

Abstract
Vibrio cholerae is a Gram-negative bacterial pathogen that exports enterotoxins to alter host cells and elicit diarrheal disease. Among the secreted toxins is the multifunctional RTX toxin, which causes cell rounding and actin depolymerization by covalently cross-linking actin monomers into dimers, trimers, and higher multimers. The region of the toxin responsible for cross-linking activity, the actin cross-linking domain (ACD), has recently been identified. In this study, we further investigate the role of the ACD in the actin cross-linking reaction. We show that the RTX toxin cross-links actin independent of tissue transglutaminase, thus eliminating an indirect model of ACD activity. We demonstrate that a fusion protein of the ACD and the N-terminal portion of Lethal Factor from B. anthracis, LFNACD, has cross-linking activity in vivo and in crude cell extracts. Furthermore, we determine that LFNACD directly catalyzes the formation of covalent linkages between actin molecules in vitro and that Mg2+ and ATP are essential cofactors for the cross-linking reaction. In addition, G-actin is proposed as a cytoskeletal substrate of the RTX toxin in vivo. Future studies of the in vitro cross-linking reaction will facilitate characterization of the enzymatic properties of the ACD and contribute to our knowledge of the novel mechanism of covalent actin cross-linking.
The causative agent of the diarrheal disease cholera is the Gram-negative bacterial pathogen Vibrio cholerae, which is transmitted to the human host following consumption of contaminated food or water. Upon colonization of the upper intestine, V. cholerae produces the major virulence factor cholera toxin (CT), which ADP-ribosylates the α-subunit of the Gs GTP-binding protein and constitutively activates the adenylate cyclase complex. The subsequent increase in the cyclic-AMP levels in intestinal epithelial cells leads to the opening of Cl- ion channels, resulting in the profuse diarrhea that is the hallmark of cholera. If untreated, the disease can progress to severe dehydration and case-fatality rates can reach as high as 50% (1).
The V. cholerae RTX toxin, which is encoded by rtxA, was discovered through a combination of genomic sequence analysis, genetic mapping, and representational difference analysis (2). The rtxA gene, which is tightly linked to the CT-encoding ctx genes, is deleted in the Classical O1 V. cholerae isolates, but expressed by both the El Tor O1 and O139 strains that are responsible for the current cholera pandemic (2,3). The RTX toxin is also produced by non-O1/non-O139 strains, and it has been suggested that the toxin may contribute to the emergence of pathogenic non-O1/non-O139 strains (4).
The full-length 4545 aa RTX toxin is predicted to be 484,000 Da in size and is secreted from the bacterium by an atypical Type I secretion system that requires two transport ATPases (5). Although related to the RTX-family of pore-forming toxins, the V. cholerae RTX toxin seems to be the founding member of a new family of RTX exoproteins. These proteins, encoded by V. cholerae, V. vulnificus, Photorhabdus luminescens, and the Xenorhabdus sp., are all larger than 3500 aa and share consensus N-terminal 20-residue glycine rich repeats in addition to 18-residue C-terminal repeats that include the nonapeptide motif GGXGXDXXX common to all RTX exoproteins. However, the central regions of these proteins vary dramatically, and it is predicted that each toxin will have a distinct repertoire of cellular activities (6).
The V. cholerae RTX toxin is multifunctional and has two distinct mechanisms for cell rounding (7). The first mechanism identified involves the covalent cross-linking of cellular actin, resulting in the depolymerization of actin stress fibers and an increase in paracellular permeability (8,9). A region of the toxin located between aa residues 1963-2375, denoted as the actin cross-linking domain (ACD), has recently been identified as the portion of the toxin responsible for cross-linking activity. Transient expression of this domain in both transformed African green monkey kidney fibroblast (COS-7) and human laryngeal epithelial (HEp-2) cells led to the formation of cross-linked actin species, demonstrating that expression of 412 aa of the toxin in the cytosol is sufficient to initiate actin cross-linking in the target cell. It remains unclear whether the ACD can bind actin and catalyze the covalent linkage directly, or if the toxin activity stimulates a host cell cross-linking enzyme.
A truncated form of the Bacillus anthracis lethal factor (LFN) has previously been shown to mediate cytosolic delivery of heterologous fusion proteins through the entry mechanism used by the anthrax toxin (10). Briefly, protective antigen (PA) binds to the anthrax toxin receptor, PA is processed to its active 63 kD form, and the PA63 fragment oligomerizes into a stable heptamer (11-13). The LFN fusion protein binds to [PA63]7, the complex enters the host cell through receptor-mediated endocytosis, and acidification of the vacuole results in PA pore formation and translocation of the LFN fusion protein to the cytosol (14-16). This system has been used to study the mechanism of action of other bacterial toxins including Corynebacterium diphtheriae diphtheria toxin and Clostridium difficile toxin B (10,17).
In this report, we further define the novel mechanism of covalent actin cross-linking by the V. cholerae RTX toxin. We find that cross-linking by the RTX toxin does not involve tissue transglutaminase (TGase), as previously suggested (9). We establish that a fusion protein of LFN and ACD (LFNACD) has cross-linking activity in vivo and in crude cell extracts. We then show that LFNACD directly catalyzes the actin cross-linking reaction in the absence of host proteins and that Mg2+ and ATP are required cofactors in vitro. Finally, we identify G-actin or actin oligomers as potential cytoskeletal substrates of the cross-linking reaction in vivo. These results contribute to our overall understanding of the mechanism of action of the V. cholerae RTX toxin and demonstrate that the ACD cross-links actin by directly catalyzing the formation of a covalent linkage between actin monomers in the absence of eukaryotic protein intermediates.
Experimental Procedures
Strains, cell lines, and reagents
Bah1P and Bah2P are strains derived from the V. cholerae El Tor Ogawa strain E7946 that either express the RTX toxin or harbor a deletion in rtxA, respectively (9). KFV119 is a ΔhapAΔhlyA derivative of the V. cholerae El Tor O1 Inaba strain N16961 that contains an intact rtxA gene (7). Bacterial cultures were grown for 18 h at 30°C in Luria broth (LB) containing streptomycin (100 μg/ml). TSA201 and HEp-2 cells were cultured at 37°C with 5% CO2 in RPMI 1640 or Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS, 50 units/ml penicillin, and 50 units/ml streptomycin. All restriction enzymes were purchased from Invitrogen. All chemicals were purchased from Sigma.
Tissue transglutaminase assay
Cells treated with bacteria for 70 min were washed with PBS, released in PBS with 0.1mM EDTA, scraped and collected in RPMI 1640 medium, and pelleted at 6000 × g. The cell pellets were resuspended in SDS-PAGE sample buffer, boiled for 10 min, and analyzed by SDS-PAGE. Tissue TGase expression was detected with a 1:400 dilution of a mouse monoclonal anti-TGase antibody (Lab Vision, Fremont, CA) followed by a 1:5000 dilution of anti-mouse IgG conjugated to horseradish peroxidase (HRP) (Sigma). Actin cross-linking was monitored by immunoblot as previously described (9).
Construction, expression, and purification of proteins
The plasmid pABII is a derivative of the pET15b plasmid (Novagen, Madison, WI) containing the DNA sequence for LFN with an N-terminal 6xHis tag (LFN). To create a fusion to the ACD, the ACD sequence was amplified from the V. cholerae chromosome using primers LFACD-F 5′-GAAAGATCTCTAAGTGCGGTACAGG-3′ and LFNACD-R 5′-AGAGATCTTAAGGAGCGGTAATTTTCGC-3′. The ∼1.3 kB PCR product was cloned into the pCR-BluntII-TOPO vector (Invitrogen) and then subcloned as a BglII fragment into the BamHI site of the pABII expression vector (17). The resulting plasmid, pTCO24, expresses the ACD protein fused to the C-terminus of LFN. DNA sequencing of the insert in pTCO24 was performed at the Northwestern University Biotechnology Laboratory to confirm the accuracy of both the coding region and the junction between the LFN and ACD sequences.
LFN and the LFNACD fusion protein were expressed and purified as described by Milne et al. and standard molecular biology protocols (10,18). Briefly, pABII or pTCO24 were transformed into E. coli BL21(DE3) cells (Novagen), and bacterial cultures were grown in LB supplemented with ampicillin (100 μg/ml) to an OD600 of 0.6-1.0. Protein expression was induced for 2 hr after the addition of 1.0 mM IPTG at 37°C and bacterial cells were pelleted at 5000 × g for 15 min at 4°C. The pellet was resuspended in Buffer A (20 mM Tris pH 8.0, 0.5 M NaCl) with 5.0 mM imidazole. Following the addition of lysozyme (1 mg/ml), the cell suspension was incubated for 30 min on ice and sonicated in the presence of 1% Triton X-100 with a Branson Digital Sonifier Cell Disruptor 450 at 35% amplitude, 15 sec pulses for 4 min total. The cell lysate was incubated with Complete EDTA-free Protease Inhibitor Cocktail (Roche), 5.0 μg/ml DNaseI, 5.0 μg/ml RNaseI, and cleared by centrifugation at 10,000 × g for 30 min at 4°C.
The soluble fraction was passed through a 0.45 μm pore-sized syringe filter, loaded onto a 1 ml HisTrap HP column (GE Healthcare, Piscataway, NJ) equilibrated in Buffer A, and purified on an ÄKTA Purifier FPLC system (GE Healthcare). The fusion protein was eluted at 107 mM imidazole using a linear gradient from 5-250 mM imidazole in Buffer A. All fractions containing the 6xHis-tagged protein were pooled, analyzed by SDS-PAGE for purity, and concentrated on a Centricon YM-30 microconcentrator (Millipore, Billerica, MA). The proteins were loaded onto a 5 ml HiTrap Desalting Column (GE Healthcare) and exchanged into Buffer A. Fractions containing the desalted protein were pooled, concentrated, and stored in 10% glycerol at -80°C.
Protective Antigen (PA) was purified using plasmid PA-pET15b as described by Voth et al. (19).
SDS-PAGE and immunoblot analysis of purified proteins
Protein samples were analyzed by SDS-PAGE and visualized with Coomassie Brilliant Blue R-250, or transferred to Hybond-ECL nitrocellulose membranes (GE Healthcare). The proteins were immunoblotted with either a 1:20,000 dilution of a mouse monoclonal anti-6xHis antibody (Sigma) followed by a 1:5000 dilution of anti-mouse IgG conjugated to HRP (Sigma), or a 1:5000 dilution of a rabbit polyclonal anti-ACD antibody followed by a 1:5000 dilution of anti-rabbit IgG conjugated to HRP (Sigma).
The ACD antibody was generated by immunization of a New England White rabbit with the synthetic peptide 1 - VESRKEAMLWLAKEFTDH-C, which corresponds to aa 2066-2083 of the RTX protein sequence (2), using standard procedures (Proteintech Group, Inc., Chicago, IL).
In vivo actin cross-linking assay
Approximately 2.5 × 105 HEp-2 cells seeded in a 12-well dish were incubated with PA and either LFN or LFNACD, or treated with PBS-washed KFV119 bacteria at an m.o.i. of ∼200. Cell rounding was monitored with a Nikon Eclipse TS100 inverted microscope. Phase contrast images were acquired at 200X magnification with an inverted Leica DMIRE2 microscope equipped with a C4742-95-12ERG digital charge-coupled device camera (Hamamatsu Photonics, Tokyo, Japan) and the Openlab software program (Improvision, Coventry, UK). For some experiments, cells were washed and scraped in PBS, centrifuged at 4,500 × g for 5 min, resuspended in SDS-PAGE sample buffer, and boiled for 10 min in preparation for the detection of actin cross-linking by immunoblot as previously described (9).
In vitro actin cross-linking assay
For the preparation of cell extracts, approximately 4 × 106 HEp-2 cells were plated onto two 100 mm dishes, washed and scraped in PBS, and sonicated with a Conoco Sonicell disrupter, 5 sec pulses for 1 min. Cell lysates were pelleted at 1000 × g for 10 min at 4°C. Protein concentrations were determined with a Bicinchoninic Acid (BCA) protein assay kit (Pierce Biotechnology, Rockford, IL), and 120 μg of cleared lysates were added to 5.0 μg of either purified LFN or LFNACD protein. Each reaction was incubated at 37°C for 90 min, then boiled in SDS-PAGE sample buffer for 5 min. Actin cross-linking was detected by immunoblot as previously described (9).
Rabbit skeletal actin was purified from rabbit back muscle as discussed in Spudich and Watt (20). Actin was added at a concentration of 10 μM to 0.018 μM of either purified LFN or LFNACD and the reactions were incubated at 37°C for 20 min in Buffer B (5 mM HEPES pH 7.5, 0.2 mM ATP, 0.2 mM CaCl2). Samples were boiled for 5 min in SDS-PAGE sample buffer and cross-linked actin bands were detected on gels by staining with Coomassie Brilliant Blue R-250. The in vitro experiments were also performed at 22°C and the results were identical.
Actin polymerization and light scattering
Actin polymerization was initiated following the addition of 2.0 mM CaCl2, 2.0 mM MgCl2, or 50 mM KCl. Light-scattering measurements were performed in a Photon Technology Industries spectrofluorometer with the emission and excitation wavelengths set at 325 nm.
Results
The RTX toxin cross-links actin independent of tissue transglutaminase activity
The formation of covalent linkages between actin monomers following the treatment of eukaryotic cells with V. cholerae strains expressing the RTX toxin may be attributed to either the activity of ACD directly, or to an ACD-mediated modification of a eukaryotic cellular enzyme. Tissue TGase, a member of the family of Ca2+-dependent enzymes that introduce ε-(γ-glutamyl)lysine side chain linkages between proteins, is the only known host enzyme that catalyzes the covalent cross-linking of cellular actin (21,22). To address a model in which the ACD functions by activating tissue TGase, actin cross-linking was monitored in TSA201 cells, a human embryonic kidney cell line with undetectable tissue TGase production by immunoblot (23) (Fig. 1A). When cells were incubated with a V. cholerae strain producing the RTX toxin, cross-linked actin species were detected by immunoblot (Fig. 1B). These data indicate that actin is cross-linked in the absence of tissue TGase, showing that tissue TGase is not required for the actin cross-linking reaction.
FIGURE 1. The RTX toxin cross-links actin through a tissue TGase-independent mechanism.
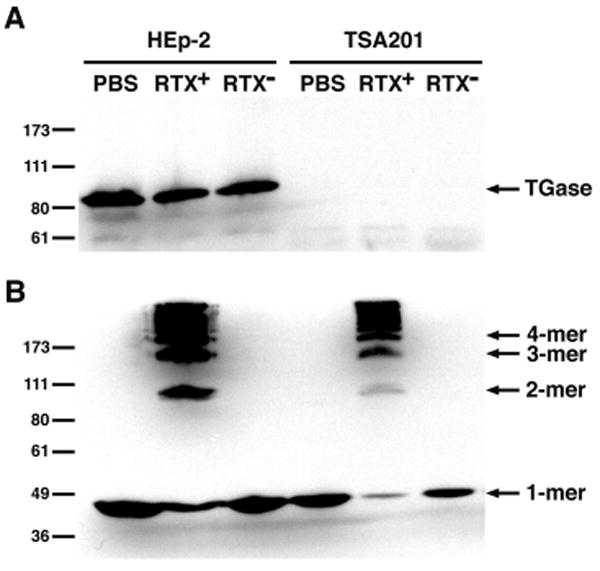
HEp-2 and TSA201 cells were incubated with either PBS-washed bacteria from an RTX-producing strain (RTX+), a strain harboring a deletion in the rtxA gene (RTX-), or PBS for 70 min. The cell pellets were boiled in SDS-PAGE sample buffer and lysates were electrophoresed on 8% SDS-polyacrylamide gels. Tissue TGase expression and actin cross-linking were detected by immunoblotting. The numbers at the left indicate the location of the Invitrogen Benchmark Pre-stained protein standards.
The site of TGase cross-linking on the actin molecule is located at the glutamine residue at position 41 of the protein sequence (Q41) (24). The involvement of this residue in the RTX-dependent cross-linking reaction was investigated by site-directed mutagenesis of the β-actin sequence from pEGFP-Actin (Clontech, Mountain View, CA). An asparagine substitution (Q41N) had no effect on cross-linking of GFP-actin in the presence of an RTX-expressing V. cholerae strain (data not shown). In addition, the TGase-specific cross-link was not detected by immunoblot with an anti-N-ε-(γ-glutamyl)lysine antibody (Abcam, Cambridge, MA) in lysates from V. cholerae-treated cells (data not shown). These data provide further evidence that the actin cross-linking reaction is tissue TGase-independent.
As there are no other enzymes known to covalently cross-link actin, an alternative model that favors a direct interaction between the ACD of the RTX toxin and cellular actin monomers was investigated.
Construction and purification of LFNACD
An in vitro assay was developed to assess whether the ACD is sufficient to cross-link actin in the absence of other cellular proteins. Previous attempts to catalyze the cross-linking reaction with partially purified holotoxin or recombinant ACD proteins were unsuccessful, and inactivation of the ACD may have been due to decreased stability or improper protein folding (9). To circumvent this problem, it was essential to first establish that the purified ACD protein had actin cross-linking activity in vivo before monitoring actin cross-linking in vitro. A fusion protein was constructed to take advantage of the entry mechanism of B. anthracis anthrax toxin, which has been used to translocate the catalytic domains of various bacterial toxins into eukaryotic cells (17).
The ACD sequence was amplified from the V. cholerae chromosome by PCR and cloned into a bacterial expression vector that expresses LFN (17). The resulting fusion protein, LFNACD, was purified on a nickel-chelating column and further desalted by gel filtration chromatography. The protein was analyzed by SDS-PAGE (Fig. 2A) and the observed mobility of the LFNACD fusion protein corresponded to an expected mol. wt. of 81 kD. This protein reacted with antibodies against both the 6xHis tag and a peptide antibody raised against ACD (Fig. 2B and 2C). The mol. wt. of the purified LFNACD protein was verified by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry to be 81,407 Da, which is accurate to 0.09% of the predicted mass. A protein containing the LFN portion alone, purified under similar conditions, reacted only to the 6xHis tag antibody and not to the ACD-specific antibody.
FIGURE 2. Characterization of LFNACD.
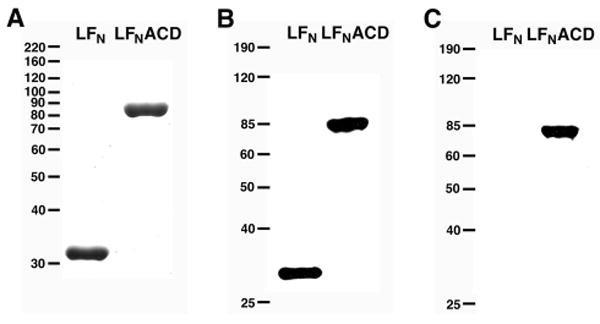
LFN (32 kD) and LFNACD (81 kD) were purified by affinity chromatography on a nickel-chelating column and desalted by gel filtration. Purified proteins were loaded onto 10% SDS-polyacrylamide gels and either A, stained with Coomassie Blue, B, immunoblotted with a mouse monoclonal anti-His antibody, or C, immunoblotted with a rabbit polyclonal anti-ACD antibody. The numbers at the left correspond to the location of either the Invitrogen A, Benchmark Unstained or B, C, Benchmark Pre-stained protein standards.
Purified LFNACD cross-links actin in vivo
Fusion of the ACD sequence to LFN allows for the cytosolic delivery of purified LFNACD proteins into target cells. To monitor the activity of LFNACD in vivo, HEp-2 cells were incubated with purified PA and either LFN or LFNACD at a molar ratio of 7:3, respectively (25). Cell rounding was observed in LFNACD-intoxicated cells, and the rounding was markedly similar to cells incubated with KFV119, a V. cholerae strain expressing the full-length RTX toxin (Fig. 3A). Actin dimers, trimers, and higher order multimers were detected in cells treated with LFNACD or KFV119 by immunoblot (Fig. 3B). These data demonstrate that the purified LFNACD protein has covalent actin cross-linking activity in vivo and is suitable for the development of an in vitro actin cross-linking assay.
FIGURE 3. LFNACD has actin cross-linking activity in vivo.
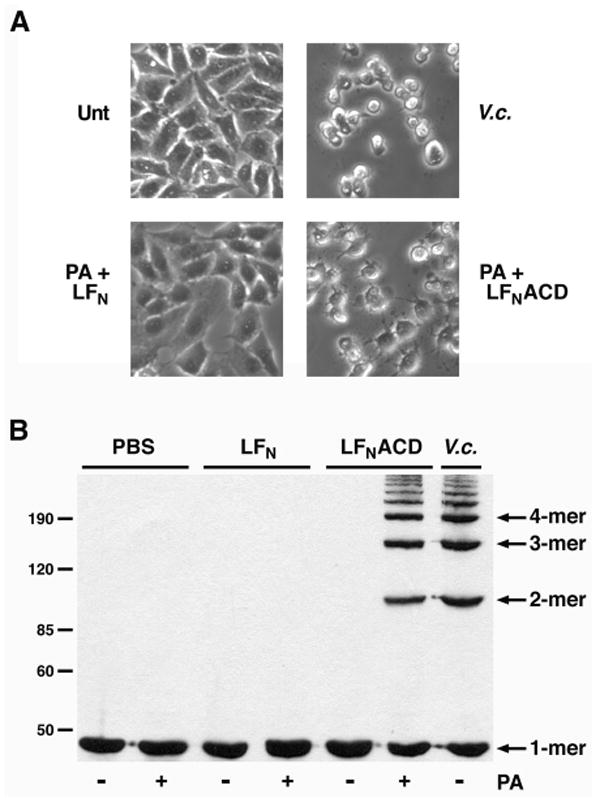
HEp-2 cells were treated with PBS (Unt), exposed to PBS-washed V. cholerae strain KFV119 (V.c.), or intoxicated with 31.7 nM purified PA and either 13.6 nM LFN or LFNACD. A, Cell rounding was observed after 90 min, and phase contrast images were acquired at 200X magnification. B, Treated cells were harvested, lysates were separated on 8% SDS-polyacrylamide gels, and the formation of cross-linked actin species was monitored by immunoblotting. The protein standards are labeled on the left side.
The results from this purified protein assay confirm previous findings based on transient transfection that the actin cross-linking activity is carried by the ACD region of the RTX toxin and that the ACD functions within the cell (7). In addition, these data show that the LFNACD protein must be translocated to the cytosol through the activity of PA, and that LFNACD alone is incapable of functioning extracellularly. These data indicate that ACD activity does not lead to actin cross-linking by stimulating an endogenous signal transduction pathway that ultimately results in the formation of cross-linked actin proteins, nor is the ACD gaining access to the target cell through a receptor interaction independent of PA. It can further be extrapolated that the ACD in the holotoxin is likely transported to the cell cytoplasm via the entry mechanism of the full-length RTX toxin and not by an ACD-specific pathway.
LFNACD has actin cross-linking activity in crude cell extracts
To further investigate the role of the ACD in the actin cross-linking reaction, purified LFN or LFNACD proteins were added to crude HEp-2 cell extracts. Cross-linked actin species were detected by immunoblot in the LFNACD-treated extracts, but not in extracts incubated with LFN (Fig. 4). To determine if the reaction requires divalent cations, cell extracts were pre-incubated with either 5.0 mM EDTA or 5.0 mM EGTA for 5 min before addition of LFNACD. The formation of cross-linked actin was inhibited in the presence of EDTA, which suggests that Mg2+ is needed for the reaction, although it is possible that addition of EDTA caused actin denaturation. The detection of actin cross-linking was inconsistent in EGTA-treated samples, a result that may be attributed to either a requirement of Ca2+ in the reaction or to a low or variable concentration of Mg2+ in different extract preparations. Taken together, these data show that the ACD can cross-link actin in vitro and that divalent cations are needed for the reaction either as a cofactor or to stabilize actin.
FIGURE 4. Divalent cations are important for actin cross-linking in crude cell extracts.
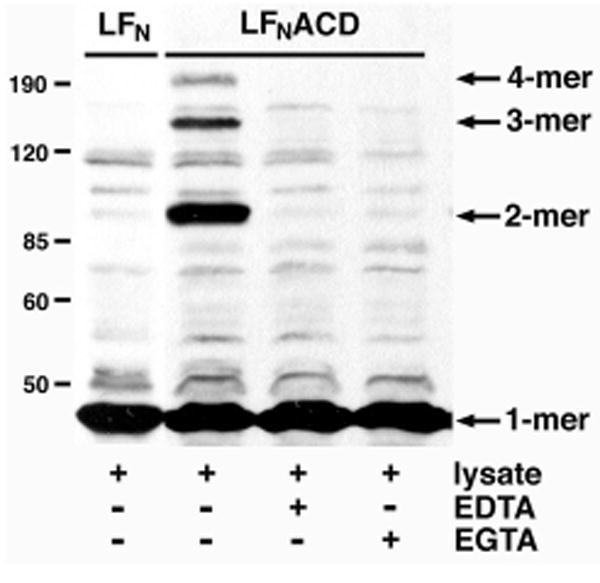
HEp-2 cells were sonicated in PBS and cleared by centrifugation at 1000 × g. 120 μg cell extract was incubated with 5 μg of purified LFN or LFNACD for 90 min. EDTA and EGTA were added to a final concentration of 5.0 mM where indicated. Actin cross-linking activity was monitored by immunoblotting. The location of the protein standards are indicated at the left.
LFNACD catalyzes cross-linking of purified actin dependent upon Mg2+
To determine if the ACD directly catalyzes the formation of covalent linkages between actin proteins, purified actin was tested as a substrate in the actin cross-linking reaction. Purified rabbit skeletal actin was incubated with LFN or LFNACD in the presence of 2.0 mM CaCl2, MgCl2, MnCl2, or ZnCl2 as the divalent cation cofactor. Upon addition of Mg2+, covalently cross-linked actin dimers, trimers, and higher multimers were detected on Coomassie Blue-stained SDS-polyacrylamide gels (Fig. 5A). Reduced activity of the ACD was observed in the presence of Mn2+ and there was no actin cross-linking following the addition of Ca2+ or Zn2+ ions.
FIGURE 5. LFNACD directly cross-links actin in the presence of Mg2+.
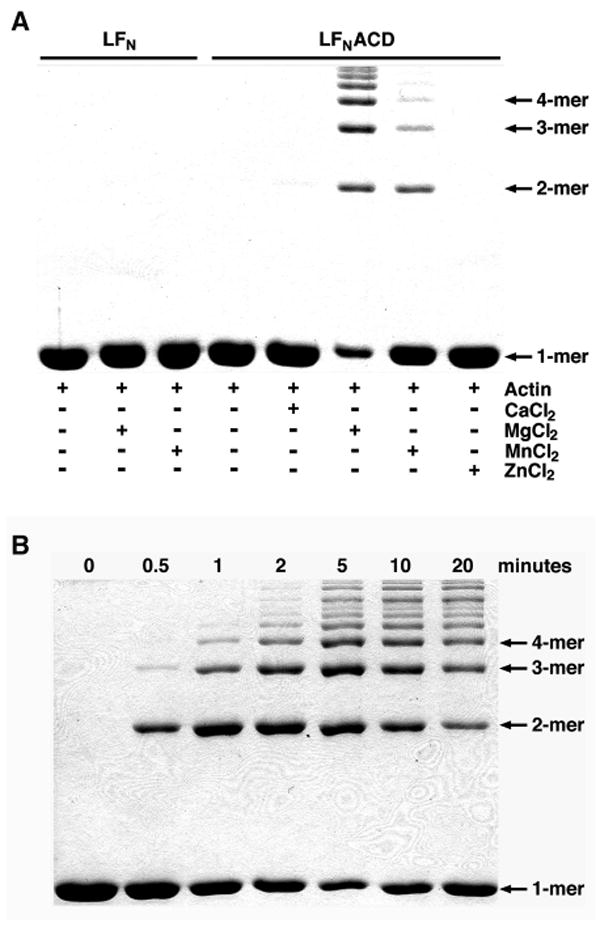
A, 10 μM purified rabbit skeletal actin and 0.018 μM LFN or LFNACD were incubated for 20 min in Buffer B. The reactions were supplemented with 2.0 mM CaCl2, MgCl2, MnCl2, or ZnCl2 as indicated. B, 0.018 μM LFNACD, 10μM actin, and 2.0 mM MgCl2 were co-incubated and the reactions were terminated at designated time points. For both A and B, samples were boiled in SDS-PAGE sample buffer for 5 min, then loaded onto 8% SDS-polyacrylamide gels. Covalent actin cross-linking was detected by Coomassie Blue staining.
To further characterize the in vitro cross-linking reaction, LFNACD, actin, and Mg2+ were co-incubated and actin cross-linking was monitored at various time points. Covalently cross-linked actin dimer proteins were detected after an incubation period of 0.5 min (Fig. 5B). The distribution of actin laddering remained unchanged after 10 min and it was concluded that the cross-linking reaction had reached completion. These data indicate that LFNACD, in the presence of Mg2+ as a cofactor, catalyzes the actin cross-linking reaction in a time-dependent manner.
Actin is present in the cell as either globular monomers (G-actin) or polymeric filaments (F-actin), with polymerization occurring under physiological salt conditions (26). However, Mg2+ alone causes efficient polymerization of actin in vitro and the requirement for Mg2+ in the actin cross-linking reaction might suggest that actin polymerization needs to be initiated prior to the formation of covalent linkages by the ACD. To investigate the contribution of Mg2+, G-actin was polymerized into F-actin following the addition of 2.0 mM CaCl2, 2.0 mM MgCl2, or 50mM KCl. As shown in Fig. 6A, all conditions resulted in actin polymerization as detected by light scattering. In parallel experiments, actin polymerization was initiated in the presence of LFNACD, and actin cross-linking was only detected in the reaction supplemented with Mg2+ (Fig. 6B). These data suggest that the initiation of actin polymerization is not sufficient to cause actin cross-linking by LFNACD and that the ACD-catalyzed cross-linking reaction is dependent upon Mg2+ as a cofactor, not simply to stabilize actin or promote polymerization.
FIGURE 6. Mg2+ is essential for the ACD-catalyzed actin cross-linking reaction.
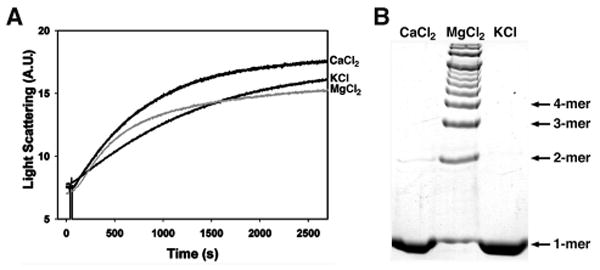
10 μM G-actin in the A, absence or B, presence of 0.018 μM purified LFNACD was incubated with 2.0 mM CaCl2, 2.0 mM MgCl2, or 50mM KCl. A, Actin polymerization was observed by light scattering. B, Actin cross-linking was monitored in samples boiled in SDS-PAGE sample buffer and electrophoresed on 8% SDS-polyacrylamide gels. Actin species were visualized by staining with Coomassie Blue R-250.
ATP is a required cofactor for ACD-catalyzed actin cross-linking
Cation binding is known to be important for the association of ATP molecules to proteins including actin (27). The requirement for Mg2+ in the cross-linking reaction may imply that ATP is also a necessary cofactor, and it was noted that ATP was a common component in all buffers used for both actin purification and in vitro actin cross-linking. To better characterize the role of ATP in the actin cross-linking reaction, Mg-ATP-actin and Mg-ADP-actin were prepared as described by Gershman et al. (28) with slight modifications (29), and tested as substrates for the cross-linking reaction. In addition, actin purified in the presence of ATP was passed over a PD10 gel filtration column to remove ATP, then used as a substrate in cross-linking reactions supplemented with either free ATP or the non-hydrolysable ATP analog adenyl-5-yl imidodiphosphate (AMP-PNP). Actin polymerization was maintained in each of these conditions as monitored by light scattering (data not shown). Covalent actin cross-linking was detected in the presence of LFNACD and Mg-ATP-actin, as well as LFNACD, Mg2+, and free ATP (Fig. 7, lanes 2 and 4). However, cross-linked actin proteins were not observed following the incubation of LFNACD and Mg-ADP-actin, despite an incubation period of 60 min to ensure adequate polymerization (Fig. 7, lane 1). The combination of free AMP-PNP with Mg2+ and LFNACD led to the production of a small amount of the cross-linked dimer, which is most likely due to contamination with traces of ATP released from the nucleotide pocket of the actin molecule (Fig. 7, lane 3). Only in the presence of both Mg2+ and ATP was ACD-catalyzed actin cross-linking detected through the higher multimer protein species. Since the structure of AMP-PNP-actin is not substantially different from ATP-actin (30) and its polymerization properties are not perturbed (31), this experiment strongly suggests that ATP is an essential cofactor for ACD-catalyzed actin cross-linking rather than for maintaining actin structure.
FIGURE 7. The actin cross-linking reaction requires ATP as a cofactor.
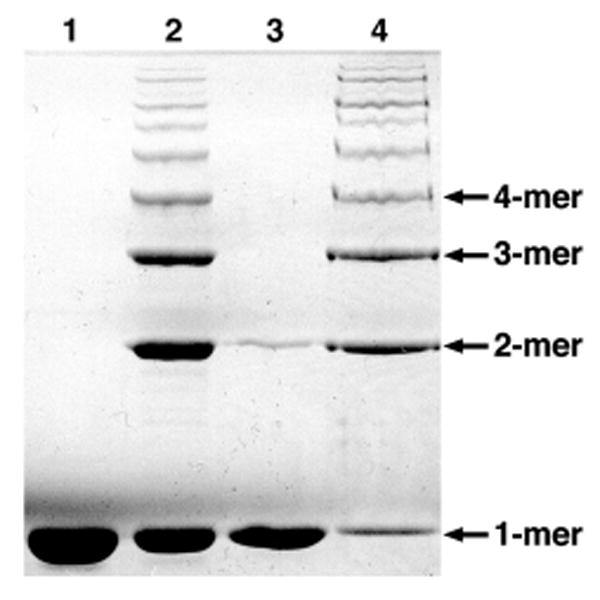
Purified LFNACD was incubated with (1) 30 μM Mg-ADP-actin or (2) 30 μM Mg-ATP-actin at a molar ratio of 1:500 in 2mM MgCl2, and actin cross-linking was assayed after 60 min. 10 μM actin, further purified by gel filtration, was added to LFNACD and Mg2+, supplemented with either (3) 0.5 mM AMP-PNP or (4) 0.5 mMATP, and the reaction was terminated after 12 min. All samples were analyzed by SDS-PAGE, and cross-linked actin species were detected by Coomassie Brilliant Blue R-250.
G-actin is a substrate of the actin cross-linking reaction
As both Mg2+ and ATP are involved in the initial stages of actin polymerization, it is difficult to distinguish from these in vitro studies whether the cytoskeletal substrate of the cross-linking reaction is G-actin or F-actin. Covalently cross-linked proteins have been detected as early as 0.5 min after the initiation of polymerization (see above), at which point actin is present as a mixed pool of monomers and polymers. Therefore, an in vivo approach was used to further clarify the actin substrate.
Previously, cytochalasin D was used to depolymerize actin in vivo prior to incubation with a strain of V. cholerae that expresses the RTX toxin and G-actin was proposed as the substrate for the actin cross-linking reaction (9). However, cytoskeletal inhibition with cytochalasin D does not fully eliminate F-actin as a possible target for actin cross-linking, as there may have been partially polymerized F-actin in the treated cells. Two additional cytoskeletal inhibitors were used in vivo to further support the idea that G-actin is the substrate of the cross-linking reaction. Latrunculin B (LatB) is an inhibitor of actin polymerization that binds and sequesters G-actin (32). Dolastatin 11 (Dol11), a depsipeptide of Dolabella auricularia, hyperpolymerizes actin monomers and stabilizes F-actin (33). HEp-2 cells were incubated with either LatB or Dol11 for 90 min to ensure sufficient disruption of the actin cytoskeleton (data not shown). Following treatment, the cells were exposed to V. cholerae strain KFV119 and the formation of cross-linked actin proteins was monitored by immunoblot (Fig. 8). These experiments were performed with RTX-expressing V. cholerae bacteria, since PA translocation of LFNACD would be inhibited by LatB (34). Actin cross-linking was observed in cells pre-treated with LatB, which implies that the inhibition of actin polymerization had no effect on the cross-linking reaction. However, in the presence of Dol11, a molecule that depletes the G-actin pool by increasing F-actin assembly, actin cross-linking by the RTX toxin was inhibited. During actin polymerization, actin monomers self-associate into trimers and short oligomers and F-actin is formed following the addition of monomers to the ends of the growing filament (26,35). These data suggest that either G-actin or short actin oligomers may be the cytoskeletal substrates of the actin cross-linking reaction in vivo.
FIGURE 8. Dol11, not LatB, inhibits actin cross-linking in vivo.
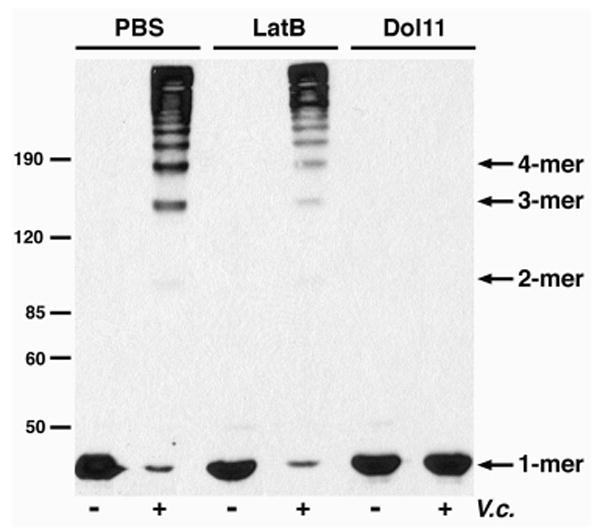
HEp-2 cells were pre-treated with either 5.0 μM LatB or 5.0 μM Dol11 for 90 min. Cells were incubated with V. cholerae strain KFV119 (V.c.) for an additional 90 min, harvested, and boiled in SDS-PAGE sample buffer. The lysates were separated on 8% SDS-polyacrylamide gels and covalent actin cross-linking was detected by immunoblot with an anti-actin antibody. Arrows at the left correspond to the protein standards.
Discussion
In this study, we show that the ACD from the V. cholerae RTX toxin is directly responsible for the covalent cross-linking of actin. We have shown that LFNACD, a fusion protein between LFN and the ACD, catalyzes the formation of actin dimer, trimer, and higher multimer proteins both in vivo and in vitro, and that Mg2+ and ATP are essential cofactors of the cross-linking reaction. In addition, we identify G-actin or short actin oligomers as potential substrates of the RTX toxin in vivo.
These results augment our understanding of the mechanism of action for the RTX toxin. After the original discovery of RTX actin cross-linking activity, a number of models were considered to explain how the holotoxin causes the covalent cross-linking of cellular actin (9). These models included both a direct model in which the RTX toxin binds actin and catalyzes the cross-linking reaction, and indirect models wherein the toxin functions by interacting with cytoplasmic host proteins to initiate actin cross-linking or binding to extracellular receptors to stimulate a signaling cascade that generates cross-linked actin proteins.
Previously, we demonstrated that expression of the ACD as a transgene in vivo resulted in actin cross-linking, suggesting that the ACD is active in the cell cytoplasm and not initiating a signal transduction pathway at the plasma membrane (7). Here, we show that cytosolic delivery of LFNACD, a protein dependent upon uptake by receptor-mediated endocytosis and transfer to the cytoplasm by anthrax toxin PA, is sufficient to cause actin cross-linking. Taken together, these data establish that the mechanism of actin cross-linking by the RTX toxin requires cytosolic delivery of the ACD into the target cell. In addition, we eliminate an indirect model in which the RTX toxin activates tissue TGase, the only known eukaryotic enzyme with actin cross-linking activity, which suggests that the ACD catalyzes the cross-linking reaction.
For proof of a direct model of RTX action, it was necessary to determine that the ACD had actin cross-linking activity in vitro, in the absence of other host proteins. We demonstrate that LFNACD cross-links actin in vitro and the reaction requires only Mg2+ and ATP. In subsequent studies, a 6xHis-ACD fusion protein was also shown to have actin cross-linking activity in the presence of Mg2+ and ATP, demonstrating that LFN does not enhance the enzymatic function of the ACD (our unpublished results). Taken together, these data indicate that the ACD can bind actin and directly introduce a covalent linkage between monomers. Although we have shown that the ACD is sufficient to cross-link actin, the chemical nature of the covalent bond remains elusive. Extensive efforts have been made to purify cross-linked actin from cells treated with V. cholerae bacteria producing the RTX toxin in order to identify the cross-linked peptide by mass spectrometry. However, the presence of eukaryotic protein contaminants complicated our analysis. The development of an in vitro actin cross-linking assay, using only purified actin, LFNACD, Mg2+, and ATP has simplified the generation of cross-linked actin proteins. Yet, only 80% of predicted actin peptides were detected by LC-MS/MS after in-gel trypsin digestion and there were no peptides unique to the dimer, trimer, tetramer, and pentamer actin species (data not shown). These results suggest that cross-linked actin may need to be purified and digested in-solution in order to isolate the covalently cross-linked actin peptide. The time-dependent actin cross-linking reaction can be manipulated to increase the overall concentration of dimeric proteins, and the peptides generated from in-solution proteolytic digestion of the purified dimer would then be analyzed by mass spectrometry.
Data from the cytoskeletal inhibitor study with LatB and Dol11 suggests that G-actin is the substrate of the actin cross-linking reaction in vivo. Although these results confirm the previous finding with cytochalasin D (9), the possibility that actin oligomers or even F-actin can also serve as substrates for the ACD cannot be fully excluded. Dol11 inhibits actin depolymerization by binding to F-actin between two long-pitch strands of diagonal monomer subunits (36). This interaction may prevent access to actin protomers in the filament that would otherwise be cross-linked by the ACD. In future studies, synthetic actin substrates and our in vitro actin cross-linking assay will be used to fully clarify the substrate of the cross-linking reaction.
The requirement for Mg2+ and ATP for actin cross-linking in vitro may indicate that the initiation of actin polymerization is necessary for the ACD-catalyzed reaction. Actin polymerization is dependent upon a thermodynamically unfavorable nucleation step that results from the self-association of actin monomers into a non-covalent trimer complex (26). It is possible that Mg2+ and ATP are important for the actin cross-linking reaction to promote the formation of nucleated actin and the ACD can either catalyze isopeptide linkages between actin monomers in the process of nucleating or bind to the trimer complex and covalently attach additional monomers during the elongation phase of polymerization.
This model, while plausible, would contrast our in vivo identification of G-actin as a substrate. Further, we have found that neither cross-linked actin nor LFNACD can serve to nucleate actin either in the presence or absence of LFNACD (our unpublished results). Hence, it is unlikely that LFNACD functions at the point of actin elongation. Instead, we favor the possibility that Mg2+ and ATP are essential for the enzymatic activity of the ACD, rather than for the regulation of actin dynamics. Tissue TGase is a bifunctional enzyme that covalently cross-links actin, among other protein substrates, and has also been shown to bind and hydrolyze GTP and ATP in the presence of Mg2+ (37). Although we have established that tissue TGase is not involved in the ACD-dependent cross-linking reaction, the requirement for Mg2+ and ATP in our in vitro cross-linking assay may indicate that the cross-linking activity of the ACD is energy consuming and coupled to ATPase activity. Sequence analysis of the ACD revealed a putative ATP binding site at aa 2125-2127. The sequence SxxAxGKT conforms to the consensus ATP-binding Walker type A consensus sequence GxxGxGK(T/S) (38), assuming conservative Ser and Ala changes for the Gly residues. A linker insertion mutant near this site disrupted function of the ACD when expressed as a transgene, indicating this region is required for cross-linking activity (our unpublished results). The presence of this site suggests that the ACD is capable of binding and hydrolyzing ATP. Ongoing experiments are focused on the role of Mg2+ and ATP in the ACD-catalyzed actin cross-linking reaction.
As a mechanism, we suggest that the covalent cross-linking of cellular actin leads to the disassembly of actin filaments due to the irreversible chemical modification of the actin monomer and its subsequent inability to undergo polymerization. Depletion of the G-actin pool leads to a shift in the critical concentration of actin within the cell and ultimately results in the depolymerization of actin stress fibers. It has been shown that microinjection of covalently cross-linked actin proteins into PtK2 cells disrupts the steady state between G-actin and F-actin, resulting in disassembly of the endogenous actin filament network (39). We hypothesize that a recently observed second cell rounding activity of the multifunctional RTX toxin also increases the overall amount of G-actin in the cell by an as yet uncharacterized mechanism (7), which subsequently provides more substrate for the actin cross-linking reaction catalyzed by the ACD.
The ACD sequence is 59% identical to the C-terminal portion of VgrG-1, also known as VC1416, a V. cholerae protein we have previously shown to cross-link actin when transiently expressed in mammalian cells (7). Recently, Pukatzki et al. (40) reported that VgrG-1 is a putative effector protein of the newly identified type VI secretion system, which is predicted to translocate potential virulence factors into target cells. Our investigation of the mechanism of actin cross-linking by the ACD may contribute to the understanding of VgrG-1 activity, as the strong sequence conservation suggests that VgrG-1 has a cross-linking mechanism similar to that of the ACD for its function in the rounding of J774 macrophage cells.
We have established that the ACD from the V. cholerae RTX toxin is a cross-linking enzyme that catalyzes the formation of covalent linkages between actin monomers. Ongoing study of the actin cross-linking reaction will further determine the enzymatic properties of the ACD and allow us to pursue its structure-function interactions with two molecules of actin. As a whole, this work will advance our knowledge of this unique mechanism of actin depolymerization and clarify the role of the RTX toxin in the pathogenesis of cholera disease.
Acknowledgments
We thank J. Ballard for providing plasmids pABII and PA-pET15b, J. Lomasney for supplying TSA201 cells, and R. Bates for the gift of dolastatin 11. We thank K.-L. Sheahan for microscopy assistance and A. Bonebrake for technical assistance. We also thank J. Loo and his laboratory for helpful insight into this project. This work was supported by a Biomedical Research Support Program Award from the Howard Hughes Medical Institute and Public Health Services Grant AI051490 from the National Institute for Allergy and Infectious Diseases (to K.J.F.S.), and grants from U.S. Public Health Service (GM-077190) and the National Science Foundation (MCB 0316269) (to E.R.). C.L.C. is supported by predoctoral National Research Service Award fellowship F31-AI52490. D.S.K. is supported by the American Heart Association Western States Affiliates fellowship 0425119Y.
The abbreviations used are
- RTX
repeats in toxin
- ACD
actin cross-linking domain
- TGase
transglutaminase
- PA
protective antigen
- LFN
N-terminal portion of Lethal Factor
- IPTG
isopropyl-β-D-thiogalactopyranoside
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene-bis(oxyethylenenitrilo)tetraacetic acid
- AMP-PNP
adenyl-5-yl imidodiphosphate
- LatB
latrunculin B
- Dol11
dolastatin 11
References
- 1.Kaper JB, Morris JGJ, Levine MM. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE, Calderwood SB, Fraser C, Mekalanos JJ. Proc Natl Acad Sci USA. 1999;96:1071–1076. doi: 10.1073/pnas.96.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chow KH, Ng TK, Yuen KY, Yam WC. J Clin Microbiol. 2001;39:2594–2597. doi: 10.1128/JCM.39.7.2594-2597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalsgaard A, Serichantalergs O, Forslund A, Lin W, Mekalanos J, Mintz E, Shimada T, Wells JG. J Clin Microbiol. 2001;39:4086–4092. doi: 10.1128/JCM.39.11.4086-4092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boardman BK, Satchell KJ. J Bacteriol. 2004;186:8137–8143. doi: 10.1128/JB.186.23.8137-8143.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satchell KJF. International Journal of Medical Microbiology in press. [Google Scholar]
- 7.Sheahan KL, Cordero CL, Satchell KJ. Proc Natl Acad Sci U S A. 2004;101:9798–9803. doi: 10.1073/pnas.0401104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fullner KJ, Lencer WI, Mekalanos JJ. Infect Immun. 2001;69:6310–6317. doi: 10.1128/IAI.69.10.6310-6317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fullner KJ, Mekalanos JJ. EMBO J. 2000;19:5315–5323. doi: 10.1093/emboj/19.20.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milne JC, Blanke SR, Hanna PC, Collier RJ. Mol Microbiol. 1995;15:661–666. doi: 10.1111/j.1365-2958.1995.tb02375.x. [DOI] [PubMed] [Google Scholar]
- 11.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 12.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 13.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- 14.Elliott JL, Mogridge J, Collier RJ. Biochemistry. 2000;39:6706–6713. doi: 10.1021/bi000310u. [DOI] [PubMed] [Google Scholar]
- 15.Gordon VM, Leppla SH, Hewlett EL. Infect Immun. 1988;56:1066–1069. doi: 10.1128/iai.56.5.1066-1069.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koehler TM, Collier RJ. Mol Microbiol. 1991;5:1501–1506. doi: 10.1111/j.1365-2958.1991.tb00796.x. [DOI] [PubMed] [Google Scholar]
- 17.Spyres LM, Qa'Dan M, Meader A, Tomasek JJ, Howard EW, Ballard JD. Infect Immun. 2001;69:599–601. doi: 10.1128/IAI.69.1.599-601.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sambrook J, Russell David W. Molecular Cloning: A Laboratory Manual. Third. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2001. [Google Scholar]
- 19.Voth DE, Hamm EE, Nguyen LG, Tucker AE, Salles II, Ortiz-Leduc W, Ballard JD. Cell Microbiol. 2005;7:1139–1149. doi: 10.1111/j.1462-5822.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- 20.Spudich JA, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 21.Chen JS, Mehta K. Int J Biochem Cell Biol. 1999;31:817–836. doi: 10.1016/s1357-2725(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 22.Nemes ZJ, Adány R, Balázs M, Boross P, Fésüs L. J Biol Chem. 1997;272:20577–20583. doi: 10.1074/jbc.272.33.20577. [DOI] [PubMed] [Google Scholar]
- 23.Murthy SN, Lomasney JW, Mak EC, Lorand L. Proc Natl Acad Sci USA. 1999;96:11815–11819. doi: 10.1073/pnas.96.21.11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takashi R. Biochemistry. 1988;27:938–943. doi: 10.1021/bi00403a015. [DOI] [PubMed] [Google Scholar]
- 25.Mogridge J, Cunningham K, Collier RJ. Biochemistry. 2002;41:1079–1082. doi: 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W, Vandekerckhove J. Annu Rev Biophys Biomol Struct. 1992;21:49–76. doi: 10.1146/annurev.bb.21.060192.000405. [DOI] [PubMed] [Google Scholar]
- 27.Estes JE, Selden LA, Kinosian HJ, Gershman LC. J Muscle Res Cell Motil. 1992;13:272–284. doi: 10.1007/BF01766455. [DOI] [PubMed] [Google Scholar]
- 28.Gershman LC, Selden LA, Kinosian HJ, Estes JE. Biochim Biophys Acta. 1989;995:109–115. doi: 10.1016/0167-4838(89)90068-x. [DOI] [PubMed] [Google Scholar]
- 29.Kudryashov DS, Reisler E. Biophys J. 2003;85:2466–2475. doi: 10.1016/s0006-3495(03)74669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graceffa P, Dominguez R. J Biol Chem. 2003;278:34172–34180. doi: 10.1074/jbc.M303689200. [DOI] [PubMed] [Google Scholar]
- 31.Cooke R. Biochemistry. 1975;14:3250–3256. doi: 10.1021/bi00685a035. [DOI] [PubMed] [Google Scholar]
- 32.Spector I, Shochet NR, Kashman Y, Groweiss A. Science. 1983;219:493–495. doi: 10.1126/science.6681676. [DOI] [PubMed] [Google Scholar]
- 33.Bai R, Verdier-Pinard P, Gangwar S, Stessman CC, McClure KJ, Sausville EA, Pettit GR, Bates RB, Hamel E. Mol Pharmacol. 2001;59:462–469. doi: 10.1124/mol.59.3.462. [DOI] [PubMed] [Google Scholar]
- 34.Fujimoto LM, Roth R, Heuser JE, Schmid SL. Traffic. 2000;1:161–171. doi: 10.1034/j.1600-0854.2000.010208.x. [DOI] [PubMed] [Google Scholar]
- 35.Attri AK, Lewis MS, Korn ED. J Biol Chem. 1991;266:6815–6824. [PubMed] [Google Scholar]
- 36.Oda T, Crane ZD, Dicus CW, Sufi BA, Bates RB. J Mol Biol. 2003;328:319–324. doi: 10.1016/s0022-2836(03)00306-1. [DOI] [PubMed] [Google Scholar]
- 37.Lai TS, Slaughter TF, Peoples KA, Hettasch JM, Greenberg CS. J Biol Chem. 1998;273:1776–1781. doi: 10.1074/jbc.273.3.1776. [DOI] [PubMed] [Google Scholar]
- 38.Walker JE, Saraste M, Runswick MJ, Gay NJ. Embo J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Handel SE, Hendry KA, Sheterline P. J Cell Sci. 1990;97(Pt 2):325–333. doi: 10.1242/jcs.97.2.325. [DOI] [PubMed] [Google Scholar]
- 40.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Proc Natl Acad Sci U S A. 2006;103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]