

Abstract
Nonalcoholic fatty liver disease (NAFLD) is associated with obesity, insulin resistance, and type 2 diabetes. NAFLD represents a large spectrum of diseases ranging from (i) fatty liver (hepatic steatosis); (ii) steatosis with inflammation and necrosis; and (iii) cirrhosis. Although the molecular mechanism leading to the development of hepatic steatosis in the pathogenesis of NAFLD is complex, recent animal models have shown that modulating important enzymes in fatty acid synthesis in liver may be key for the treatment of NAFLD. This review discusses recent advances in the field.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is gaining increasing recognition as a component of the epidemic of obesity in the United States as well as in other parts of the world. NAFLD is the most common cause of liver dysfunction and affects close to 20 million patients in the US (1). The spectrum of NAFLD ranges from simple fatty liver (hepatic steatosis), with benign prognosis, to a potentially progressive form, nonalcoholic steatohepatitis (NASH), which may lead to liver fibrosis and cirrhosis, resulting in increased morbidity and mortality. All features of the metabolic syndrome, including obesity, type 2 diabetes, arterial hypertension, and hyperlipidemia (in the form of elevated triglyceride [TG] levels) are associated with NAFLD/NASH (2, 3). NAFLD is generally asymptomatic, although a minority of patients may present with progressive liver injury with complications of cirrhosis, liver failure, and hepatocellular carcinoma. While the clinical diagnosis of NAFLD is usually made based on high transaminase levels, elevated BMI, ultrasound evidence of fat, and features of metabolic syndrome, a liver biopsy is required to determine the presence of NASH and to assess the degree of fibrosis (4). Despite being potentially severe, little is known about the natural history or prognostic significance of NAFLD. Although diabetes, obesity, and age are recognized risk factors for advanced liver disease, other significant factors leading to progressive liver injury remain to be identified. Excessive accumulation of TG in hepatocytes is the hallmark of NAFLD, which is strongly associated with insulin resistance (2–4). As we will discuss, despite the existing correlation between fatty liver and insulin resistance, it remains unclear whether insulin resistance causes the excessive accumulation of TG in liver, or whether the increase in TG itself or of metabolite intermediates may play a causal role in the development of hepatic or systemic insulin resistance. Recent studies have favored the concept that the accumulation of intrahepatic lipids precedes the state of insulin resistance, while others have shown that hepatic TGs themselves are not toxic and may in fact protect the liver from lipotoxicity by buffering the accumulation of fatty acids (5, 6), suggesting in fact that hepatic steatosis is not necessarily associated with insulin resistance (7–9).
Regulation of TG synthesis in liver
Insulin is essential for the maintenance of carbohydrate and lipid homeostasis. Insulin is secreted by pancreatic β cells in response to increased circulating levels of glucose after a meal. A large fraction of glucose absorbed from the small intestine is immediately taken up by hepatocytes, which convert it into glycogen. However, when the liver is saturated with glycogen (roughly 5% of liver mass), any additional glucose taken up by hepatocytes is shunted into pathways leading to synthesis of fatty acids, which will be esterified into TG to be exported to adipose tissue as very low-density lipoproteins (VLDLs). Insulin inhibits lipolysis in adipose tissue by inhibiting hormone-sensitive lipase (HSL), the enzyme regulating FFA release from adipose tissue (10). Therefore, from a whole-body perspective, insulin has a “fat-sparing” effect by driving most cells to preferentially oxidize carbohydrates instead of fatty acids for energy. Insulin also regulates glucose homeostasis at many sites, reducing hepatic glucose production (HGP) (via decreased glucose biosynthesis [gluconeogenesis] and glycogen breakdown [glycogenolysis]) and increasing the rate of glucose uptake, primarily into skeletal muscle and adipose tissue.
Insulin action is initiated through the binding to and the activation of its cell-surface receptor through a series of intramolecular transphosphorylation reactions. Once activated, the insulin receptor phosphorylates several substrates on their tyrosine residues; among such substrates are members of the insulin receptor substrate family (IRS-1, -2, -3, -4) (see ref. 11 for review) (Figure 1). The insulin signal is further spread through a phosphorylation network involving intracellular proteins that propagate the various metabolic actions of insulin. Insulin resistance, which can be considered the result of a signaling defect, occurs when normal circulating concentrations of the hormone are insufficient to regulate key metabolic pathways in adipose tissue, skeletal muscles, and/or liver. One of the approaches used to assess hepatic insulin sensitivity involves measuring the ability of insulin to phosphorylate intracellular substrates such as IRS-1, IRS-2, or Akt (also known as protein kinase B) since defects in the activation of these molecules are known to lead to the inability of insulin to properly inhibit HGP (Figure 1). One transcription factor, Foxo1, plays a key role in the regulation of HGP, through the transcriptional control of gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase (G6Pase) (12) (Figure 1). Insulin-mediated Akt phosphorylation of Foxo1 (13) leads to its nuclear exclusion, ubiquitination, and proteasomal degradation (14). The subsequent decrease in nuclear Foxo1 reduces expression levels of PEPCK and G6Pase, thereby decreasing gluconeogenic rates and reducing blood glucose levels (Figure 1).
Figure 1. Mechanisms by which lipid metabolites affect insulin sensitivity in the liver.

Under conditions in which an adequate transduction signal is present (right panel), insulin binding to the insulin receptor results in the phosphorylation of tyrosine residues (Y) on insulin receptor substrates (IRS-1 and -2), which leads to the activation of PI3K and the subsequent phosphorylation of Akt, which are involved in mediating the metabolic effects of insulin. The transcription factor Foxo1, plays a key role in the regulation of HGP, through the transcriptional control of gluconeogenic enzymes, such as phosphoenolpyruvate carboxykinase (PEPCK). Insulin-mediated Akt phosphorylation of Foxo1 leads to its nuclear exclusion, ubiquitination, and subsequent proteasomal degradation, leading to the decreased PEPCK transcription. In turn, gluconeogenic rates and blood glucose concentrations decrease (14). Under conditions of insulin resistance (HF/HC-diet) (left panel), excess lipid metabolites such as DAG can cause insulin resistance by activating PKCε. The activated PKCε binds to the insulin receptor and inhibits its tyrosine kinase activity. The activation of PKCε may also interfere with the ability of insulin to phosphorylate IRS-2 on tyrosine residues (91). IRE, insulin-responsive element; S, serine; Ub, ubiquitination.
Metabolic pathways leading to hepatic steatosis
Under nonpathological conditions, the potential sources of fats contributing to fatty liver include peripheral fats stored in white adipose tissue that flow to the liver by way of the plasma nonesterified fatty acid (NEFA) pool, dietary fatty acids (mainly through the uptake of intestinally derived chylomicron [CM] remnants), and fatty acids newly made within the liver through de novo lipogenesis (Figure 2). After the esterification step (conversion of FAs into TGs), TGs can then be stored as lipid droplets within hepatocytes or secreted into the blood as VLDL, but they can also be hydrolyzed and the fatty acids channeled towards the β-oxidation pathway. Therefore, excessive fat accumulation in the liver can occur as a result of increased fat delivery, increased fat synthesis, reduced fat oxidation, and/or reduced fat export in the form of VLDL (Figure 2). Studies in humans and in rodents have demonstrated that the mechanisms leading to the excessive accumulation of hepatic TGs are mainly linked to increased delivery of NEFA from peripheral expanded adipose tissue to the liver and enhanced de novo lipid synthesis via the lipogenic pathway in the liver itself, while lipid disposal via β-oxidation and well VLDL export are only moderately affected (15). Strong evidence exists demonstrating that in NAFLD patients, insulin does not suppress adipose tissue lipolysis to the same extent that it does in healthy individuals (15). Because insulin has a potent suppressive effect on HSL (16), there has been some focus on determining whether resistance of HSL to insulin in insulin-resistant states or type 2 diabetes is the predominant defect accounting for the increased flux of NEFA from adipose tissue. Studies have revealed that HSL-knockout mice show increased hepatic sensitivity due to reduced plasma NEFA and hepatic TG concentrations (17, 18). These studies therefore suggest that restricted lipolysis could help prevent a “spillover” of fat from adipose tissue toward liver and therefore prevent hepatic steatosis and/or insulin resistance. Altogether, several studies have manipulated gene expression in adipose tissue and, until recently, fewer approaches had been developed to determine the contribution of de novo fat synthesis to the development of hepatic steatosis.
Figure 2. Metabolic defects leading to the development of hepatic steatosis.

Different sources of fatty acids contribute to the development of fatty liver. Under conditions of insulin resistance, insulin does not adequately inhibit HSL, and lipolysis in white adipose tissue is not suppressed. Therefore peripheral fats stored in adipose tissue flow to the liver by way of plasma NEFAs. Dietary fatty acids are also taken up by the liver through the uptake of intestinally derived chylomicron (CM). In addition, the combination of elevated plasma glucose (hyperglycemia) and insulin concentrations (hyperinsulinemia) promotes de novo fatty acid synthesis (lipogenesis) and impairs β-oxidation, thereby contributing to the development of hepatic steatosis. After the esterification step (conversion of FAs into TGs) TG can then be stored as lipid droplets within hepatocytes or secreted into the blood as VLDL. Although the hepatic accumulation of lipids is widely believed to result in insulin resistance, it remains uncertain whether a causal relationship exists. Several recent studies have even showed a clear dissociation between hepatic steatosis and insulin resistance (7, 87). FA, fatty acid.
Contribution of de novo fat synthesis to VLDL production
The synthesis of TGs in liver is nutritionally regulated, and its formation from simple carbohydrates requires multiple metabolic pathways, including glycolysis and pyruvate oxidation to generate acetyl-CoA for fatty acid synthesis, NADPH generation to supply the reductive power, packaging of fatty acids into a glycerophosphate backbone, and finally, lipoprotein packaging to export TGs. Feeding previously fasted animals a low-fat/high-carbohydrate (LF/HC) diet causes a marked induction of enzymes involved in these pathways, including (i) glucokinase (GK) (19) and liver-pyruvate kinase (L-PK) (20) for glycolysis; (ii) ATP citrate lyase (21), acetyl-CoA carboxylase (ACC) (22), and fatty acid synthase (FAS) (23) for lipogenesis; (iii) long-chain elongase (ELOVL6, also known as LCE) (24) and stearoyl-CoA desaturase 1 (SCD1) (25), catalyzing fatty acid elongation and desaturation steps; and finally (iv), mitochondrial glycerol 3-phosphate acyltransferase (GPAT) and diacylglycerol acyltransferase (DGAT) for TG synthesis (26) (Figure 3). Under nonstimulated conditions, the contribution of de novo fat synthesis to fatty acid, TG, and VLDL synthesis is small in humans, estimated to be less than 5% in the postabsorptive state (27). Nevertheless, a strong correlation exists between the rates of de novo lipogenesis and the secretion of VLDL, even under basal conditions (28). Conditions associated with high rates of lipogenesis, such as LF/HC-diet ingestion, hyperglycemia, and hyperinsulinemia are associated with a shift in cellular metabolism from lipid oxidation to TG esterification, thereby increasing the availability of liver TGs derived from VLDL synthesis and secretion (Figure 2). Indeed, elevation in malonyl-CoA, which is the product of the lipogenic enzyme ACC, inhibits liver carnitine palmitoyltransferase I (L–CPT I), the rate limiting enzyme of β-oxidation, which regulates the transfer of long-chain acyl-CoAs (LCCoAs) from the cytosol into the mitochondria, thus, resulting in a shift from an oxidative to a reesterification pathway (29) (Figure 3). Using a multiple-stable-isotope approach, Donnelly et al. (30) estimated the contribution of de novo lipid synthesis to hepatic fat content in NAFLD patients. Of the TG content accounted for in the liver of these patients, 60% arose from NEFA, a little over 10% from the diet, and close to 30% came from de novo lipogenesis, underlying the importance of fat synthesis in the pathology of NAFLD. We will discuss some of the recent genetically engineered mouse models that have helped identify the enzymes and/or transcription factors that play a key role in the control of fat synthesis.
Figure 3. Metabolic pathways leading to the synthesis of TGs in liver.

The synthesis of TGs in liver is nutritionally regulated. The ingestion of a LF/HC diet causes a marked induction of enzymes involved in key metabolic pathways, including (i) glucokinase (GK) and L-PK for glycolysis; (ii) ATP citrate lyase, ACC, and FAS for lipogenesis; (iii) ELOVL6 and SCD1 for fatty acid elongation and desaturation steps; and finally (iv) GPAT and DGAT for TG synthesis. Under these nutritional conditions, elevation in malonyl-CoA concentrations, the product of the lipogenic enzyme ACC, inhibits L–CPT I, the rate-limiting enzyme of β-oxidation (v), which regulates the transfer of long-chain acyl-CoAs from the cytosol into the mitochondria, thus resulting in a shift from an oxidative (production of ketone bodies) to an esterification pathway (TG synthesis). F6P, fructose 6-phosphate; F1, 6P2, fructose 1,6 diphosphate; G3P, glycerol 3-phosphate; G6P, glucose 6-phosphatase; PEP, phosphoenol pyruvate; LCFA, long-chain fatty acids; CPT II, carnitine palmitoyltransferase II.
Inhibiting ACC expression prevents hepatic steatosis and related syndromes
For the ACC, FAS, and SCD1 enzymes (Figure 3), animal models of their knockdown and/or knockout have been generated and have helped to provide a better understanding of important regulatory checkpoints in fat synthesis. Because ACC catalyzes the synthesis of malonyl-CoA, the metabolic intermediate between lipogenesis (31) and β-oxidation (32) (Figure 3), this lipogenic enzyme has garnered significant attention over recent years. In mammals, two ACC isoforms exist, each with distinct tissue distribution and physiological roles: ACC1 is cytosolic and participates in de novo lipogenesis, while ACC2 is mitochondrial and is thought to be involved in the negative regulation of mitochondrial β-oxidation by modulating local malonyl-CoA levels. ACC1 is highly expressed in liver and adipose tissue, whereas ACC2 is predominantly expressed in heart and skeletal muscle and, to a lesser extent, in liver (33). It is believed that only ACC1, but not ACC2, is committed to de novo lipogenesis in liver (34), since it has been shown that malonyl-CoA synthesized by ACC2 cannot be accessed by the FAS enzyme due to a strict mitochondrial compartmentalization of the metabolite (33). Global ACC2-knockout mice (Acc2–/– mice) are leaner than controls due to increased fat β-oxidation in heart and skeletal muscle but also in liver, suggesting that lowering local malonyl-CoA concentrations in the vicinity of mitochondria is indeed important to control L–CPT I activity (35). In addition, the protection of Acc2–/– mice against the development of high-fat/high-carbohydrate (HF/HC) diet–induced obesity and diabetes is due to enhanced fatty acid oxidation in these mice (36).
Global inactivation of ACC1 was also performed but led to embryonic lethality, indicating that de novo fatty acid synthesis is essential for embryonic development (37). Clarification of the role of ACC1 in the control of de novo lipogenesis was attempted by the generation of two liver-specific ACC1-knockout mouse models (38, 39). However, these models led to moderate phenotypes and did not clearly elucidate the role of ACC1, since, in both cases, a compensatory upregulation in ACC2 expression occurred due to the lack of functional ACC1. Despite this compensatory effect, liver-specific ACC1-knockout mice (LACC1KO) mice generated by Mao et al. (38) showed a 70% reduction in hepatic malonyl-CoA concentrations compared with controls, a 50% decrease in de novo fatty acid synthesis (as measured by the incorporation of 14C-acetate into isolated hepatocytes), and a 40% decrease in hepatic TG concentration. Surprisingly, despite the significant decrease in lipid accumulation in liver, LACC1KO mice failed to be protected against the development of HF/HC diet–induced obesity, fatty liver, or insulin-resistance. Similarly, and mainly due to the compensatory effect of ACC2, liver-specific ACC1 mice generated by Harada et al. (39) showed virtually no alteration in lipogenic rates in liver. Interestingly, in this mouse model, hepatic malonyl-CoA levels were not decreased, suggesting that malonyl-CoA synthesized by ACC2 is involved, not only in mitochondrial fatty acid oxidation but also in cytosolic de novo lipogenesis, at least under the artificial conditions induced by ACC1 disruption. An alterative approach to gene knockout, in which intraperitoneal injection of antisense oligonucleotide (ASO) inhibitors was used to knock down ACC1 and ACC2 expression, either independently or synergistically, helped resolve the respective roles of these enzymes in the control of de novo lipogenesis (40). While this approach did not allow for a complete depletion in protein levels (20% of residual ACC1 and ACC2 expression was measured after ASO treatment), no compensatory increase in the nontargeted isoform was observed. In vitro suppression of ACC1 inhibited lipogenesis in primary rat hepatocytes whereas ACC2 reduction had no effect. However, under HF-diet conditions in vivo, the synergic inhibition of ACC1 and ACC2 was required to significantly reduce hepatic malonyl-CoA concentrations, lower hepatic lipids, and improve hepatic insulin sensitivity. ACC1 and ACC2 ASO therapy significantly reduced HGP during the phase of the euglycemic-hyperinsulinemic clamp study. This beneficial effect was associated with increases in both Akt and Foxo1 phosphorylation. Altogether, this study has clearly revealed that targeting ACC has beneficial effects on both hepatic steatosis and insulin resistance (40).
Global and liver-specific inactivation of FAS
Because FAS catalyzes the last step in the fatty acid biosynthetic pathway (Figure 3), it is believed to be a determinant of the maximal capacity of a tissue, and liver in particular, to synthesize fatty acids by de novo lipogenesis. Similarly to the global inactivation of ACC (37), deletion of the gene coding for FAS results in embryonic lethality (41). Chakravarthy et al. (42) showed that liver-specific FAS knockout in mice (FASKOL mice) results in mutant mice that possess a similar phenotype to controls animals when fed normal chow. Surprisingly, the lack of FAS did not protect against the development of fatty liver but rather exacerbated it (under specific nutritional conditions). Indeed, when fed a LF/HC diet for 4 weeks, FASKOL mice developed hepatic steatosis due to a reduction in β-oxidation, as evidenced by a 3-fold increase in hepatic malonyl-CoA concentrations and a significant decrease in blood ketone bodies (42). In fact, this mouse model led to the novel and interesting concept that “new fat” synthesized via FAS activity (mainly saturated palmitate [16:0]) would specifically activate a pool of nuclear receptors (e.g., PPARα) and would in turn lead to enhanced β-oxidation. PPARα is a common mediator of the transcriptional effects of long-chain fatty acids (LCFAs). However, the transcriptional effect of PPARα on L–CPT I remains controversial since studies have suggested that LCFA regulates L–CPT I expression through a PPARα-independent pathway (43, 44). In addition, since unsaturated fats are known to be preferential ligands for PPARα, the exact mechanism by which fatty acids, generated through FAS activity, may activate PPARα in liver remains to be determined.
Deficiency in SCD1 protects against adiposity and hepatic steatosis
Also based on data derived from animal models, SCD1 has recently become a target of interest for the reversal of hepatic steatosis and insulin resistance (45). SCD1 catalyzes the synthesis of monounsaturated fatty acids, particularly oleate (C18:1n-9) and palmitoleate (C16:1n-7) (Figure 3), which are the major components of membrane phospholipids, TGs, and cholesterol esters. Mice with a global knockout of SCD1 (Scd1–/– mice) show decreased lipogenic gene expression and increased β-oxidation and are protected from diet-induced obesity and insulin resistance when fed a HC/HF diet (46, 47). Inhibition of SCD1 using an ASO strategy (targeting SCD1 in both liver and adipose tissues) also prevents many of the HF/HC-diet metabolic complications, including hepatic steatosis and postprandial hyperglycemia (48, 49). In these studies, the protective effect of SCD1 on hepatic steatosis is attributed to a combined decrease in lipogenic rates and to the activation of the β-oxidation pathway, underlying once again the importance of modulating both pathways in vivo. However, it is not clear how SCD1 affects and/or regulates lipogenic rates in liver. Additional information was recently obtained via the liver-specific knockout of SCD1 (LKO mice) (50). These mice are protected from LF/HC-diet–induced obesity and hepatic steatosis (50). Under both short-term and long-term conditions, LKO mice exhibit reduced rates of fatty acid synthesis in liver and decreased expression of key genes of the lipogenic pathway (such as Fas and Acc). In fact, hepatic SCD1 deficiency reduces the nuclear content of two key factors (carbohydrate-responsive element–binding protein [ChREBP] and SREBP-1c) (50) involved in the transcriptional control of lipogenic gene expression in response to glucose and insulin, respectively, as we will discuss below (Figure 4). The exact mechanism by which SCD1 affects the maturation and/or the translocation of these two transcription factors is not clear but could be linked to oleate concentrations in hepatocytes. Indeed, supplementation of oleate in the LF/HC diet restores both nuclear SREBP-1c and ChREBP content in LKO livers (50). These results are not in agreement with studies from our laboratory, in which oleate, when tested both in vivo and in vitro (e.g., in primary cultures of hepatocytes), failed to induce ChREBP and SREBP-1c gene expression (51).
Figure 4. Enzymes of TG synthesis are transcriptionally regulated by ChREBP, SREBP-1c, and LXRs in liver.
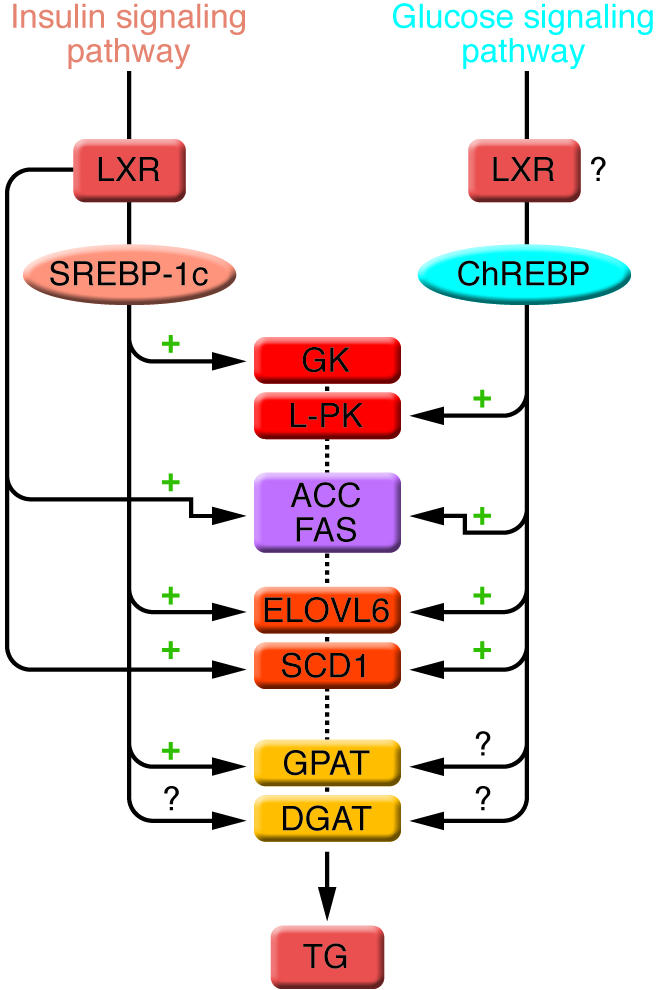
The conversion of glucose into TGs is nutritionally regulated, and both insulin and glucose signaling pathways are activated in response to dietary carbohydrates to synergistically induce gene expression. The transcription factor SREBP-1c mediates the effect of insulin on GK, ACC, FAS, ELOVL6, SCD1, and GPAT. LXRs are also important regulators of TG synthesis, through the direct transcriptional activation of ACC, FAS, and SCD1 but also indirectly via the insulin-mediated transcriptional activation of SREBP1-c. ChREBP, which is induced by glucose, is required for the induction of L-PK, which is exclusively dependent on glucose. The induction of ACC, FAS, ELOVL6, and SCD1 genes is under the synergistic action of ChREBP and SREBP-1c in response to glucose and insulin, respectively. The direct effect of ChREBP on GPAT expression is not clearly established. ChREBP is also a direct transcriptional target of LXRs, and glucose was recently shown to bind and activate LXRs, thereby indicating that LXRs are part of the glucose signaling pathway. However, in our point of view, the relevance of a potential regulation by LXRs of glucose-sensitive genes needs to be demonstrated in a physiological context. The transcriptional regulation of DGAT in liver is, to our knowledge, still largely unknown.
Transcriptional control of fat synthesis
If modulating the lipogenic pathway is interesting for the treatment of hepatic steatosis and insulin resistance, a better knowledge of the transcriptional, posttranslational, and/or pharmacological regulation of enzymes involved in fatty acid synthesis appears important for the development of potential therapeutic approaches. These enzymes are acutely regulated by posttranslational and allosteric mechanisms but are mainly controlled on a long-term basis by a modulation of their transcription rate. It is now well accepted that the transcription factor SREBP-1c, itself stimulated by insulin, mediates the transcriptional effect of this hormone on the enzymes involved in fatty acid synthesis (e.g., ACC, FAS, SCD1, ELOVL6) (52–54) and TG synthesis (e.g., GPAT) (55) (Figure 4). At least two independent mammalian DGAT enzymes are expressed. DGAT1 likely plays a role in intestinal repackaging of FFA, whereas DGAT2 (mainly expressed in liver) functions primarily in TG synthesis (56). To our knowledge, little is known about DGAT2 transcriptional control in liver. DGAT2 was not reported as a potential target of SREBP-1 in the microarray analysis of SREBP-1 transgenic mice (53).
In addition to SREBP-1c, liver X receptors (LXRs) are also important regulators, since they directly regulate lipogenic genes (FAS, ACC, and SCD1) (57–59) but are also required for the transcriptional control of SREBP-1c by insulin (60) (Figure 4). However, SREBP-1c activity alone is not sufficient to account for the stimulation of gene expression in response to carbohydrate since deletion of the gene encoding SREBP-1c in mice only results in a 50% reduction in fatty acid synthesis (58). More importantly, L-PK, one of the rate-limiting enzymes of glycolysis, is exclusively dependant on glucose (61) and is not subjected to SREBP-1c regulation (62) (Figure 4). Until recently, the nature of the glucose-signaling compound was not known, but the identification of a glucose-responsive basic helix-loop-helix/leucine zipper (bHLH/LZ) transcription factor named ChREBP has shed light on the mechanism whereby glucose affects gene transcription (63, 64). ChREBP is a large protein (864 amino acids and 94,600 Da) that contains several domains, including a nuclear localization signal (NLS) near the N-terminus polyproline domains, a bHLH/LZ, and a LZ–like (Zip-like) domain. Glucose activates ChREBP by regulating its entry from the cytosol into the nucleus (51, 65), thereby promoting its binding to the carbohydrate-responsive element (ChoRE) present in the promoter regions of glycolytic and lipogenic genes (reviewed in ref. 66). ChREBP is also a target of LXRs (67), thereby placing LXRs at the crossroad of the insulin and glucose pathways via SREBP-1c and ChREBP, respectively (Figure 4). Interestingly, glucose was also recently shown to bind and activate LXRs, leading to the activation of their target genes, including ChREBP (68). While the results of this study posited LXRs as master regulators of the glucose signaling pathway in liver, several concerns were raised (69), including the fact that the experiments were performed in HepG2 cells, a hepatoma cell line that responds poorly to glucose, and that phosphorylated sugars, which cannot be transported inside the cell, were reported to induce LXR promoter activity with a similar affinity as glucose, when directly added to the culture medium (68). Therefore, the relevance of a potential regulation of LXRs by glucose needs to be demonstrated in a physiological context.
Studies by Towle and colleagues have revealed that ChREBP binds as a heterotetramer, together with its functional partner Mlx (Max-like protein X) on carbohydrate-response elements (70, 71). Target genes of ChREBP/Mlx have been identified using a microarray profiling strategy in primary hepatocytes, and most of the lipogenic genes were found to be regulated by the ChREBP-Mlx complex (70). Aside from previously known targets of ChREBP, such as L-PK, ACC, FAS, and SCD1 (70, 72, 73), novel targets were also identified, including enzymes of the pentose phosphate pathway necessary for the generation of NAPDH (e.g., glucose-6-phosphate dehydrogenase [G6PDH], transketolase), the enzyme catalyzing the formation of glycerol 3-phosphate necessary for TG synthesis and glycerol 3-phosphate dehydrogenase (GPDH) as well as microsomal TG transfer protein (MTP), the rate-limiting enzyme of VLDL production (70). In addition, the observation that hepatic ELOVL6, the elongase that catalyzes the conversion of palmitate to stearate (Figure 3), is also transcriptionally regulated by ChREBP and Mlx, further underlines the importance of ChREBP in the control of the entire lipogenic program (74).
A role for ChREBP in the prevention of hepatic steatosis and insulin resistance
Based on these recent findings, we have chosen to determine the role of ChREBP in the pathophysiology of hepatic steatosis using the ob/ob mouse model. While most models of hepatic steatosis are created through HF/HC-diet feeding in rodents, the ob/ob mouse is also commonly studied not only as a model of early onset of severe obesity and insulin resistance but also as an excellent model of fatty liver. These mice have a mutation in the ob gene that prevents the synthesis of leptin (75). Leptin, a satiety hormone synthesized by the white adipose tissue, inhibits feeding behavior and increases energy expenditure by acting on anorexigenic neurons in the ventral median nucleus of the hypothalamus (76). In the absence of leptin, ob/ob mice are hyperphagic, inactive, and become obese. These animals are also insulin resistant and hyperinsulinemic, with resultant hyperglycemia and hyperlipidemia. Most importantly, ob/ob mice spontaneously develop fatty livers, due in part to an exacerbated glycolytic and lipogenic pathway (77). Lipid droplet accumulation is clearly seen in this obese model and is characterized by TG deposition in the liver (78). The ob/ob mouse model was previously used to determine the role of SREBP-1c in the development of hepatic steatosis. Interestingly, while SREBP-1c–/– ob/ob mice showed a significant improvement of their hepatic steatosis, systemic insulin resistance was not prevented (79). It should also be noted that SREBP-1c expression was not only inhibited in liver but also in white adipose tissue of ob/ob mice, thereby preventing the liver-specific contribution of SREBP-1c to be determined.
To specifically target liver and to determine the relative contribution of ChREBP in the control of lipogenesis, we have used an adenovirus-mediated RNA interference technique to inhibit ChREBP expression in the liver of ob/ob mice (80). Adenoviral delivery of short hairpin ChREBP–RNA (shChREBP-RNA) in vivo efficiently knocked down ChREBP expression in liver of ob/ob mice under both short- (2 days) and long-term (7 days) conditions (80). At the molecular level, ChREBP knockdown led to the expected inhibition of L-PK, ACC, FAS, and SCD1 as well as GPAT. While a carbohydrate-response element was previously identified in the promoter region of the GPAT gene (81), its expression was found to be unaffected in liver of ChREBP-knockout mice upon refeeding (67). It is possible that the nutritional regulation of GPAT may be more sensitive to insulin via SREBP-1c than to glucose via ChREBP. Nevertheless, following ChREBP knockdown, a resultant decrease in lipogenic rates was observed in shChREBP-RNA–treated ob/ob mice, leading to a 50% reduction in hepatic and circulating TG concentrations (80). ChREBP knockdown not only affected the rate of de novo lipogenesis but also had consequences on β-oxidation. Therefore, similarly to the liver-specific knockout of SCD1 (LKO mice) (50), the coordinated modulation in fatty acid synthesis and oxidation in liver led to an overall improvement of lipid homeostasis in ChREBP-deficient mice. In addition, in agreement with our data, the decrease in lipogenic rates observed in LKO mice was at least partially attributed to a decrease in ChREBP nuclear protein content (50). Clearly, ChREBP needs now to be considered as a key determinant of the molecular regulation of the lipogenic pathway (82).
Correction of hepatic steatosis via ChREBP knockdown also led to decreased levels of plasma TG and NEFA (80). As a consequence, insulin signaling was improved in liver, skeletal muscles, and white adipose tissue, and systemic glucose tolerance and insulin sensitivity were restored in ob/ob mice after a 7-day treatment with the shChREBP-RNA (80). With the discovery of ChREBP, our understanding of the long-term regulation of glucose and lipid metabolism in liver has recently made considerable progress. ChREBP deficiency overcomes the fatty liver phenotype, and improves glucose tolerance and insulin resistance in ob/ob mice, suggesting that a reduction of ChREBP activity may have a beneficial effect in the treatment of metabolic diseases associated with hyperglycemia and dyslipidemia. The study by Uyeda and coworkers (83) further underlined the importance of ChREBP in the development of obesity and type 2 diabetes by intercrossing ChREBP knockout mice (72) with ob/ob mice. Similarly to what we observed, hepatic fat accumulation was prevented, and the hyperlipidemic phenotype was significantly improved (83). The implication of ChREBP in the development of hepatic steatosis in NAFLD needs to be determined in future studies.
Role of hepatic steatosis per se in the development of insulin resistance
As discussed above, an association between hepatic steatosis and insulin resistance exists, and several rodent models have shown that decreasing hepatic TG pools correlates with improved insulin sensitivity (40, 80, 84). However, it remains uncertain whether a causal relationship exists. Several lines of evidence suggest that lipid metabolites, such as acyl-CoAs and diacylglycerol (DAG), rather that TGs themselves, are determinants for the development of insulin resistance (Figure 1). Studies have shown that cellular TG accumulation per se is not initially toxic (5, 6). In fact, the accumulation of excess fatty acids into TG pools may divert fatty acids from pathways that could be cytotoxic, such as the generation of ROS (85) or ceramides, and this leads to subsequent alteration of mitochondrial function (86). Using a mouse model of hepatic steatosis induced by a methionine- and choline-deficient diet (methionine and choline are essential for the export of TGs as VLDL), Yamaguchi et al. (5) showed that while the inhibition of TG synthesis (mediated through the reduction in DGAT2 activity) led to the improvement of liver steatosis, it also increased liver damage. Indeed, levels of hepatic fatty acids, cytochrome P450, and markers of lipid peroxidation and oxidant stress were markedly increased as well as fibrosis. Interestingly, liver damage occurred despite a significant amelioration in systemic insulin sensitivity in the treated mice, suggesting that TG synthesis may in fact protect against lipotoxicity by buffering the accumulation of fatty acids in liver (5). Dissociation between hepatic steatosis and insulin resistance was also observed in transgenic mice overexpressing DGAT2 in liver (Liv-dgat2 mice) (7). Despite a significant hepatic steatosis, Liv-dgat2 mice (fed a standard diet) show normal in vivo glucose and insulin tolerance. In hyperinsulinemic-euglycemic clamp studies, Liv-dgat2 mice also had a similar HGP to wild-type mice, demonstrating that Liv-dgat2 mice are not insulin-resistant. In agreement with the clamp studies, PEPCK expression as well as insulin-mediated phosphorylation of Akt were identical in livers of Liv-dgat2 animals compared with controls (7).
Lipotoxicity has been almost exclusively attributed to saturated fat. For example, oleate (C18:1) supplementation is well tolerated in Chinese hamster ovary (CHO) cells, because it leads to synthesis of TGs, while an excess of palmitate (C16:0), characterized by a lower incorporation rate into the TG pool, leads to subsequent apoptosis (6). An unexpected phenotype was obtained from the analysis of mice deficient for ELOVL6 (Elovl6–/– mice) (Figure 3). Paradoxically, Elovl6–/– mice are protected against the development of hepatic insulin resistance when fed a HF/HC diet, despite the accumulation of palmitate concentrations. Improvement in insulin signaling (as evidence by the restoration in insulin-mediated Akt phosphorylation) occurred despite hepatic steatosis and marked obesity in Elovl6–/– mice (87). While these results are somehow surprising given the role of palmitate as a potent inducer of insulin resistance (at least in primary cultures of hepatocytes) (88), they are also interesting since they indicate that the hepatic fatty acid composition, and particularly the conversion of palmitate to stearate, is crucial for insulin sensitivity. It should be noted that the reduced expression in SCD1 expression observed in livers of Elovl6–/– mice could have also contributed to the amelioration of insulin resistance in these mice (87).
The concept that lipid metabolites may contribute to the development of insulin resistance emerged from elegant studies mostly published by the Shulman laboratory. Lipid metabolites including acyl-CoA, lysophosphatidic acid (LPA), and DAG are provided through the glycerol 3-phosphate pathway, and GPAT, by catalyzing the formation of LPA, is considered to be one of the rate-limiting enzymes (55) (Figures 1 and 3). Both GPAT-knockout mice (84) and mice overexpressing GPAT in liver (89) have been studied, providing evidence for an important role of this enzyme in the development of hepatic steatosis. GPAT-knockout mice show reduced levels of both DAG and TGs in liver, and as a result were protected against HF/HC-diet–induced hepatic insulin resistance. Interestingly, while the inhibition of GPAT led to a significant accumulation in hepatic acyl-CoA content, GPAT-knockout mice did not exhibit hepatic insulin resistance, suggesting that DAG is most likely the better candidate to account for insulin resistance. It should be noted, however, that overexpressing key enzymes of TG synthesis (GPAT vs. DGAT2) leads to quite divergent phenotypes, suggesting that modulating TG synthesis at different steps may be a determinant for the outcome of insulin resistance. Indeed, the overexpression of GPAT in rat liver is associated with hepatic steatosis and insulin resistance, while Liv-dgat2 mice, despite elevated concentrations DAG in liver, remain sensitive to the action of insulin (7). Nevertheless, follow-up studies showed that excess DAG causes insulin resistance by activating a specific isoform of protein kinase C, PKCε (90, 91) (Figure 1). PKCε is a serine-threonine kinase that when activated binds to the insulin receptor and inhibits its tyrosine kinase activity (91). The activation of PKCε may also interfere with the ability of insulin to phosphorylate IRS-2 on tyrosine residues. Adenoviral expression of GPAT in liver of rats also supported the importance of DAG versus acyl-CoAs in the development of hepatic insulin resistance. In GPAT-overexpressing rats, hepatic insulin resistance was associated with elevated levels of LPA, DAG, and TGs but not of acyl-CoAs. In addition, a 30% increase in DAG-mediated activation of PKCε was observed. Finally, ASO-mediated PKCε knockdown protected rats against HF-diet–induced hepatic insulin resistance and restored the insulin-mediated inhibition of HGP.
Importance of the metabolic zonation in liver
Evidence has accumulated over the years supporting the concept that metabolic pathways undergo specific zonation in the liver (92–94). Two different zones may be distinguished within the liver acinus, namely an afferent periportal area and an efferent perivenous region. While the periportal region is dedicated to gluconeogenesis, the perivenous region is the preferential site for glycolysis and lipogenesis. Concerning fatty acid oxidation, higher rates of oxidation are detected in periportal hepatocytes as evidenced by higher L–CPT I activity as well as a lower sensitivity of the enzyme to inhibition by malonyl-coA. In contrast, no major difference between the two pools of hepatocytes seems to exist in relation to VLDL secretion (92). Therefore, the periportal zone is less likely exposed to fatty acid accumulation, while TG pools probably preferentially accumulate within perivenous hepatocytes. Therefore, according to this zonation concept, one can wonder whether it is relevant to correlate excessive hepatic TG accumulation with the failure of insulin to inhibit gluconeogenesis, since the two metabolic processes are probably located in different liver zones (e.g., TG accumulation in perivenous and gluconeogenesis in periportal hepatocytes). While difficult to achieve, it would be important that studies addressing the causal relationship between excessive hepatic TG accumulation and insulin resistance be carried out separately in the two different hepatocyte subpopulations instead of being performed in vivo or using classical heterogeneous liver cell preparations.
Current treatment options for NAFLD
There is currently no generally accepted treatment for NAFLD. To date the only effective treatments of NAFLD are lifestyle changes (diet, weight reduction, and exercise). As NAFLD seems to be caused and worsened by insulin resistance, the most promising drugs are agents that restore insulin sensitivity. Thiazolidinediones (TZDs) are a class of oral antidiabetic drugs that improve insulin sensitivity by acting as a selective agonist of the nuclear PPARγ. TZDs reduce hepatic and peripheral insulin resistance, decrease hepatic steatosis, and attenuate the inflammatory response (95–97). TZDs exert insulin-sensitizing actions directly on adipocytes (increase number and differentiation, stimulation of glucose uptake) and indirectly via decreased lipolysis and altered release of adipokines. TZDs decrease the secretion of antiinsulin adipokines (TNF-α and resistin) and increase the secretion of insulin-like adipokine (adiponectin) by adipocytes (98). However, the main side effect of TZDs is weight gain and increased body adiposity. Until recently, increased body adiposity was considered a negative factor. This must be reconsidered in the light of recent experiments reporting that increased expression of circulating levels of adiponectin completely rescued the diabetic phenotype in ob/ob mice (99). In the study by Kim et al. (99), ob/ob mice were crossed with transgenic mice overexpressing adiponectin in adipose tissue. These mice displayed increased expression of PPARγ target genes and a reduction in macrophage infiltration in adipose tissue. As a result, these mice, while morbidly obese, showed reduced TG levels in liver and muscle, and showed an improvement of insulin sensitivity. It has been proposed that it is ectopic fat accumulation that induces insulin resistance, and that the capacity of adipose tissue to expand affects metabolic homeostasis (99, 100).
Concluding remarks
Although rodent models of hepatic steatosis and/or insulin resistance do not always perfectly reproduce the human pathology of NAFLD, the use of transgenic, knockout, and knockdown mouse models have helped over the past years to better our understanding of the molecular determinants of NAFLD. Key enzymes of fatty acid synthesis, such as ACC, ELOVL6, SCD1, GPAT, or DGAT (7, 40, 46–49, 87), have been shown, when knocked down, to reverse many of the metabolic defects associated with hepatic steatosis and/or insulin resistance, indicating that decreased TG synthesis in liver is a potential and interesting target for the treatment of NAFLD. Therefore, a better knowledge of the function and/or regulation of the transcription factors that control the activity of the enzymes modulating fatty acid synthesis in liver, namely ChREBP, SREBP-1c, and LXRs (Figure 4), may in the future help the development of potential therapeutic approaches for this disease.
Acknowledgments
The authors would like to thank all the members of the “Insulin-glucose signaling” team for helpful comments and discussion. The work from our laboratory was supported by grants from Alfediam/Sanofi-Synthelabo, from the Agence Nationale pour la Recherche (ANR-05-PCOD-035-02), and from the Programme National de Recherche sur le Diabète (PNRD-2005).
Footnotes
Nonstandard abbreviations used: ACC, acetyl-CoA carboxylase; ASO, antisense oligonucleotide; ChREBP, carbohydrate-responsive element–binding protein; DAG, diacylglycerol; DGAT, DAG acyltransferase; ELOVL6, long-chain elongase; FAS, fatty acid synthase; GPAT, mitochondrial glycerol 3-phosphate acyltransferase; HF/HC, high fat/high carbohydrate; HPG, hepatic glucose production; HSL, hormone-sensitive lipase; L–CPT I, liver–carnitine palmitoyltransferase I; LF/HC, low fat/high carbohydrate; L-PK, liver-pyruvate kinase; LXR, liver X receptor; Mlx, Max-like protein X; NAFLD, nonalcoholic fatty liver disease; NEFA, nonesterified fatty acid; SCD1, stearoyl-CoA desaturase-1; TG, triglyceride; TZD, thiazolidinedione; VLDL, very LDL.
Conflict of interest: J. Girard has served as an advisor or consultant within the last year to Sanofi-Aventis, Novartis Pharmaceuticals, Novo Nordisk, and GlaxoSmithKline.
Citation for this article: J. Clin. Invest. 118:829–838 (2008). doi:10.1172/JCI34275.
References
- 1.Ahmed M.H., Byrne C.D. Modulation of sterol regulatory element binding proteins (SREBPs) as potential treatments for non-alcoholic fatty liver disease (NAFLD). Drug Discov. Today. 2007;12:740–747. doi: 10.1016/j.drudis.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Abdelmalek M.F., Diehl A.M. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med. Clin. North Am. 2007;91:1125–1149. doi: 10.1016/j.mcna.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Charlton M. Nonalcoholic fatty liver disease: a review of current understanding and future impact. Clin. Gastroenterol. Hepatol. 2004;2:1048–1058. doi: 10.1016/s1542-3565(04)00440-9. [DOI] [PubMed] [Google Scholar]
- 4.Adams L.A., Lindor K.D. Nonalcoholic fatty liver disease. Ann. Epidemiol. 2007;17:863–869. doi: 10.1016/j.annepidem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi K., et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 6.Listenberger L.L., et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monetti M., et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Rinella M.E., Green R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 9.Buettner R., Ottinger I., Scholmerich J., Bollheimer L.C. Preserved direct hepatic insulin action in rats with diet-induced hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2004;286:E828–E833. doi: 10.1152/ajpendo.00453.2003. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Yanes C., Sanchez-Margalet V. Signalling mechanisms regulating lipolysis. Cell. Signal. 2006;18:401–408. doi: 10.1016/j.cellsig.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Pessin J.E., Saltiel A.R. Signaling pathways in insulin action: molecular targets of insulin resistance. J. Clin. Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puigserver P., et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 13.Nakae J., Kitamura T., Ogawa W., Kasuga M., Accili D. Insulin regulation of gene expression through the forkhead transcription factor Foxo1 (Fkhr) requires kinases distinct from Akt. Biochemistry. 2001;40:11768–11776. doi: 10.1021/bi015532m. [DOI] [PubMed] [Google Scholar]
- 14.Matsuzaki H., Daitoku H., Hatta M., Tanaka K., Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. U. S. A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis G.F., Carpentier A., Adeli K., Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002;23:201–229. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 16.Ginsberg H.N., Zhang Y.L., Hernandez-Ono A. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch. Med. Res. 2005;36:232–240. doi: 10.1016/j.arcmed.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Voshol P.J., et al. Increased hepatic insulin sensitivity together with decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology. 2003;144:3456–3462. doi: 10.1210/en.2002-0036. [DOI] [PubMed] [Google Scholar]
- 18.Park S.Y., et al. Hormone-sensitive lipase knockout mice have increased hepatic insulin sensitivity and are protected from short-term diet-induced insulin resistance in skeletal muscle and heart. Am. J. Physiol. Endocrinol. Metab. 2005;289:E30–E39. doi: 10.1152/ajpendo.00251.2004. [DOI] [PubMed] [Google Scholar]
- 19.Iynedjian P.B., Ucla C., Mach B. Molecular cloning of glucokinase cDNA. Developmental and dietary regulation of glucokinase mRNA in rat liver. J. Biol. Chem. 1987;262:6032–6038. [PubMed] [Google Scholar]
- 20.Vaulont S., Munnich A., Decaux J.F., Kahn A. Transcriptional and post-transcriptional regulation of L-type pyruvate kinase gene expression in rat liver. J. Biol. Chem. 1986;261:7621–7625. [PubMed] [Google Scholar]
- 21.Elshourbagy N.A., et al. Rat ATP citrate-lyase. Molecular cloning and sequence analysis of a full-length cDNA and mRNA abundance as a function of diet, organ, and age. J. Biol. Chem. 1990;265:1430–1435. [PubMed] [Google Scholar]
- 22.Katsurada A., et al. Effects of nutrients and hormones on transcriptional and post-transcriptional regulation of acetyl-CoA carboxylase in rat liver. Eur. J. Biochem. 1990;190:435–441. doi: 10.1111/j.1432-1033.1990.tb15593.x. [DOI] [PubMed] [Google Scholar]
- 23.Katsurada A., et al. Effects of nutrients and insulin on transcriptional and post-transcriptional regulation of glucose-6-phosphate dehydrogenase synthesis in rat liver. Biochim. Biophys. Acta. 1989;1006:104–110. doi: 10.1016/0005-2760(89)90329-9. [DOI] [PubMed] [Google Scholar]
- 24.Jakobsson A., Westerberg R., Jacobsson A. Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog. Lipid Res. 2006;45:237–249. doi: 10.1016/j.plipres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Ntambi J.M. Dietary regulation of stearoyl-CoA desaturase 1 gene expression in mouse liver. . J. Biol. Chem. 1992;267:10925–10930. [PubMed] [Google Scholar]
- 26.Coleman R.A., Lee D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 27.Diraison F., Beylot M. Role of human liver lipogenesis and reesterification in triglycerides secretion and in FFA reesterification. Am. J. Physiol. 1998;274:E321–E327. doi: 10.1152/ajpendo.1998.274.2.E321. [DOI] [PubMed] [Google Scholar]
- 28.Gibbons G.F., Wiggins D. The enzymology of hepatic very-low-density lipoprotein assembly. Biochem. Soc. Trans. 1995;23:495–500. doi: 10.1042/bst0230495. [DOI] [PubMed] [Google Scholar]
- 29.McGarry J.D. The mitochondrial carnitine palmitoyltransferase system: its broadening role in fuel homoeostasis and new insights into its molecular features. Biochem. Soc. Trans. 1995;23:321–324. doi: 10.1042/bst0230321. [DOI] [PubMed] [Google Scholar]
- 30.Donnelly K.L., et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. . J. Clin. Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wakil S.J., Stoops J.K., Joshi V.C. Fatty acid synthesis and its regulation. Annu. Rev. Biochem. 1983;52:537–579. doi: 10.1146/annurev.bi.52.070183.002541. [DOI] [PubMed] [Google Scholar]
- 32.McGarry J.D., Brown N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- 33.Abu-Elheiga L., et al. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1444–1449. doi: 10.1073/pnas.97.4.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim K.H. Regulation of mammalian acetyl-coenzyme A carboxylase. Annu. Rev. Nutr. 1997;17:77–99. doi: 10.1146/annurev.nutr.17.1.77. [DOI] [PubMed] [Google Scholar]
- 35.Abu-Elheiga L., Matzuk M.M., Abo-Hashema K.A., Wakil S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 36.Abu-Elheiga L., Oh W., Kordari P., Wakil S.J. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc. Natl. Acad. Sci. U. S. A. 2003;100:10207–10212. doi: 10.1073/pnas.1733877100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abu-Elheiga L., et al. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12011–12016. doi: 10.1073/pnas.0505714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao J., et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. U. S. A. 2006;103:8552–8557. doi: 10.1073/pnas.0603115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harada N., et al. Hepatic de novo lipogenesis is present in liver-specific ACC1-deficient mice. Mol. Cell. Biol. 2007;27:1881–1888. doi: 10.1128/MCB.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savage D.B., et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J. Clin. Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chirala S.S., et al. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6358–6363. doi: 10.1073/pnas.0931394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakravarthy M.V., et al. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005;1:309–322. doi: 10.1016/j.cmet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 43.Louet J.F., et al. Long-chain fatty acids regulate liver carnitine palmitoyltransferase I (L-CPT I) gene expression through a PPARα-independent pathway. Biochem. J. 2001;354:189–197. doi: 10.1042/0264-6021:3540189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le May C., et al. Fatty acids induce L-CPT I gene expression through a PPARalpha-independent mechanism in rat hepatoma cells. J. Nutr. 2005;135:2313–2319. doi: 10.1093/jn/135.10.2313. [DOI] [PubMed] [Google Scholar]
- 45.Flowers M.T., Miyazaki M., Liu X., Ntambi J.M. Probing the role of stearoyl-CoA desaturase-1 in hepatic insulin resistance. J. Clin. Invest. 2006;116:1478–1481. doi: 10.1172/JCI28774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen P., et al. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 47.Ntambi J.M., et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11482–11486. doi: 10.1073/pnas.132384699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang G., et al. Prevention of obesity in mice by antisense oligonucleotide inhibitors of stearoyl-CoA desaturase-1. J. Clin. Invest. 2005;115:1030–1038. doi: 10.1172/JCI23962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gutierrez-Juarez R., et al. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Invest. 2006;116:1686–1695. doi: 10.1172/JCI26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyazaki M., et al. Hepatic Stearoyl-CoA Desaturase-1 deficiency protects mice from carbohydrate-induced adiposity abd hepatic steatosis. Cell Metab. 2007;6:484–496. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 51.Dentin R., et al. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Invest. 2005;115:2843–2854. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimano H., et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. . J. Biol. Chem. 1999;274:35832–35839. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 53.Horton J.D., et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moon Y.A., Shah N.A., Mohapatra S., Warrington J.A., Horton J.D. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. . J. Biol. Chem. 2001;276:45358–45366. doi: 10.1074/jbc.M108413200. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez-Baro M.R., Lewin T.M., Coleman R.A. Regulation of triglyceride metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292:G1195–G1199. doi: 10.1152/ajpgi.00553.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turkish A., Sturley S.L. Regulation of triglyceride metabolism. I. Eukaryotic neutral lipid synthesis: “Many ways to skin ACAT or a DGAT”. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292:G953–G957. doi: 10.1152/ajpgi.00509.2006. [DOI] [PubMed] [Google Scholar]
- 57.Schultz J.R., et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang G., et al. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 59.Chu K., Miyazaki M., Man W.C., Ntambi J.M. Stearoyl-coenzyme A desaturase 1 deficiency protects against hypertriglyceridemia and increases plasma high-density lipoprotein cholesterol induced by liver X receptor activation. Mol. Cell. Biol. 2006;26:6786–6798. doi: 10.1128/MCB.00077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen G., Liang G., Ou J., Goldstein J.L., Brown M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Decaux J.F., Antoine B., Kahn A. Regulation of the expression of the L-type pyruvate kinase gene in adult rat hepatocytes in primary culture. J. Biol. Chem. 1989;264:11584–11590. [PubMed] [Google Scholar]
- 62.Stoeckman A.K., Towle H.C. The role of SREBP-1c in nutritional regulation of lipogenic enzyme gene expression. J. Biol. Chem. 2002;277:27029–27035. doi: 10.1074/jbc.M202638200. [DOI] [PubMed] [Google Scholar]
- 63.Yamashita H., et al. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9116–9121. doi: 10.1073/pnas.161284298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uyeda K., Repa J.J. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 65.Kawaguchi T., Takenoshita M., Kabashima T., Uyeda K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13710–13715. doi: 10.1073/pnas.231370798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Towle H.C. Glucose as a regulator of eukaryotic gene transcription. Trends Endocrinol. Metab. 2005;16:489–494. doi: 10.1016/j.tem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 67.Cha J.Y., Repa J.J. The liver X receptor (LXR) and Hepatic lipogenesis: The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007;282:743–751. doi: 10.1074/jbc.M605023200. [DOI] [PubMed] [Google Scholar]
- 68.Mitro N., et al. The nuclear receptor LXR is a glucose sensor. Nature. 2007;445:219–223. doi: 10.1038/nature05449. [DOI] [PubMed] [Google Scholar]
- 69.Lazar M.A., Willson T.M. Sweet dreams for LXR. Cell Metab. 2007;5:159–161. doi: 10.1016/j.cmet.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 70.Ma L., Robinson L.N., Towle H.C. ChREBP/Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006;281:28721–28730. doi: 10.1074/jbc.M601576200. [DOI] [PubMed] [Google Scholar]
- 71.Ma L., Sham Y.Y., Walters K.J., Towle H.C. A critical role for the loop region of the basic helix-loop-helix/leucine zipper protein Mlx in DNA binding and glucose-regulated transcription. Nucleic Acids Res. 2007;35:35–44. doi: 10.1093/nar/gkl987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iizuka K., Bruick R.K., Liang G., Horton J.D., Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dentin R., et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 74.Wang Y., et al. Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J. Lipid Res. 2006;47:2028–2041. doi: 10.1194/jlr.M600177-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Friedman J.M., Leibel R.L., Bahary N. Molecular mapping of obesity genes. Mamm. Genome. 1991;1:130–144. doi: 10.1007/BF00351059. [DOI] [PubMed] [Google Scholar]
- 76.Campfield L.A., Smith F.J., Burn P. The OB protein (leptin) pathway — a link between adipose tissue mass and central neural networks. Horm. Metab. Res. 1996;28:619–632. doi: 10.1055/s-2007-979867. [DOI] [PubMed] [Google Scholar]
- 77.Hron W.T., Sobocinski K.A., Menahan L.A. Enzyme activities of hepatic glucose utilization in the fed and fasting genetically obese mouse at 4-5 months of age. Horm. Metab. Res. 1984;16(Suppl. 1):32–36. doi: 10.1055/s-2007-1014893. [DOI] [PubMed] [Google Scholar]
- 78.Lindstrom P. The physiology of obese-hyperglycemic mice [ob/ob Mice]. ScientificWorldJournal. 2007;7:666–685. doi: 10.1100/tsw.2007.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yahagi N., et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J. Biol. Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 80.Dentin R., et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 81.Jerkins A.A., Liu W.R., Lee S., Sul H.S. Characterization of the murine mitochondrial glycerol-3-phosphate acyltransferase promoter. . J. Biol. Chem. 1995;270:1416–1421. doi: 10.1074/jbc.270.3.1416. [DOI] [PubMed] [Google Scholar]
- 82.Postic C., Dentin R., Denechaud P.D., Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 83.Iizuka K., Miller B., Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006;291:E358–E364. doi: 10.1152/ajpendo.00027.2006. [DOI] [PubMed] [Google Scholar]
- 84.Neschen S., et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 85.Kohli R., Pan X., Malladi P., Wainwright M.S., Whitington P.F. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J. Biol. Chem. 2007;282:21327–21336. doi: 10.1074/jbc.M701759200. [DOI] [PubMed] [Google Scholar]
- 86.Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007;22(Suppl. 1):S20–S27. doi: 10.1111/j.1440-1746.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- 87.Matsuzaka T., et al. Crucial role of a long-chain fatty acid elongase, ELOVL6, in obesity-induced insulin resistance. Nat. Med. 2007;13:1193–1202. doi: 10.1038/nm1662. [DOI] [PubMed] [Google Scholar]
- 88.Mordier S., Iynedjian P.B. Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochem. Biophys. Res. Commun. 2007;362:206–211. doi: 10.1016/j.bbrc.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 89.Nagle C.A., et al. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase 1 in rats causes insulin resistance. J. Biol. Chem. 2007;282:14807–14815. doi: 10.1074/jbc.M611550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Samuel V.T., et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 91.Samuel V.T., et al. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guzman M., Castro J. Zonation of fatty acid metabolism in rat liver. Biochem. J. 1989;264:107–113. doi: 10.1042/bj2640107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jungermann K., Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu. Rev. Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- 94.Gumucio J.J. Hepatocyte heterogeneity: the coming of age from the description of a biological curiosity to a partial understanding of its physiological meaning and regulation. Hepatology. 1989;9:154–160. doi: 10.1002/hep.1840090124. [DOI] [PubMed] [Google Scholar]
- 95.Belfort R., et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 96.Tiikkainen M., et al. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes. 2004;53:2169–2176. doi: 10.2337/diabetes.53.8.2169. [DOI] [PubMed] [Google Scholar]
- 97.Promrat K., et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188–196. doi: 10.1002/hep.20012. [DOI] [PubMed] [Google Scholar]
- 98.Hammarstedt A., Andersson C.X., Rotter Sopasakis V., Smith U. The effect of PPARgamma ligands on the adipose tissue in insulin resistance. Prostaglandins Leukot. Essent. Fatty Acids. 2005;73:65–75. doi: 10.1016/j.plefa.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 99.Kim J.Y., et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gray S.L., Vidal-Puig A.J. Adipose tissue expandability in the maintenance of metabolic homeostasis. Nutr. Rev. 2007;65:S7–S12. doi: 10.1111/j.1753-4887.2007.tb00331.x. [DOI] [PubMed] [Google Scholar]