p42 and p44 MAP kinase. Densitometric analyses are shown as mean ± S.E.M. of six independent experiments. * P < 0.05 vs. NS.
3.2. ERK 1/2 phosphorylation by ONOO− occurs via a MEK 1-dependent pathway
Treatment of cells with 500 μM ONOO− for 15 min resulted in a marked phosphorylation of MEK 1 (Fig. 3A) with no changes in the level of non-phosphorylated MEK. The MEK inhibitor PD98059 was used to investigate whether ERK phosphorylation induced by ONOO− depends on MEK 1 phosphorylation. As shown in Fig. 3B, treatment of cells with 50 μM PD98059 for 1 h resulted in a significant inhibition of ERK 1/2 phosphorylation. These data indicate that ERK phosphorylation by ONOO− depends completely on the upstream phosphorylation of MEK 1 by ONOO−.
Fig. 3.
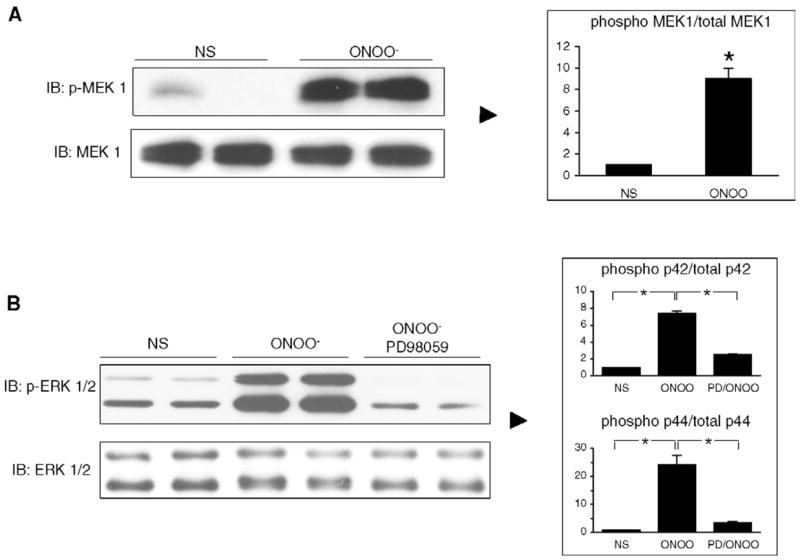
ONOO− mediates ERK activation via MEK phosphorylation. A. MEK phosphorylation induced in H9C2 cells exposed to ONOO− (500 μM) for 15 min. B. Cells were pretreated with the MEK inhibitor PD98059 for 1 h and then activated with ONOO− (500 μM) for 15 min. The phosphorylation of ERK 1/2 induced by ONOO− was significantly reduced by PD98059. NS: not stimulated. Densitometric analyses are shown as mean ± S.E.M. of at least three independent experiments. * P < 0.05.
3.3. Role of EGF receptor and Raf-1 kinase in the process of ERK 1/2 phosphorylation by ONOO−
The typical signaling cascade leading to ERK activation by growth factors involves the activation of growth factor receptors, Raf-1 kinase and MEK 1. This cascade was observed in H9C2 cells upon stimulation with EGF (0.1 μg ml−1): after 15 min, ERK 1/2 was phosphorylated, and this effect was abrogated when cells were pretreated withAG1478, an EGFR inhibitor, BAY 43–9006, a Raf-1 kinase inhibitor, or PD98059 (Fig. 4A). In cells treated with ONOO−, ERK phosphorylation was not influenced by AG1478 whereas it was prevented by BAY 43–9006 and significantly reduced by PD 98059 (Fig. 4B). Also, the phosphorylation of MEK 1 (Fig. 5A) and the kinase activity of ERK 1/2 (Fig. 5B) triggered by ONOO− remained unaffected in the presence of AG1478 but was abolished by BAY 43–9006. These data indicate that ONOO− triggers a signaling cascade involving Raf-1 kinase and MEK 1 to activate ERK, independently from EGFR activation.
Fig. 4.
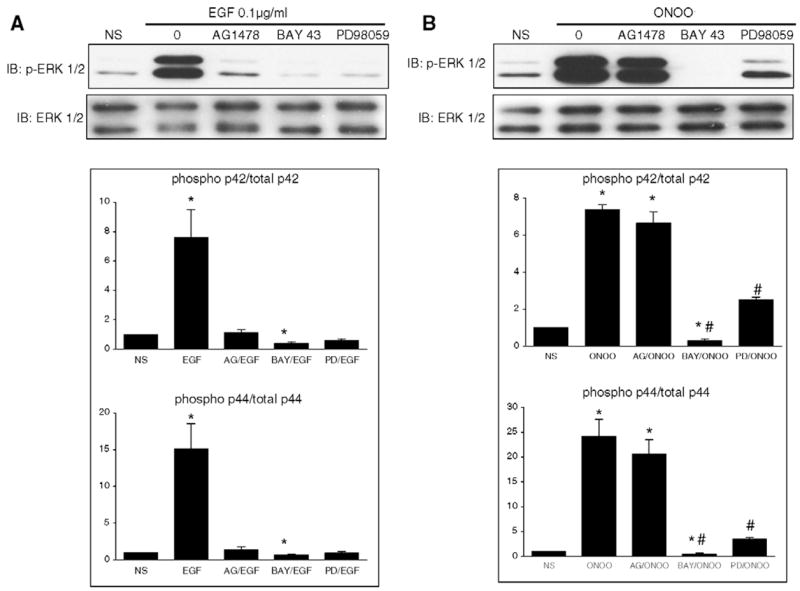
Distinct signaling pathways are involved in ERK phosphorylation elicited by EGF or ONOO−. A. H9C2 cells were NS or treated with 0.1 μg ml−1 EGF for 15 min in the absence or in the presence of AG1478 (100 nM), BAY 43–9006 (5 μM) or PD98059 (50 μM). The phosphorylation of ERK 1/2 (p44/p42) induced by EGF was prevented by AG1478, BAY 43–9006 or PD98059. B. Cells were NS or treated with ONOO− (500 μM, 15′) with or without inhibitors. The activation of ERK 1/2 (p44/p42) induced by ONOO− was not influenced by AG1478, but was prevented by BAY 43–9006 and PD98059. Densitometric analyses are shown as mean ± S.E.M. of at least three independent experiments. * P < 0.05 vs. NS. † P < 0.05 vs. EGF. # P < 0.05 vs. ONOO−.
Fig. 5.
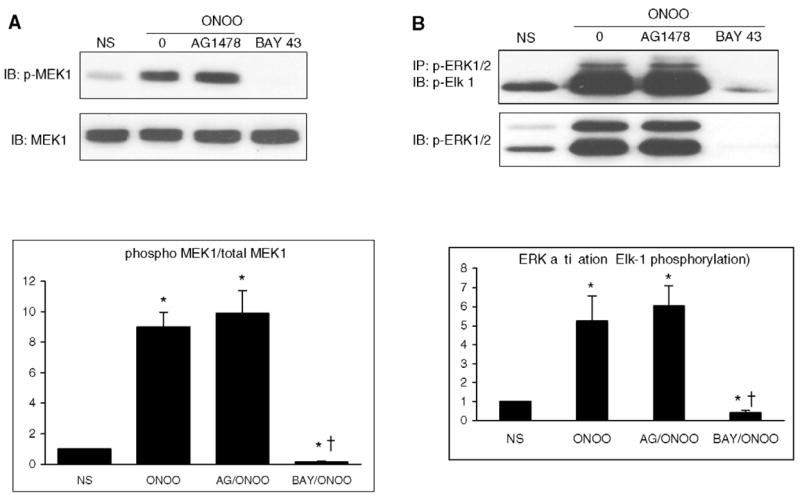
MEK phosphorylation and ERK 1/2 activity induced by ONOO− are not dependent on EGFR but depend on Raf-1 activation. Cells were either NS or were exposed to ONOO− (500 μM, 15 min) in the absence or in the presence of AG1478 (100 nM) or BAY 43–9006 (5 μM). MEK phosphorylation (A) and ERK activity (B) were not influenced by AG1478, whereas they were both suppressed in the presence of BAY 43–9006. Densitometric analyses are mean ± S.E.M. of at least three independent experiments. * P < 0.05 vs. NS. † P < 0.05 vs. ONOO−.
3.4. ONOO− does not induce the activation of p21Ras
P21Ras has been shown to act as a common signaling target of reactive free radicals and cellular redox stress and some investigators reported that NO and NO-related species could stimulate guanine nucleotide exchange on P21Ras [20,23,24]. Using a Ras activation assay, we found that P21Ras was activated constitutively in H9C2 cells (Fig. 6A). Stimulation with ONOO− did not activate further P21Ras at time-points ranging from 2 to 15 min. Furthermore, the activation of ERK by ONOO− was not inhibited by FPT II, a specific inhibitor of P21Ras (Fig. 6B). Thus, P21Ras does not represent a relevant intermediate in the pathway linking ONOO− stimulation to ERK activation in H9C2 cells.
Fig. 6.

ONOO− does not activate Ras in H9C2 cells. Cells were NS or exposed to decomposed ONOO− (DC, 5 min) or authentic ONOO− (500 μM, 2–15 min). Ras activation was assessed as described in Section 2. A. A baseline activation of Ras was noted in untreated cells, that was not further increased by ONOO−stimulation, whereas ERK 1/2 was phosphorylated in these conditions. B. The phosphorylation of ERK triggered by ONOO− for 15 min was not influenced by FPTII, a potent and selective inhibitor of Ras. C+, positive control; C– negative control. Densitometric analyses are mean ± S.E.M. of four independent experiments. * P < 0.05 vs. NS.
3.5. ONOO− activates ERK 1/2 independently of the nitration of tyrosine residues
As illustrated in Fig. 7, stimulation with 500 μM ONOO−for 15 min induced a widespread nitration of cellular proteins. Nitration was completely inhibited by epicatechin, at concentrations of 100 μM and above (Fig. 7A). However, pretreatment of cells with 100 μM epicatechin did not affect ERK phosphorylation, implying that the latter was not due to ONOO−-mediated tyrosine nitration (Fig. 7B, C).
Fig. 7.

ONOO− phosphorylates ERK 1/2 independently from tyrosine nitration. A. H9C2 cells were stimulated with 500 μM ONOO− for 15 min in the absence or in the presence of increasing concentrations of epicatechin (EC), an inhibitor of tyrosine nitration. ONOO− induced a widespread nitration of tyrosine residues, that was dose-dependently inhibited by epicatechin. B and C. Effects of epicatechin (100 μM) on the phosphorylation of ERK 1/2 by ONOO−. Cells were either NS or stimulated with 500 μM ONOO− (15 min) with or without epicatechin (100 μM) pretreatment (1 h). ERK 1/2 (p44/p42) phosphorylation was not influenced by epicatechin. Densitometric analyses are mean ± S.E.M. of four independent experiments. * P < 0.05 vs. NS.
3.6. ONOO− activates the phosphorylation of JNK and p38 in H9C2 cells
As illustrated in Fig. 8A. Incubation of H9C2 cells for 15 min with ONOO− activated the phosphorylation of JNK p46 and p54, an effect that was significant at all concentrations of the oxidant, whereas the levels of non-phosphorylated JNK1 were not influenced. We then examined the time-course of this effect by stimulating cells with 500 μM ONOO−for periods ranging from 2 to 60 min. As shown in Fig. 8B, JNK phosphorylation was significantly induced from 5 to 60 min (p54) and from 2 to 60 min (p46) after ONOO−. Similarly to JNK, ONOO− also significantly activated the phosphorylation of p38 MAP kinase at all time-points and all concentrations (50 to 1000 μM) studied, while it did not change the cellular levels of non-phosphorylated p38 (Fig. 9). Thus, ONOO− acts as a potent signaling molecule able to activate all members of the MAP kinase family in H9C2 cardiomyocytes.
Fig. 8.
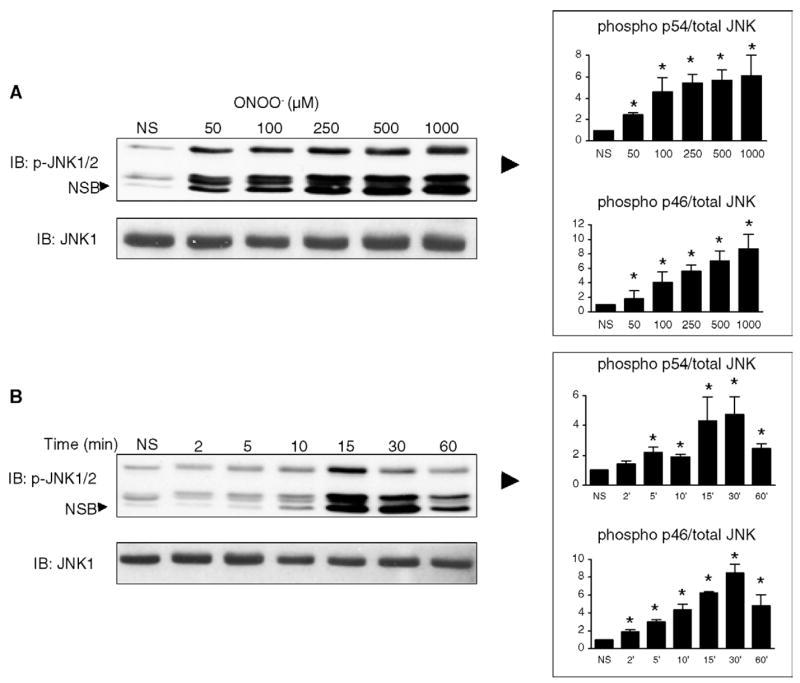
ONOO− activates the phosphorylation of JNK in rat H9C2 cardiomyocytes. A. Cells were either NS or treated with ONOO− at concentrations ranging from 50 to 1000 μM for 15 min. JNK activation was monitored by Western blotting using specific antibodies against phosphorylated JNK p46 and p54. Phosphorylation of JNK was triggered by ONOO− at concentrations of 100 μM and above, with a maximum at 1000 μM. Total JNK1 was not influenced by any of the conditions. B. Cells were NS or exposed to 500 μM ONOO− for 2 to 60 min. JNK phosphorylation started at 2 (p46) and 5 min (p54), peaked at 15–30 min and then steadily declined at 60 min. NSB: non-specific band. Densitometric analyses are shown as means of at least four independent experiments. * P < 0.05 vs. NS.
Fig. 9.
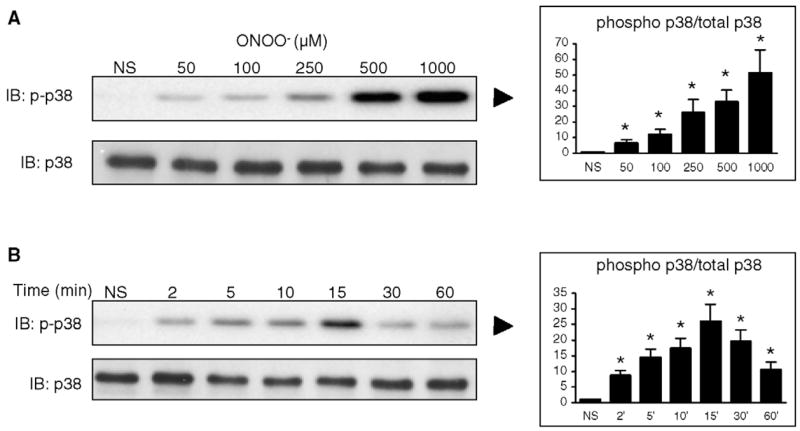
ONOO− activates p38 phosphorylation in H9C2 cells. A. Cells were either NS or treated with increasing concentrations of ONOO− and the level of phosphorlyated p38 was determined by western blot. ONOO− dose-dependently phosphorylated p38 in a range of concentrations from 50 to 1000 μM, whereas it did not change the cell content in total p38. B. Treatment of cells with 500 μM ONOO− activated p38 phosphorylation already after 2 min, with a peak at 15 min and a progressive decline up to 60 min, with no changes in total p38. Densitometric analyses are shown as means of at least four independent experiments. * P < 0.05 vs. NS.
4. Discussion
In the present study, we provide the novel demonstration that ONOO− acts as a potent inducer of ERK1/2 phosphorylation and ERK kinase activity in H9C2 cardiomyocytes (Fig. 1). ONOO− also induced the phosphorylation of ERK in primary mouse ventricular cardiomyocytes (Fig. 2), indicating that this effect was not an artifact related to the use of clonal cell line. We then dissected the cascade leading to ERK activation in these conditions and found an uncommon signaling mechanism triggered by ONOO−. In the typical pathway of ERK activation by growth factors such as the EGF, ERK1/2 phosphorylation occurs through a cascade involving EGF receptor (EGFR) tyrosine kinase and the downstream activation of Raf-1 and MEK1/2. EGF stimulation of H9C2 cells resulted in this typical cascade (Fig. 4), as EGF-dependent activation of ERK 1/2 was inhibited by AG1478, a specific EGFR inhibitor, by BAY 43–9006, an inhibitor of Raf-1 kinase, and by PD 98059, an inhibitor of MEK1. Upon stimulation with ONOO−, we found that ERK activation was associated with MEK phosphorylation and was completely prevented by PD98059 (Fig. 3). Also, both MEK phosphoryation and ERK activity were inhibited by BAY 43–9006 (Fig. 5), which indicates that ONOO−-mediated ERK activation totally depends on MEK 1 phosphorylation, itself being dependent on Raf-1 kinase. In contrast to EGF, the effects of ONOO− on ERK were not influenced by AG1478, which supports that EGFR is not a relevant signaling intermediate in the process of ERK activation by ONOO− in H9C2 cardiomyocytes. It is noteworthy that ONOO− has also been found to activate ERK in fibroblasts, but through signaling pathways independent from Raf-1 [2], or even independent from MEK-1, via a mechanism involving protein kinase C [14]. Also, a previous report indicated that ONOO− could activate EGFR in PC 12 cells, triggering the downstream activation of ERK [25]. Our data are in clear contrast with these findings and thus indicate that the cellular targets of ONOO− critically depend on the cell type studied.
We also examined the possibility that ONOO− might activate ERK via p21Ras, which has been proposed to serve as a general signaling target of reactive free radicals and as a cellular sensor of redox stress [20,24]. At variance with previous findings that ONOO− or SIN-1, a peroxynitrite generator, induced the activation of p21Ras in human neutrophils [26] and murine neural cells [27], we could not detect any increase in p21Ras activation upon ONOO− exposure in H9C2 cells (Fig. 6).Also, we did not find any influence of FPTII, a highly selective and potent inhibitor of Ras farnesyltransferase, on ERK phosphorylation by ONOO− indicating that Ras is dispensable for ERK activation by peroxynitrite. This observation contrasts with the requirement of Ras of other known activators of ERK in cardiomyocytes, such as endothelin-1, phenylephrine and phorbol esters [6,28]. Thus, our findings introduce the novel concept that an unusual, alternative pathway of ERK activation, which is independent from p21Ras signaling, is triggered by ONOO− in cardiac muscle cells. If ONOO− can activate the Raf-1/MEK/ERK pathway in the absence of p21Ras activation, one may wonder which signaling intermediate uspstream from Raf-1 is activated by ONOO−. Raf-1 is a serine-threonine kinase governed by complex regulatory mechanisms, which notably involve phosphorylation at multiple amino acid residues [29]. Several kinases have been implicated in the process of Raf-1 phosphorylation and activation, including src-family tyrosine kinases, the phosphatidyl inositol 3 kinase (PI3K) and protein kinase C (PKC) [29,30]. It is here worth to mention that recent investigations have indicated that ONOO− can activate src-tyrosine kinase in endothelial and neural cells [31,32], PI3K in endothelial cells, fibroblasts and adipocytes [13,31,33], and PKC in fibroblasts and cardiomyocytes [14,34]. Thus, one can hypothesize that ONOO− acted via one of these signaling intermediates to activate the Raf-1/MEK/ERK pathway in our experimental conditions.
The signaling properties of ONOO− may depend either on oxidative type of chemistry or on the nitration of tyrosine residues, resulting in the formation of protein-bound 3-nitrotyrosine [7]. Tyrosine nitration has been shown to impair tyrosine phosphorylation, thereby altering various signaling processes (either activation or down-regulation) relying on tyrosine phosphorylation [35]. The relative contributions of nitration and oxidation reactions in the signaling events triggered by ONOO− can be distinguished using the flavanol (−)-epicatechin, a compound inhibiting tyrosine nitration several orders of magnitude more efficiently than oxidation reactions by ONOO− [15]. Stimulation of H9C2 cells with ONOO− induced a marked nitration of cellular proteins that was dose-dependently inhibited by epicatechin (Fig. 7), in agreement with previous findings [15]. However, epicatechin failed to inhibit ERK 1/2 phosphorylation, implying that activation was not due to tyrosine nitration, but rather to an oxidative chemistry. This is consistent with the recent proposal that oxidative inactivation of tyrosine phosphatases by ONOO− [36] may be an important mechanism of signal transduction triggered by ONOO−, by allowing kinase activities to become predominant [35].
In addition to ERK, we also show, for the first time, that ONOO− is a potent activator of the phosphorlyation of two other MAPKs, JNK and p38, in cardiomyocytes (Figs. 8 and 9). Similarly to the activation of ERK, p38 and JNK were phosphorylated in response to ONOO− in a time- and concentration-dependent way. Both p38 and JNK have been correlated with cardiomyocyte apoptosis [37,38], whereas ERK activation has emerged as an important mechanism of cytoprotection able to delay or offset an apoptotic program [5,6]. Due to their opposite effects, it is likely that the balance of activity of these distinct signaling pathways is critical for the decision between cell demise or cell survival [6]. Importantly, all three subclasses of MAP kinases have been shown to be activated in the human failing heart [39], as well as in the heart of experimental animals following ischemia–reperfusion [40], conditions in which ONOO− is currently considered a potential culprit of myocardial damage and dysfunction [10,41]. The current data provide the new concept that ONOO− may not only be a direct toxic effector in these circumstances, but may also be instrumental in the activation of signal transduction pathways playing central roles in the mechanisms of apoptosis.
In summary, the present results represent the first experimental evidence that peroxynitrite mediates ERK 1/2 activation in cardiomyocytes through a signaling cascade involving Raf-1 kinase and MEK 1, but which is independent from EGFR and p21Ras. The activation of MEK 1-ERK 1/2 by peroxynitrite is mediated by an oxidative, instead of nitrative, type of chemistry. Our data also show that, in addition to ERK, peroxynitrite is able to activate p38 and JNK, indicating that it can simultaneoulsy trigger anti- and pro-apoptotic signaling cascades in cardiomyocytes. Given that myocardial peroxynitrite generation and MAP kinase activation occur in similar pathological conditions, our findings indicate that peroxynitrite may represent a previously unrecognized signaling molecule in a wide range of cardiac diseases.
Acknowledgments
This work was supported by grants from the Swiss National Fund for Scientific Research (Nr. 3100-65005.01 and PP00B-68882/1) to L.L.
Abbreviations
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ERK
extracellular signal-regulated kinase
- JNK
C-Jun NH2-terminal kinase
- MAPK
mitogen-activated protein kinase
- MEK
MAPK kinase
- NO
nitric oxide
- ONOO−
peroxynitrite
- O2−
superoxide anion
References
- 1.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 2.Zhang P, Wang YZ, Kagan E, Bonner JC. Peroxynitrite targets the epidermal growth factor receptor, Raf-1, and MEK independently to activate MAPK. J Biol Chem. 2000;275:22479–86. doi: 10.1074/jbc.M910425199. [DOI] [PubMed] [Google Scholar]
- 3.Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–81. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 4.Yue TL, Wang C, Gu JL, et al. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res. 2000;86:692–9. doi: 10.1161/01.res.86.6.692. [DOI] [PubMed] [Google Scholar]
- 5.Lips DJ, Bueno OF, Wilkins BJ, et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004;109:1938–41. doi: 10.1161/01.CIR.0000127126.73759.23. [DOI] [PubMed] [Google Scholar]
- 6.Vlahos CJ, McDowell SA, Clerk A. Kinases as therapeutic targets for heart failure. Nat Rev Drug Discov. 2003;2:99–113. doi: 10.1038/nrd1009. [DOI] [PubMed] [Google Scholar]
- 7.Liaudet L, Soriano FG, Szabo C. Biology of nitric oxide signaling. Crit Care Med. 2000;28:N37–N52. doi: 10.1097/00003246-200004001-00005. [DOI] [PubMed] [Google Scholar]
- 8.Liaudet L, Szabo G, Szabo C. Oxidative stress and regional ischemia–reperfusion injury: the peroxynitrite-poly(ADP-ribose) polymerase connection. Coron Artery Dis. 2003;14:115–22. doi: 10.1097/00019501-200304000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Pacher P, Liaudet L, Bai P, et al. Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation. 2003;107:896–904. doi: 10.1161/01.cir.0000048192.52098.dd. [DOI] [PubMed] [Google Scholar]
- 10.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–16. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 11.Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res. 1991;69:1476–86. doi: 10.1161/01.res.69.6.1476. [DOI] [PubMed] [Google Scholar]
- 12.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–50. [PubMed] [Google Scholar]
- 13.Klotz LO, Schieke SM, Sies H, Holbrook NJ. Peroxynitrite activates the phosphoinositide 3-kinase/Akt pathway in human skin primary fibroblasts. Biochem J. 2000;352(Pt 1):219–25. [PMC free article] [PubMed] [Google Scholar]
- 14.Bapat S, Verkleij A, Post JA. Peroxynitrite activates mitogen-activated protein kinase (MAPK) via a MEK-independent pathway: a role for protein kinase C. FEBS Lett. 2001;499:21–6. doi: 10.1016/s0014-5793(01)02511-x. [DOI] [PubMed] [Google Scholar]
- 15.Schroeder P, Klotz LO, Buchczyk DP, Sadik CD, Schewe T, Sies H. Epicatechin selectively prevents nitration but not oxidation reactions of peroxynitrite. Biochem Biophys Res Commun. 2001;285:782–7. doi: 10.1006/bbrc.2001.5210. [DOI] [PubMed] [Google Scholar]
- 16.van der Vliet A, Hristova M, Cross CE, Eiserich JP, Goldkorn T. Peroxynitrite induces covalent dimerization of epidermal growth factor receptors in A431 epidermoid carcinoma cells. J Biol Chem. 1998;273:31860–6. doi: 10.1074/jbc.273.48.31860. [DOI] [PubMed] [Google Scholar]
- 17.Ischiropoulos H, Zhu L, Beckman JS. Peroxynitrite formation from macrophage-derived nitric oxide. Arch Biochem Biophys. 1992;298:446–51. doi: 10.1016/0003-9861(92)90433-w. [DOI] [PubMed] [Google Scholar]
- 18.Upmacis RK, Deeb RS, Resnick MJ, et al. Involvement of the mitogen-activated protein kinase cascade in peroxynitrite-mediated arachidonic acid release in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2004;286:C1271–C1280. doi: 10.1152/ajpcell.00143.2003. [DOI] [PubMed] [Google Scholar]
- 19.Bollag G, Freeman S, Lyons JF, Post LE. Raf pathway inhibitors in oncology. Curr Opin Investig Drugs. 2003;4:1436–41. [PubMed] [Google Scholar]
- 20.Oliveira CJ, Schindler F, Ventura AM, Morais MS, Arai RJ, Debbas V, et al. Nitric oxide and cGMP activate the Ras-MAP kinase pathway-stimulating protein tyrosine phosphorylation in rabbit aortic endothelial cells. Free Radic Biol Med. 2003;35:381–96. doi: 10.1016/s0891-5849(03)00311-3. [DOI] [PubMed] [Google Scholar]
- 21.Foschi M, Chari S, Dunn MJ, Sorokin A. Biphasic activation of p21ras by endothelin-1 sequentially activates the ERK cascade and phosphatidylinositol 3-kinase. EMBO J. 1997;16:6439–51. doi: 10.1093/emboj/16.21.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–8. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 23.Lander HM, Jacovina AT, Davis RJ, Tauras JM. Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem. 1996;271:19705–9. doi: 10.1074/jbc.271.33.19705. [DOI] [PubMed] [Google Scholar]
- 24.Lander HM, Ogiste JS, Teng KK, Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270:21195–8. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 25.Jope RS, Zhang L, Song L. Peroxynitrite modulates the activation of p38 and extracellular regulated kinases in PC12 cells. Arch Biochem Biophys. 2000;376:365–70. doi: 10.1006/abbi.2000.1728. [DOI] [PubMed] [Google Scholar]
- 26.Zouki C, Zhang SL, Chan JS, Filep JG. Peroxynitrite induces integrin-dependent adhesion of human neutrophils to endothelial cells via activation of the Raf-1/MEK/Erk pathway. FASEB J. 2001;15:25–7. doi: 10.1096/fj.00-0521fje. [DOI] [PubMed] [Google Scholar]
- 27.Kaji T, Kaieda I, Hisatsune T, Kaminogawa S. 3-Morpholino-sydnonimine hydrochloride induces p53-dependent apoptosis in murine primary neural cells: a critical role for p21(ras)-MAPK-p19(ARF) pathway. Nitric Oxide. 2002;6:125–34. doi: 10.1006/niox.2001.0389. [DOI] [PubMed] [Google Scholar]
- 28.Chiloeches A, Paterson HF, Marais R, Clerk A, Marshall CJ, Sugden PH. Regulation of Ras.GTP loading and Ras-Raf association in neonatal rat ventricular myocytes by G protein-coupled receptor agonists and phorbol ester. Activation of the extracellular signal-regulated kinase cascade by phorbol ester is mediated by Ras. J Biol Chem. 1999;274:19762–70. doi: 10.1074/jbc.274.28.19762. [DOI] [PubMed] [Google Scholar]
- 29.Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–9. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- 30.King WG, Mattaliano MD, Chan TO, Tsichlis PN, Brugge JS. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol Cell Biol. 1997;17:4406–18. doi: 10.1128/mcb.17.8.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, et al. Activation of 5′-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite J Biol Chem. 2003;278:34003–10. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 32.Minetti M, Mallozzi C, Di Stasi AM. Peroxynitrite activates kinases of the src family and upregulates tyrosine phosphorylation signaling. Free Radic Biol Med. 2002;33:744–54. doi: 10.1016/s0891-5849(02)00891-2. [DOI] [PubMed] [Google Scholar]
- 33.Guzman-Grenfell AM, Garcia-Macedo R, Gonzalez-Martinez MT, Hicks JJ, Medina-Navarro R. Peroxynitrite activates glucose uptake in 3T3-L1 adipocytes through a PI3-K-dependent mechanism. Front Biosci. 2005;10:47–53. doi: 10.2741/1505. [DOI] [PubMed] [Google Scholar]
- 34.Balafanova Z, Bolli R, Zhang J, Zheng Y, Pass JM, Bhatnagar A, Tang XL, et al. Nitric oxide (NO) induces nitration of protein kinase Cepsilon (PKCepsilon), facilitating PKCepsilon translocation via enhanced PKCepsilon–RACK2 interactions: a novel mechanism of no-triggered activation of PKCepsilon. J Biol Chem. 2002;277:15021–7. doi: 10.1074/jbc.M112451200. [DOI] [PubMed] [Google Scholar]
- 35.Klotz LO, Schroeder P, Sies H. Peroxynitrite signaling: receptor tyrosine kinases and activation of stress-responsive pathways. Free Radic Biol Med. 2002;33:737–43. doi: 10.1016/s0891-5849(02)00892-4. [DOI] [PubMed] [Google Scholar]
- 36.Takakura K, Beckman JS, MacMillan-Crow LA, Crow JP. Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1B, CD45, and LAR by peroxynitrite. Arch Biochem Biophys. 1999;369:197–207. doi: 10.1006/abbi.1999.1374. [DOI] [PubMed] [Google Scholar]
- 37.Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–50. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Huang S, Sah VP, Ross J, Jr, Brown JH, Han J, et al. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem. 1998;273:2161–8. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- 39.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–7. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 40.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–36. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 41.Szabo G, Liaudet L, Hagl S, Szabo C. Poly(ADP-ribose) polymerase activation in the reperfused myocardium. Cardiovasc Res. 2004;61:471–80. doi: 10.1016/j.cardiores.2003.09.029. [DOI] [PubMed] [Google Scholar]