

Abstract
Alterations in the state of excitability of midbrain dopamine (DA) neurons from the ventral tegmental area (VTA) may underlay changes in the synaptic plasticity of the mesocorticolimbic system. Here, we investigated norepinephrine’s (NE) regulation of VTA DA cell excitability by modulation of the hyperpolarization-activated cation current, Ih in whole cell recordings. Current clamp recordings show that NE (40 µM) hyperpolarizes spontaneously firing VTA DA cells (11.23 ±4 mV; n = 8). In voltage clamp, NE (40 µM) induces an outward current (100 ± 24 pA; n = 8) at − 60 mV that reverses at about the Nernst potential for potassium (−106 mV). In addition, NE (40 µM) increases the membrane cord conductance (179 ± 42%; n = 10) and reduces Ih amplitude (68 ± 3% of control at −120 mV; n = 10). The noradrenergic alpha-1 antagonist prazosin (40 µM; n = 5) or the alpha-2 antagonist yohimbine (40 µM; n = 5) did not block NE effects. All NE-evoked events were blocked by the D2 antagonists sulpiride (1 µM) and eticlopride (100 nM) and no significant reduction of Ih took place in the presence of the potassium channel blocker BaCl2 (300 µM). Therefore, it is concluded that NE inhibition of Ih was due to an increase in membrane conductance by a non-specific activation of D2 receptors that induce an outward potassium current and not a result of a second messenger system acting on h-channels. The results also suggest that Ih channels are mainly located at dendrites of VTA DA cells and thus their inhibition may facilitate the transition from single spike firing to burst firing and vice versa.
Keywords: VTA, Ih, Norepinephrine, Dopamine receptors, Adrenergic receptors
Introduction
The ventral tegmental area (VTA) is the source of dopaminergic projections that innervate structures in the ventral forebrain and cortical areas, a system collectively known as the mesocorticolimbic (Dahlstrom and Fuxe, 1964; Ungerstedt, 1971). The pattern of activity of VTA dopamine cells ranges from single action potential pacemaker-like firing to multiple action potential burst firing (Cooper, 2002). Bursting activity causes a greater increase in extracellular dopamine (DA) concentration per action potential than does the low frequency pacemaker-like firing (Kitai et al., 1999; Overton and Clark, 1997; Grace and Bunney, 1984). Therefore, transitions between these two states of excitability may provide a physiological mechanism by which DA neurons modulate the mesocorticolimbic system.
The state of excitability of midbrain DA cells depends on the modulation of the conductances that generate and affect the different firing modes of these neurons (Kitai et al., 1999; Overton and Clark, 1997; Grace and Bunney, 1984). Among these conductances is the hyperpolarization-activated cation current, Ih (Cathala and Paupardin-Tritsch, 1999; Jiang et al., 1993; Mercuri et al., 1995; Watts et al., 1995). The Ih is a depolarizing current that affects cell excitability in three interrelated ways: it helps to determine the resting potential and firing frequency; reduces the amplitude and duration of hyperpolarizing inputs; and generates pacemaker activity when coupled to other voltage-dependent conductances (e.g., IT, a voltage dependent calcium current) (Luthi and McCormick, 1998; Pape, 1996). In the lateral geniculate nucleus of the thalamus (McCormick and Pape, 1990), in primary sensory neurons of the dorsal root ganglia (Yagi and Sumino, 1998), and in neurons of the medial nucleus of the trapezoid body (Banks et al., 1993), norepinephrine (NE) affects the state of excitability by modulation of the voltage sensitivity of Ih. The possibility of noradrenergic modulation of Ih in VTA DA cells has not been studied. Here we explore the effects of NE in Ih voltage and time dependence in this nucleus.
Methods
Preparation of horizontal midbrain slices
Male Sprague-Dawley rats (15–20 days postnatal) were anesthetized with a chloral hydrate intraperitoneal injection (15mg/Kg) and then decapitated. The brain was removed from the skull in less than 1.5 minutes and put into ice-cold (2°C – 4°C) dissection solution of composition (mM): NaCl 126; KCl 2.5; NaH2PO4 1.2; MgCl2 7.0; CaCl2 0.5; Glucose 25; NaHCO3 25; saturated with 95% O2 and 5% CO2. A brain section containing the ventral and rostral half of the brainstem was obtained as follows. Two coronal cuts were performed to remove the cerebellum and the rostral half of the cerebral hemispheres. The remaining piece was placed with the caudal end up and a transverse cut was made through the cerebral aqueduct. The portion containing the ventral half of the cerebral aqueduct was positioned with the ventral surface up and two parasagittal cuts were made to remove the temporal lobes. The resulting block was glued with cyanoacrylate (Krazy Glue™) to the stage of a Leica VT 1000S vibratome (Nussloch, Germany) immersed in ice-cold dissection solution and cut into horizontal slices (thickness: 220 µm) starting from the ventral surface. To reduce shear forces on the tissue, a block of agar was glued to the stage and placed against the caudal end of the tissue (opposite to the vibratome blade). Slices were cut and discarded until the floor of the interpeduncular fossa was reached. The next three slices were placed in an incubation chamber containing oxygenated artificial cerebrospinal fluid (ACSF) (mM: NaCl 126; KCl 2.5; NaH2PO4 1.2; MgCl2 1.0; CaCl2 2.0; Glucose 25; NaHCO3 25; saturated with 95% O2 and 5% CO2) at 35° C. Slices remained in the chamber between 30 to 60 minutes before recording. One half of the slice was placed in the recording chamber (500 °L) and superfused with ACSF (35°C) at a rate of 2 mL/min. Slices not used for recording were kept in the incubation chamber until the end of the experiments.
Whole cell recordings
The recording site in the brain slice was visually identified with an Olympus BX51WI microscope equipped with infrared light and Nomarski optics. The VTA was identified as the region lateral to the fasciculus retroflexus and medial to the medial terminal nucleus of the accessory optic tract (Paxinos and Watson 1986; Johnson and North, 1992). The substantia nigra compacta (SNc), identified as the regions rostral and caudal to the medial terminal nucleus of the accessory optic tract, was avoided. Whole cell recordings were made using borosilicate glass pipettes pulled in two stages by a vertical pipette puller (PP-830; Narishige, Japan). When the recording pipette was filled with the internal solution ((mM) K-methyl sulfate 115; KCl 20; MgCl2 1; HEPES 10; EGTA 0.1; ATP 2; GTP 0.3; creatine phosphate 10), the tip of the pipette had a resistance of 2–3 MΩ. The recording pipette was advanced towards the brain slice and a seal was obtained by applying a pulse of suction. After gigaseal formation between the pipette and the cell membrane the holding potential was set to − 60 mV and the fast capacitance was compensated. In order to obtain the whole cell configuration the membrane was ruptured by further suction. Once in whole cell configuration the slow capacitance was corrected and the access resistance compensated. Recordings with high access resistance values (10–20 MΩ) were rejected if the access resistance changed by more than 15%.
Whole cell recordings were done using the Axopatch 1-D amplifier (Axon Instruments, CA, USA). Membrane currents were filtered at 10 kHz and digitized at 5 kHz (Digidata 1322A; Axon Instruments, CA, USA). The digitized current was displayed and analyzed on a MacIntosh G4 computer using Axograph 4.6 software (Axon Instruments, CA, USA).
Data Analysis
Ih was evoked by family of 6 hyperpolarizing voltage steps (step amplitude: −10 mV; step duration: 1 s) from the holding potential of −60 mV to −120 mV. The steps were applied in 7s intervals. Ih was calculated as the difference between the membrane current at the end of the voltage step (Iss) and the instantaneous current (Iinst) measured after the settling of the capacitative transient. To determine Ih voltage dependence, activation curves were constructed from Ih tail currents, which were calculated by subtracting the pre-step holding current from the peak of the tail current. The activation curves were fitted by the Boltzmann function to estimate the potential of half-activation (V1/2) and the slope factor (k):
Ih time dependence was studied by fitting current traces (from − 90 to − 120 mV) with an exponential function of the form:
where τh is the activation time constant that gives the time (ms) required to reach 63% of Ih,SS, which is the steady state Ih amplitude for the fitted current trace. Membrane cord conductance was estimated by the slope obtained from linear fits to the plot of Iins vs. command voltage. Averaged data are expressed as means ± S.E.M. Statistical significance of data from different drug conditions was assessed with 1-way or 2-way ANOVA as appropriate.
Compounds
Drugs were applied (2 min.) to the slice by superfusion. Complete exchange of the bath solution occurred in less than 1 min. All drugs and salts were acquired from Sigma (St. Louis, Missouri).
Results
The data presented here were based on in vitro whole cell recordings from 78 putative VTA dopaminergic neurons. Recorded cells were located lateral to the fasciculus retroflexus and medial to the medial terminal nucleus of the accessory optic tract (Paxinos and Watson 1986; Johnson and North, 1992). They were considered putative VTA dopamine cells if they exhibited a pronounced Ih (i.e., the smallest Ih amplitude was 500 pA at − 120 mV) and a slow spontaneous activity (Mercuri et al., 1994; Paladini and Williams, 2004; Paladini et al., 2004).
Margolis et al. (2006) clearly shows that if a VTA cell is dopaminergic then it has Ih, although the converse is not necessarily true since according to same researchers approximately 1/3 of VTA cells that have Ih are not DA cells. Therefore, immunohistochemestry is the only reliable method for identifying Ih containing cells as dopaminergic (Margolis et al. 2006). Nevertheless, our electrophysiological criteria for identification VTA DA cell is satisfactory by current standards (Paladini and Williams, 2004; Paladini et al., 2004; Neuhoff et al., 2002). Indeed, convincing evidence had been presented about the cellular physiology of DA cells base only on this criteria (Fiorillo and Williams, 1998; Fiorillo and Williams, 2000; Paladini et al., 2001, Morikawa et al., 2003; Paladini and Williams, 2004; Paladini et al., 2004).
Noradrenergic effects on VTA dopamine cells
Norepinephrine (NE; 40 µM) application evokes an outward current (100 ± 24 pA; n = 8) at − 60 mV (n = 11; Fig. 1B). The activation of this current was associated with an increase in membrane conductance (179 ± 42%; n = 10, data not shown) and membrane hyperpolarization (11.23±4 mV; n = 8; Fig. 1A and 1C). The NE-evoked current reversed at − 96 ± 8 mV (n = 6). This value was close to the reversal potential of K+ (−106 mV) as predicted by the Nernst equation (Fig. 1D). These effects were blocked by 300 µM of external barium (data not shown). The NE effects on membrane conductance and membrane potential were not seen in the presence of the D2 antagonist eticlopride (100 nM) or sulpiride (1 µM), (n = 19; data not shown). These results are in agreement with those of Grenhoff et al. (1995) who have shown that NE induced hyperpolarization in VTA DA cells was due to activation of D2 receptors.
Fig. 1. Effects of norepinephrine (NE; 40 µM) on membrane potential, holding current and membrane conductance of VTA dopamine (DA) cells.

A. Bath application of NE hyperpolarizes spontaneously firing VTA DA cells.
B. NE evokes an outward current on VTA DA cells when held at − 60 mV.
C. Current vs. voltage curves showing the effect of NE on membrane cord conductance. Observe that NE increases the membrane conductance (increased slope). The curves also show what is evident from parts A and B. That is, NE evokes an outward current at − 60 mV (reduction in the current amplitude) and hyperpolarizes (change of intercepts on the potential axis) the probed cells. The membrane potential coordinate of the point of intersection of the two IV-curves represents the reversal potential of the conductance activated by NE (see part D). The curves were constructed by plotting the instantaneous membrane current against the command potentials. Points on each curve represent the average ± s.e.m. from a sample of six cells. The smooth lines show linear fits.
D. Current vs. voltage curve obtained by subtracting the NE curve from the control curve for each of the six cells in part C. Points on the curve represent average ± s.e.m.. This curve represents the current vs. voltage relation of the conductance activated by NE. The reversal potential (intercept on the potential axis) for this conductance is − 96 ± 8 mV (n = 6), which is very close to the predicted Nernst potential for K+ (− 106 mV). The smooth line shows a linear fit.
NE inhibits Ih
The effects of NE on Ih were studied with whole cell voltage clamp recordings in a total of 35 putative VTA DA cells.
Concomitant to the NE evoked increase in potassium conductance there was a decrease in Ih amplitude (n =10; Fig. 2A and 2C). This inhibition was apparent from the reduction in the difference between ISS and Iins, which resulted mainly from an increase in Iins (Fig. 2A and 2B). The increased Iins was seen at all potentials where Ih inhibition was present and the time course of Ih inhibition was parallel to the time course of the increase in Iins (Fig. 2B). Therefore, the time course of Ih inhibition is parallel to the time course of the increase in potassium conductance (Fig. 1 and Fig. 2B).
Fig. 2. NE (40 µM) inhibits Ih.
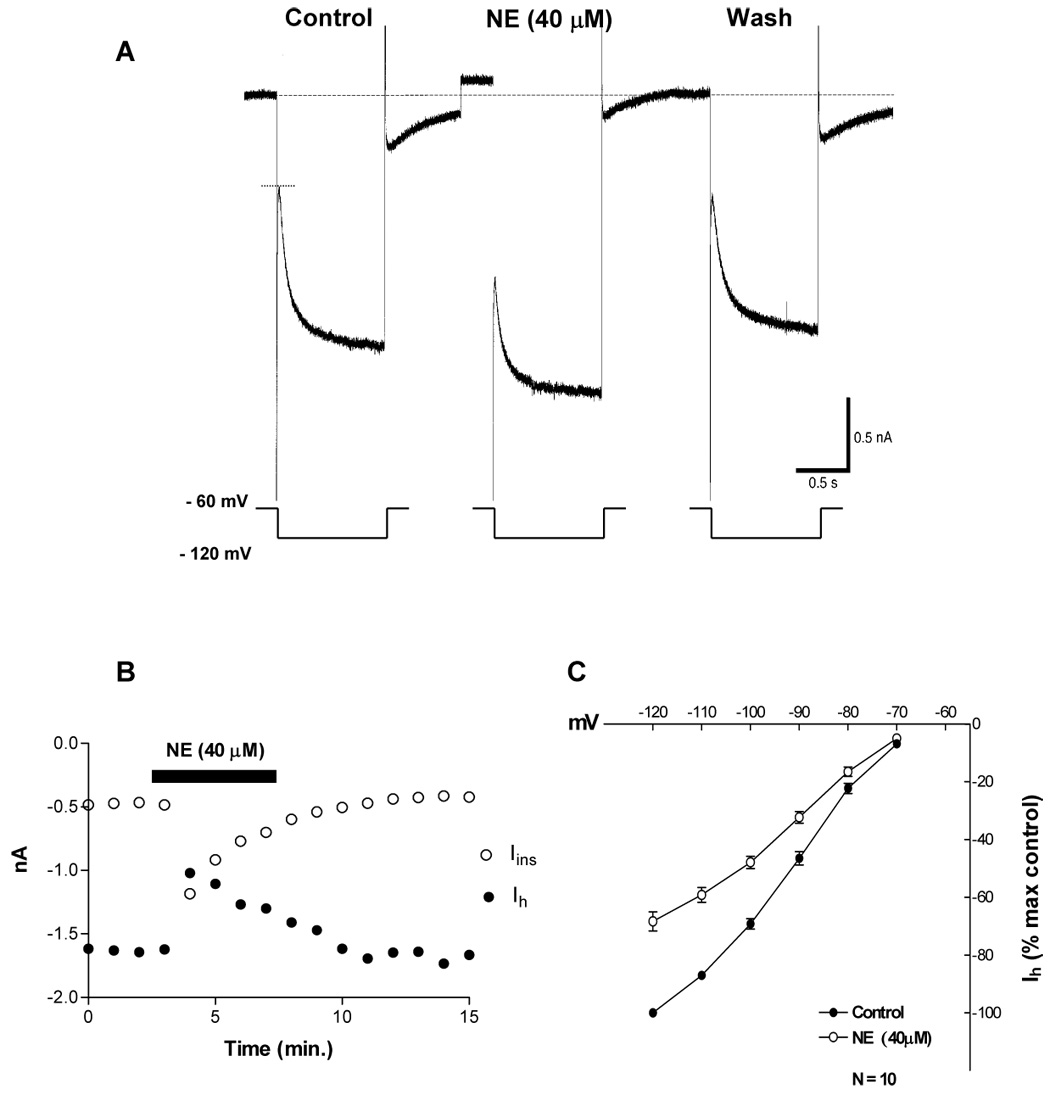
A. Voltage clamp recording showing the current response of a VTA DA cell when held at − 60 mV and commanded to − 120 mV for 1 second. Bath application of NE (2 min.) induced an outward current of 96 pA (change in holding current), a 1.7-fold increase in membrane conductance and a 30% reduction in Ih amplitude. The membrane conductance was calculated from the amplitude of the instantaneous current (between …. and ----).
B. The time course of Ih inhibition is parallel to that of the NE-induced increase in membrane conductance (increase in instantaneous current (Iins) amplitude). The data used to plot the graph is from a recording similar to the one shown in part A.
C. NE significantly reduced Ih amplitude (2-way ANOVA, F= 249, df = 1, 108; P < 0.0001). Data points represent average ± s.e.m. of ten cells.
To permit the comparison of the responses of the recorded neurons, Ih was expressed as a percentage of the maximal Ih during the control condition and plotted against the command potentials. As seen in Figure 2C, the percentage of Ih inhibition increases with the amplitude of the hyperpolarizing voltage step and reaches a maximal value of 32% at −120 mV.
Ih inhibition is not mediated by alpha adrenergic receptors
The amplitude of Ih can be reduced by activation of protein kinase C (PKC) (Zhaoping et al, 2003; Cathala and Paupardin-Tritsch, 1997) or by a decrease in cytoplasmic levels of cAMP (Ulens and Siegelbaum, 2003). The adrenergic metabotropic receptors alpha-1 and alpha-2 are known to exert these actions. For example, all subtypes of alpha-1 receptors are known to activate PKC (Zhong and Minneman, 1999), whereas alpha-2 receptors tend to decrease intracellular levels of cAMP (Raymond et al, 1994). In order to determine if the NE-evoked Ih inhibition was caused by the activation of one of these receptors, the following experiments were performed. First, Ih was recorded in the presence of NE plus the alpha-1 antagonist prazosin (n = 5). Under these conditions Ih inhibition was significant and not different from the inhibition evoked by NE alone (Fig. 3C, 3E and 3G). Second, Ih was recorded in the presence of NE plus the alpha-2 antagonist yohimbine (n = 5). Nevertheless in the presence of yohimbine, Ih inhibition was similar to the inhibition evoked by NE alone (Fig. 3D, 3F and 3G). Therefore, the inhibition of Ih does not appear to result from the activation of these adrenergic receptors.
Fig. 3. Alpha adrenergic antagonists did not block the NE-induced outward current, increase in membrane conductance or Ih inhibition.
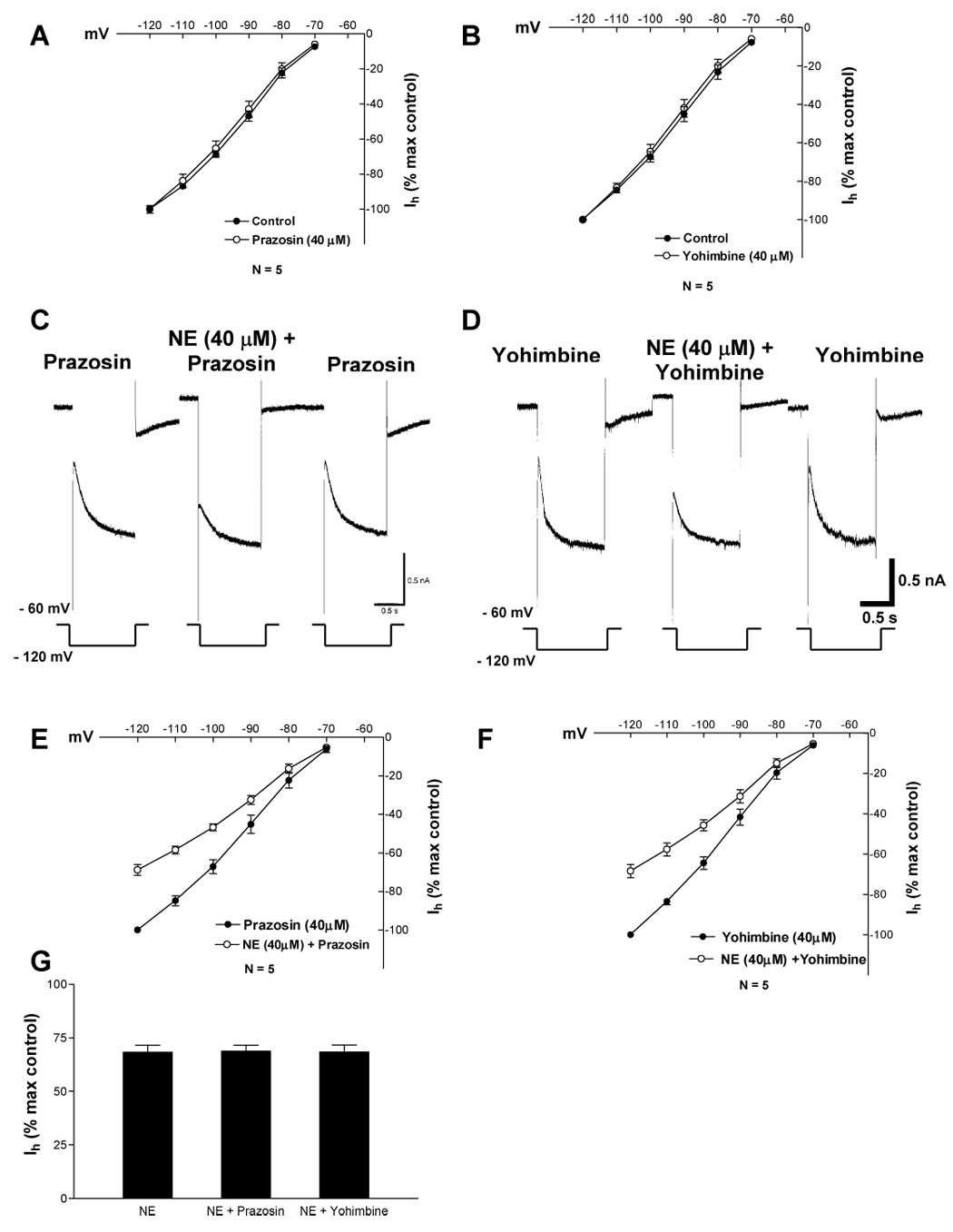
Data points on each graph represent average ± s.e.m. of five cells.
A. At 40 µM alpha-1 antagonist prazosin did not affect Ih amplitude.
B. At 40 µM alpha-2 antagonist yohimbine did not affect Ih amplitude.
C. In the presence of prazosin, NE evokes an outward current, increases the membrane conductance and inhibits Ih.
D. In the presence of yohimbine, NE evokes an outward current, increases the membrane conductance and inhibits Ih.
E. NE significantly reduced Ih amplitude in the presence of prazosin (2-way ANOVA, F=534.9, df = 1, 48;P < 0.0001).
F NE significantly reduced Ih amplitude in the presence of yohimbine (2-way ANOVA, F=99.72, df = 1, 48;P < 0.0001).
G. Maximal Ih inhibition (− 120 mV) in the presence of NE and prazosin or NE and yohimbine was not different from that of NE alone (1-way ANOVA, MS= 0.3481, df = 2, 19;P = 0.9956).
The non-specific activation of D2 receptors by NE inhibits Ih
The effects of NE on membrane potential and holding current were caused by the non-specific activation of D2 receptors and not by the activation of adrenergic receptors (Fig. 1). Furthermore, the time course of the D2 mediated effects was parallel to that of Ih inhibition (Fig. 2A and 2B). These findings suggest that Ih inhibition could have resulted from the activation of D2 receptors. To determine if this was the case, Ih was recorded in the presence of NE plus the specific D2 antagonist eticlopride. Under these conditions, NE was unable to evoke Ih inhibition (n = 10; Fig. 4). It is therefore concluded that Ih inhibition resulted from the non-specific activation of D2 receptors by NE.
Fig. 4. In presence of D2 antagonist eticlopride (100 nM) NE did not affect Ih.
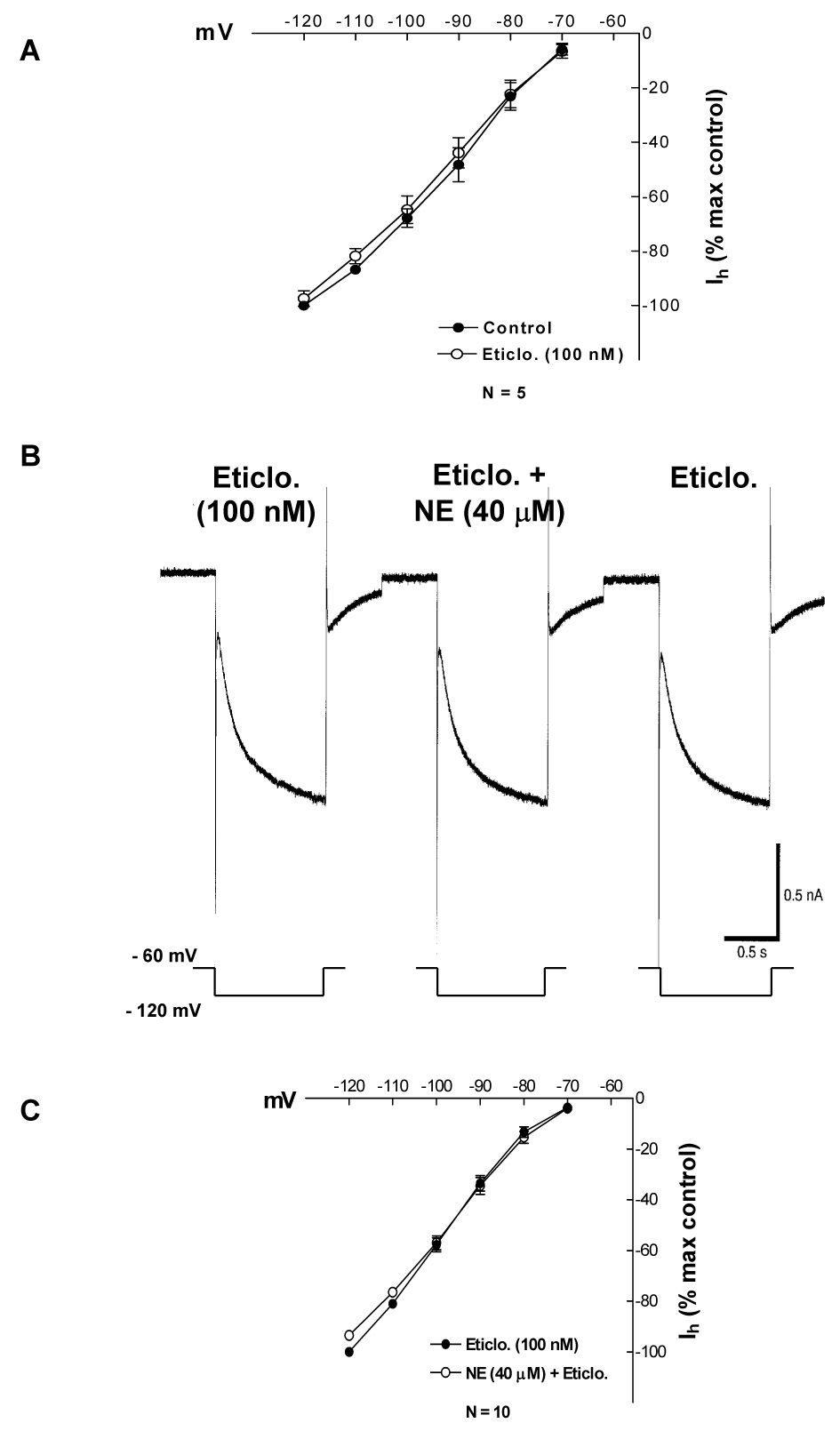
Data points in part A and C represent average ± s.e.m. of five and ten cells, respectively.
A. At 100 nM eticlopride has no effect on Ih amplitude.
B. Eticlopride blocks the NE-induced outward current, increase in membrane conductance and Ih inhibition.
C. Summary of NE effects on Ih in the presence of eticlopride.
NE inhibits Ih without affecting its voltage and time dependence
Dopamine D2 receptors are known to lower the intracellular concentration of cAMP (Kebabian and Calne, 1979). Hence the sensitivity of h-channels to hyperpolarization should be diminished when these receptors are activated (Ulens and Siegelbaum, 2003). If this is the case for the D2 mediated Ih inhibition, then the Ih activation curve should be shifted to the left on the potential axis, and the activation time constants at the interval of potentials where the inhibition is present should be increased. This type of inhibition is called voltage dependent inhibition because at maximal Ih activation, the inhibition is not present. However, if the inhibition is present without a negative shift in the activation curve and increase in the activation time constants, the inhibition is called voltage independent.
To determine if Ih inhibition was voltage dependent or independent, activation curves were constructed from the tail currents recorded in both the presence and absence of NE for five cells. A control activation curve was constructed by averaging the five curves in the absence of NE. A treatment curve was constructed by averaging the five curves in the presence of NE. Both curves were fitted by the Boltzmann function to estimate the average potential of half-activation (V1/2) and the average slope factor (k) under each condition. This analysis revealed that V1/2 and k were not affected by the presence of NE and that there was no significant difference between the two curves (2-way ANOVA) (Fig. 5A). The same cells used in the previous analysis were also inspected for changes in time dependence. For each cell, current traces from − 90 to − 120 mV were well fitted by single exponential functions in the presence and absence of NE. Current traces at − 70 and − 80 mV were not used for curve fitting since the time dependence at these potentials was not exponential for all cells. A control and treatment graph of the activation time constants vs. command potentials was constructed for each cell. Control and treatment graphs were averaged into single plots and statistically compared (2-way ANOVA). This analysis showed that the activation time constants were not significantly affected by the presence of NE (Fig. 5B).
Fig. 5. NE (40 µM) inhibits Ih without affecting its voltage and time dependence.
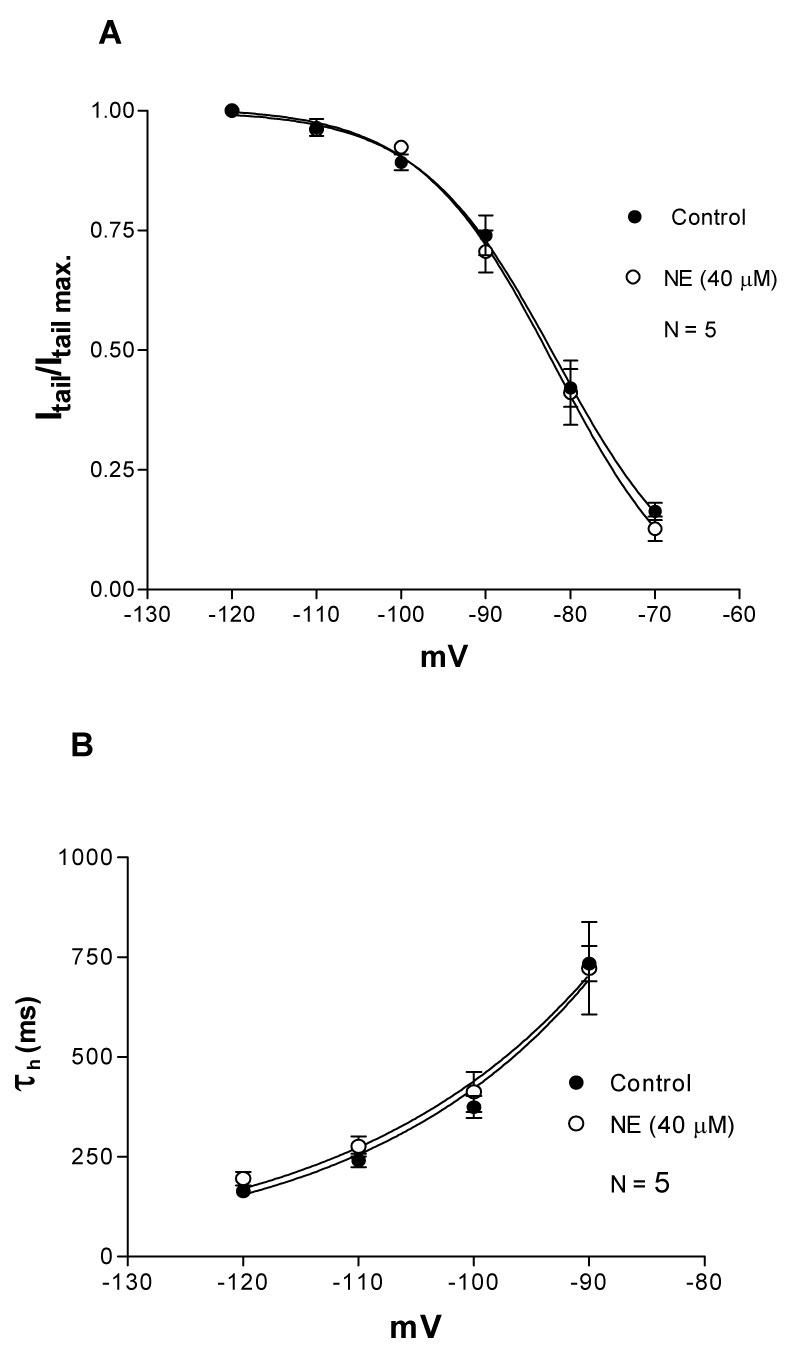
A. NE inhibits Ih without affecting its activation curve, indicating a voltage independent inhibition (2-way ANOVA, F= 0.2384, df = 1, 48; P = 0.6276). The level of Ih activation for a given command potential and condition were calculated as the ratio between the Ih tail (Itail) current at the command step and the tail current at − 120 mV (Itail max.). The half activation voltage and slope factor (k) were − 82 mV and k = 8 mV in control and in NE. The smooth curves show the Boltzmann fit. Data points represent average ± s.e.m. of five cells.
B. The time course of Ih was fitted by single exponential function. The activation time constants (τh) obtained from the fit are plotted against command potentials. NE did not affect the time dependence of Ih, suggesting a voltage independent inhibition (2-way ANOVA, F=0.4951, df = 1, 32; P = 0.4868). The smooth curves show a single exponential fit. Data points represent average ± s.e.m. of five cells.
These results show that Ih inhibition was voltage independent since in the presence of NE the activation curve was similar to the curve in the control condition and there was no significant change in the activation time constants at all tested potentials (Fig. 5A and 5B).
Ih inhibition is dependent upon the increase in membrane conductance
The results presented show that Ih inhibition is voltage independent (Fig. 5). However, recordings showed that Ih inhibition and the increase in conductance share a parallel time course (Fig. 2B). Thus the inhibition could be dependent upon the D2 mediated increase in potassium conductance. In midbrain DA cells the D2 mediated increase in potassium conductance has been shown to be blocked by barium at 300 µM (Cathala and Paupardin-Tritsch, 1999). Therefore, to test if Ih inhibition was dependent on the increase in potassium conductance, Ih was recorded in the presence of NE plus 300 µM of external barium. Under these conditions NE failed to inhibit Ih (n = 5) suggesting that the inhibition is dependent upon the increase in membrane conductance (Fig. 6).
Fig. 6. In the presence of the K+ channel blocker BaCl2 (300 µM), NE did not affect Ih.
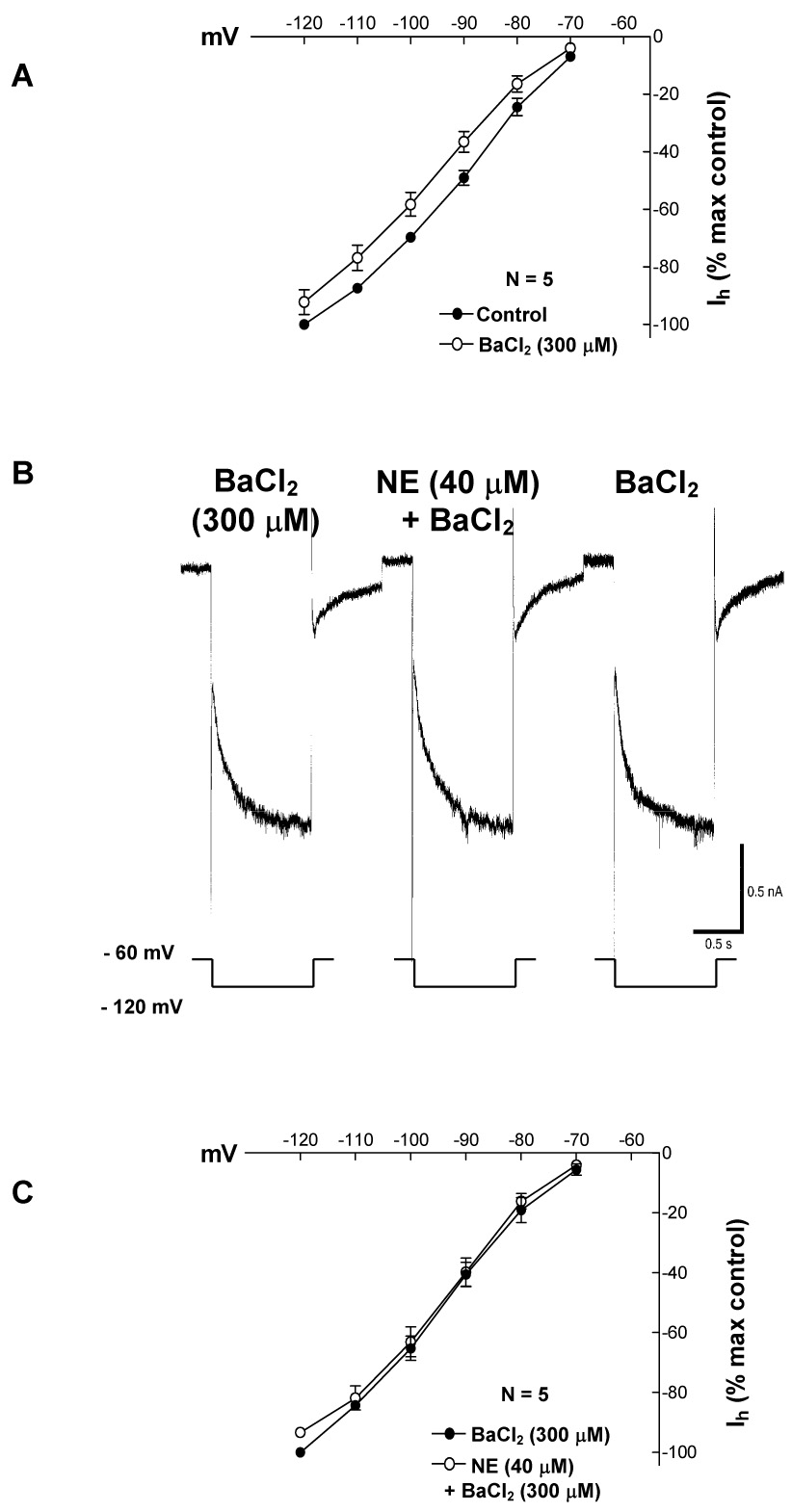
Data points in part A and C represent average ± s.e.m. of five cells.
A. At 300 µM BaCl2 causes a small diminution of Ih amplitude.
B. BaCl2 blocks the NE-induced outward current, increase in membrane conductance and Ih inhibition.
C. Summary of NE effects on Ih in the presence of BaCl2.
Discussion
The present study shows that NE hyperpolarizes VTA DA cells by increasing the membrane conductance to potassium through a non-specific activation of dopamine D2 receptors. Parallel to the time course of the D2-mediated hyperpolarization, NE also inhibits Ih. This effect of NE does not appear to arise from a specific modulatory action of second messengers on h-channels since the inhibition is present without a hyperpolarizing shift in the activation curve nor an increase in the activation time constants. Ih inhibition appears to be dependent upon the D2-mediated opening of potassium channels; that is, the inhibition probably resulted from inefficient space clamp of the neuron caused by the increase in conductance. A similar mechanism has been reported in SNc DA cells where Ih inhibition by NE or baclofen results from the increase in conductance and is not caused by a distinct effect of these agonists on Ih (Cathala and Paupardin-Tritsch, 1999; Watts et al, 1995). Our results differ from the above in two important ways. First, the VTA is functionally different to the SNc in terms of synaptic inputs, functional specificity and gene expression profiles (Sesack and Orr, 2002; Grace et al. 2007 and Greene, 2006). For example, burst firing in VTA DA cells is triggered by glutamatergic afferents that originate from the prefrontal cortex, pedunculopintine tegmentun and the lateral preoptic-rostral hypothalamic area. In contrast, burst firing in SNc DA cells is evoked by glutamatergic afferents from the subthalamic nucleus (Grace et al., 2007). Hence, VTA DA cells are exposed to a neurochemical environment quite different from that of DA cells in SNc. In addition, concerted differences in gene expression appear to underlie the unique properties of distinct dopamine neurons. VTA DA cells prominently express genes related to synaptic plasticity and neuropeptides, suggesting a role of VTA dysfunction in addiction and mood disorders. On the other hand, SNc DA neurons which have a tendency to degenerate in Parkinson's disease, express high levels of transcripts related to energy metabolism, mitochondria and phosphate signalling pathways (Greene, 2006). Thus, the need to identify differences and similarities at the cellular level between these anatomical structures is imperative. Second, the present work clearly shows that NE evoked Ih inhibition is voltage independent.
Ideal space clamp is achieved when the charge density imposed by the command potential is uniform across the cell capacitance. This means that the membrane potential equals the command potential at any location of the neuron. In reality, due to the cable-like geometry of neurons, the charge deposited by the command potential escapes across the membrane conductance as one moves away from the electrode, leading to a non-uniform charging of the cell capacitance and thus poor voltage clamp of sites distal to the electrode. Therefore, during a significant increase in membrane conductance, the voltage control of dendrites is severely reduced because of the increase in electrotonic length of these neural processes (Spruston et al., 1994).
If the channels that generate the whole cell Ih have a dendritic location, then the attenuation of voltage control of dendrites as a consequence of the increase in conductance will underlay the apparent Ih inhibition without NE having any modulatory action on h-channels. In support of this hypothesis is the finding that Ih inhibition in VTA DA cells cannot be isolated from the increase in conductance when barium is used to block potassium channel opening. Blocking the increase in conductance with barium abolishes Ih inhibition. It can be argued that external barium may prevent the binding of NE to the D2 receptor and hence blocks Ih inhibition. Notwithstanding, barium blocks the pore of potassium channels (Kubo et al., 1993) and is not known to affect the activation of the D2 receptors (Cathala and Paupardin-Tritsch, 1999). Taken together, these results make it difficult to support the possibility that Ih inhibition by NE is caused by a distinct mechanism acting on h-channels. They rather suggest that Ih inhibition is a consequence of inefficient space clamp caused by the increase in potassium conductance.
Norepinephrine is known to modulate the Ih of thalamic relay neurons (McCormick and Pape, 1990), neurons from the medial nucleus of the trapezoid body (MNTB) (Banks et al., 1993) and primary sensory neurons of the dorsal root ganglia (Yagi and Sumino, 1998). In the thalamus, NE activates beta-adrenergic receptors elevating intracellular cAMP levels and shifting Ih voltage dependence to more depolarized potentials. Similarly, in neurons of MNTB, NE also evokes a cAMP-dependent positive shift on the Ih activation curve. On the other hand, activation of alpha-2 adrenergic receptors in dorsal root ganglion (DRG) neurons evokes a significant reduction in Ih amplitude. In view of this data the lack of noradrenergic modulation of Ih in VTA DA cells suggests the absence of both types of adrenergic receptors. In support of this contention is the in vitro intracellular recording study of Grenhoff et al. (1995) which did not find alpha-2 or beta noradrenergic responses to NE bath application in VTA and SNc DA cells.
A more recent study in SNc DA cells has shown the functional presence of alpha-2 adrenergic receptors (Cathala et al., 2002). However, the activation of these receptors was not associated with Ih inhibition as in DRG neurons. A possible explanation for this paradox (i.e., alpha-2 receptors that do not affect Ih) is that alpha-2 receptors in DA cells activate a second messenger pathway that does not interact with h-channels. That is, although the activation of alpha-2 receptors may result in Ih inhibition (e.g., reduction in cAMP levels), these metabotropic receptors could be localized in a cell compartment distant or isolated from h-channels. A similar argument would also explain our results, i.e., the activation of dopamine D2 receptors does not affect Ih voltage and time dependence in VTA DA cells. Therefore, the spatial separation of alpha-2 and D2 receptors from h-channels may explain why Ih in midbrain DA cells is insensitive to the activation of these receptors.
The noradrenergic effects on VTA DA cells described here differ from the effects observed by others in that we did not record alpha-1 adrenergic responses to NE bath application, i.e. inward current (Grenhoff et al., 1995; Paladini et al., 2001; Paladini and Williams, 2004). Such a discrepancy could be the result of different experimental conditions. For example, the DA cells used in our study were obtained from rats of different age range and species than those used in other studies. On the other hand, studies have revealed a specific role of alpha-1B receptors in the modulation of activity of nigrostriatal DA neurons (Battaglia et al., 2003). Due to the lack of specific antagonists for alpha-1 receptor subtypes, it is possible that some of the observed NE effects could still be mediated by alpha-1 receptors. Prazosin, being a non-subtype selective antagonist, might not be able to block such actions.
All subtypes of alpha-1 adrenergic receptors are known to activate PKC (Zhong and Minneman, 1999). The activation of PKC in VTA and SNc DA cells inhibits Ih (Zhaoping et al., 2003; Cathala and Paupardin-Tritsch, 1997). Interestingly, studies that clearly show the functional presence of alpha-1 receptors in the midbrain do not report alpha-1 effects on Ih (Grenhoff et al., 1995; Paladini et al., 2001; Paladini and Williams, 2004). Since PKC is known to exist in numerous isoforms (Zhaoping et al., 2003), it is possible that the absence of alpha-1 effects on Ih results from the activation of a PKC isoform that does not phosphorylate h-channels or interacts with them indirectly.
In addition, it is worth noting that the DA cell response to the activation alpha-1 adrenergic receptors is sensitive to the duration of the activation. For instance, an outward current is observed when these receptors are activated by a transient iontophoretic pulse but an inward current is seen when a prolonged activation is evoked by bath application of an agonist. This dual action is due to fact that apha-1 receptors are couple to phosphoinositide hydrolysis (Paladini and Williams, 2004). Here we show that bath application of NE activates D2 receptors evoking an outward current identical to the outward current evoked by iontophoretic activation of D2 receptors reported by Paladini and Williams (2004). Thus, suggesting that extrasynaptic DA may have similar effects as those evoked by rapid synaptic release of the neurotransmitter.
The reduction in Ih amplitude due to a significant drop in input conductance suggests that h-channels are mainly located at the dendrites of DA cells in comparison to the soma. This places the functional role of Ih in the excitability of midbrain DA cells in a position quite different from the function most commonly ascribed to this conductance (i.e., a positive shift in the voltage dependence of Ih increases DA cell excitability). In particular, burst firing in DA cells evoked by activation of a dendritically located high threshold calcium spike (HTCaS) may be prevented by a depolarizing shift of Ih activation curve and vice versa.
In a DA cell the low threshold sodium spike is initiated in the axon hillock, whereas the HTCaS is generated somewhere in the dendrite. Somatic sodium spikes propagate forward into the axon and back into the dendrites. During back-propagation somatic spikes interact with dendritic synaptic potentials, leading to the temporal summation of both events. If this summation reaches the threshold of HTCaS then the activation of this calcium spike provides a long lasting somatic supra-threshold depolarization that initiates bursts of sodium action potentials in the axon hillock (Overton and Clark, 1997). Consequently, the interaction of back-propagating somatic spikes with dendritic synaptic inputs may facilitate the transition from single spike firing to burst firing in DA cells.
The temporal summation of dendritic synaptic inputs with back-propagating action potentials depends on the electrotonic properties of the dendrites. For example, the presence of leak conductances such as potassium channels or Ih makes the temporal summation of these events more difficult (Berger et al., 2001). Ih activation in layer V pyramidal neurons of the somatosensory cortex may prevent the back-propagation of somatic action potentials and thereby their ability to initiate burst firing when temporally coupled to synaptic inputs (Berger et al., 2003). Moreover, pyramidal bursting neurons in the Subiculum of the Hippocampus, which had a large Ih, exhibit a reduced temporal summation of synaptic inputs when stimulated at high frequency (van Wilie et al., 2006). This property provides the neuron with the ability to discriminate between inputs in terms of their frequencies. It is possible that dendritically located Ih in midbrain DA cells may play similar roles. Therefore, neuromodulators that negatively shift the voltage dependence of Ih may facilitate burst firing whereas those that evoke a positive shift may prevent burst firing.
Finally, the mesocorticolimbic system is a brain region rich in DA receptors and DA is a more potent agonist of D2 receptors than NE. For example, GTPase activity evoked by D2 activation in rat striatal membranes has an EC50 of 3.7 µM versus 14 µM for NE (Odagaki et al., 1995). This may suggest that NE plays a minor role in the physiology of this complex neural circuit; however, empirical evidence indicates a much bigger role for this catecholamine. Experiments have demonstrated that the loss of a trophic influence of neurons form the locus coeruleus might be crucial in Parkinson’s disease (Gesi et al., 2000). On the other hand, NE may decrease DA cell excitability through synaptic activation of alpha-1 adrenergic receptors in the VTA (Paladini and Williams, 2004). Also, the rewarding properties of cocaine and morphine are reduced in mice lacking alpha-1 adrenergic receptors (Drouin et al., 2002). In addition, blockade of alpha-adrenergic receptors with prazosin protects against methamphetamine toxicity (Battaglia et al., 2003). In light of this evidence, our results should not be view as validation of the idea that the principal neuromodulator of the mesocorticolimbic system is DA. Instead, they are a reminder that in the same way DA can modulate the mesocorticolimbic system by activation of receptors that are not their pharmacological counterparts (Paladini et al., 2001), NE may modulate this circuit through activation of DA receptors. In turn, this requires a reevaluation of a popular model in receptor physiology that stays that there is strict correspondence between an agonist and its receptor.
In conclusion, it appears that Ih inhibition by NE in VTA DA cells resulted from inefficient space clamp rather than arising from a modulatory action of second messengers on h-channels. This result also suggests that h-channels are mostly located in the dendrites and thus their activation may prevent the transition from single spike firing to burst firing.
Acknowledgements
This work was supported by the NINDS and NCRR SNRP # NS39408, GM-50695 and GM-08224 to C.A. Jiménez-Rivera. We thank Mr. Rafael Vázquez for his excellent technical assistance and Drs. Priscilla Sanabria and Amelia Rivera for critical review of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Battaglia G, et al. Alpha-1B adrenergic receptor knockout mice are protected against methamphetamine toxicity. Journal of Neurochemistry. 2003;86:413–421. doi: 10.1046/j.1471-4159.2003.01867.x. [DOI] [PubMed] [Google Scholar]
- Berger T, et al. Hyperpolarization-activated current Ih disconnects somatic and dendritic spike initiation zones in layer V pyramidal neurons. Journal of Neurophysiology. 2003;90:2428–2437. doi: 10.1152/jn.00377.2003. [DOI] [PubMed] [Google Scholar]
- Berger T, et al. High Ih channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. Journal of Neurophysiology. 2001;85:855–868. doi: 10.1152/jn.2001.85.2.855. [DOI] [PubMed] [Google Scholar]
- Cathala L, et al. Alpha-2 adrenoreceptor activation increases a cationic conductance and spontaneous gabaergic synaptic activity in dopaminergic neurones of the rat substantia nigra. Neuroscience. 2002;115(4):1059–1065. doi: 10.1016/s0306-4522(02)00542-0. [DOI] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Effect of catecholamines on the hyperpolarization-activated Ih and the inward rectifying potassium Ikir currents in the rat’s substantia nigra pars compacta. European J. of Neuroscience. 1999;11:398–406. doi: 10.1046/j.1460-9568.1999.00452.x. [DOI] [PubMed] [Google Scholar]
- Cathala L, Paupardin-Tritsch D. Neurotensin inhibition of the hyperpolarizationcation current (Ih) in rat substantia nigra pars compacta implicates the protein kinase C pathway. Journal of Physiology. 1997;503(1):87–97. doi: 10.1111/j.1469-7793.1997.087bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC. The significance of action potential bursting in the brain reward circuit. Neurochemistry International. 2002;41:333–340. doi: 10.1016/s0197-0186(02)00068-2. [DOI] [PubMed] [Google Scholar]
- Drouin C, et al. α-1-b-Adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. Journal of Neuroscience. 2002;22:2873–2884. doi: 10.1523/JNEUROSCI.22-07-02873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Cholinergic inhibition of ventral midbrain dopamine neurons. Journal of Neuroscience. 2000;20:7855–7860. doi: 10.1523/JNEUROSCI.20-20-07855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–82. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- Gesi M, et al. The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci. Biobehav. Rev. 2000;24:655–668. doi: 10.1016/s0149-7634(00)00028-2. [DOI] [PubMed] [Google Scholar]
- Grace AA, et al. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends in Neurosciences. 2007;30:220–227. doi: 10.1016/j.tins.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Grace AA, Buney BS. The control of firing pattern in dopamine neurons: burst firing. Journal of Neuroscience. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JG. Gene expression profiles of brain dopamine neurons and relevance to neuropsychiatric disease. Journal of Physiology. 2006;575:411–416. doi: 10.1113/jphysiol.2006.112599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenhoff J, et al. Alpha-1 adrenergic effects on dopamine neurons recorded intracellularly in rat midbrain slice. European J. of Neuroscience. 1995;7:1707–1713. doi: 10.1111/j.1460-9568.1995.tb00692.x. [DOI] [PubMed] [Google Scholar]
- Dahlstrom A, Fuxe K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol. Scand. 1964;62:1–55. [PubMed] [Google Scholar]
- Jiang ZG, et al. Dopamine and baclofen inhibit the hyperpolarization-activated cation current in rat ventral tegmental neurones. Journal of Physiology. 1993;462:753–764. doi: 10.1113/jphysiol.1993.sp019580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. Journal of Physiology. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature. 1979;277:93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- Kitai ST, et al. Afferent modulation of dopamine neuron firing patterns. Current Opinion in Neurobiology. 1999;9:690–697. doi: 10.1016/s0959-4388(99)00040-9. [DOI] [PubMed] [Google Scholar]
- Kubo Y, et al. Primary structure and functional expression of mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Luthi A, McCormick DA. H-current: Properties of neuronal and network pace maker. Neuron. 1998;21:9–12. doi: 10.1016/s0896-6273(00)80509-7. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, et al. Ih Channels Contribute to the Different Functional Properties of Identified Dopaminergic Subpopulations in the Midbrain. The Journal of Neuroscience. 2002;22(4):1290–1302. doi: 10.1523/JNEUROSCI.22-04-01290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, et al. K opiods selectively control dopaminergic neurons projecting to the prefrontal cortex. PNAS. 2006;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Noradrenergic and serotonergic modulation of a hyperpolarization-activated cation current in the thalamic relay neurones. Journal of Physiology. 1990;431:319–342. doi: 10.1113/jphysiol.1990.sp018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, et al. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. European J. of Neuroscience. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- Morikawa H, et al. Inositol 1, 4, 5-triphosphate-evoked responses in midbrain dopamine neurons. Journal of Neuroscience. 2000;20(RC103):1–5. doi: 10.1523/JNEUROSCI.20-20-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odagaki Y, et al. Additivity and non-additivity between dopamine-, norepinephrine-, carbachol- and GABA-stimulated GTPase activity. Europ. J of Pharmacol. 1995;291:245–243. doi: 10.1016/0922-4106(95)90064-0. [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res. Rev. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Williams JT. Noradrenergic Inhibition of midbrain Dopamine Neurons. The Journal of Neuroscience. 2004;24(19):4568–4575. doi: 10.1523/JNEUROSCI.5735-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, et al. Cocaine Self-administration Selectively Drecreases Noradrenergic Regulation of Metabotropic Glutamete Receptor-Mediated Inhibition in Dopamine Neurons. The Journal of Neuroscience. 2004;24(22):5209–5215. doi: 10.1523/JNEUROSCI.1079-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, et al. Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nature Neuroscience. 2001;4(3):275–281. doi: 10.1038/85124. [DOI] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annual Review of Physiology. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1986. [Google Scholar]
- Raymond JR, et al. Alpha 2A adrenergic receptors inhibit cAMP accumulation in embryonic stem cells which lack Gi alpha 2. J. Biol. Chem. 1994;269(18):13073–13075. [PubMed] [Google Scholar]
- Sesack SR, Carr DB. Selective prefrontal cortex inputs to dopamine cells: implications for schizophrenia. Physiol. Behav. 2002;77:513–517. doi: 10.1016/s0031-9384(02)00931-9. [DOI] [PubMed] [Google Scholar]
- Spruston N, et al. Dendritic attenuation of synaptic potentials and currents: the role of passive membrane properties. Trends Neuroscience. 1994;17:161–166. doi: 10.1016/0166-2236(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Ulens C, Siegelbaum SA. Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron. 2003;40:959–970. doi: 10.1016/s0896-6273(03)00753-0. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U. Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol. Scand. 1971;367 (Suppl.):1–48. doi: 10.1111/j.1365-201x.1971.tb10998.x. [DOI] [PubMed] [Google Scholar]
- van Welie I, et al. Different levels of Ih dtermine distinct temporal integration in bursting and regular spiking neurons in rat subiculum. Journal of Physiology. 2006;576(Pt 1):203–214. doi: 10.1113/jphysiol.2006.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AE, et al. Baclofen inhibition of the hyperpolarization-activated cation current, Ih, in rat substantia nigra zona compacta neurons may be secondary to potassium current activation. Journal of Neurophysiology. 1996;76(4):2–10. doi: 10.1152/jn.1996.76.4.2262. [DOI] [PubMed] [Google Scholar]
- Yagi J, Sumino R. Inhibition of a hyperpolarization-activated current by clonidine in rat dorsal root ganglion neurons. Journal of Neurophysiology. 1998;80:1094–1104. doi: 10.1152/jn.1998.80.3.1094. [DOI] [PubMed] [Google Scholar]
- Zhaoping L, et al. Serotonin reduces the hyperpolarization-activated current Ih in ventral tegmental area dopamine neurons: involvement of 5-HT2 receptors and protein kinase C. Journal of Neurophysiology. 2003;90:3201–3212. doi: 10.1152/jn.00281.2003. [DOI] [PubMed] [Google Scholar]
- Zhong H, Minneman KP. Alpha-1 adrenoceptor subtypes. European J. Pharmacology. 1999;375:261–276. doi: 10.1016/s0014-2999(99)00222-8. [DOI] [PubMed] [Google Scholar]