

Abstract
Mutations in α-actinin-4 have been linked to familial focal segmental glomerulosclerosis (FSGS), a common renal disorder in humans, and produce an apparent increase in the actin-binding affinity of α-actinin-4 in vitro. One of the mutations in particular, Lys255Glu, falls in the middle of the actin-binding interface of the ABD (actin-binding domain). The ABD consists of tandem calponin-homology domains (CH1 and CH2). The crystal structures of most ABDs display a compact conformation, characterized by extensive inter-CH interactions. However, the conformation of F-actin bound ABDs is unsettled. Some EM studies find that the compact conformation is preserved upon binding to F-actin, whereas other studies suggest that the CHs separate and the ABD becomes extended. The Lys255Glu mutation in CH2 is significant in this regard since it removes a crucial inter-CH interaction with Trp147 of CH1, thought to stabilize the compact conformation. Together, the increased actin-binding affinity and the removal of this important inter-CH contact suggested that the Lys255Glu mutation might facilitate the transition toward the open ABD conformation proposed by some of the EM studies. However, the crystal structure of the ABD of α-actinin-4 Lys255Glu mutant described here displays the canonical compact conformation. Furthermore, the sedimentation coefficients by analytical ultracentrifugation of wild type and FSGS mutant ABDs (Lys255Glu, Ser262Pro and Thr259Ile) are nearly identical (2.50 ± 0.03 S), and are in good agreement with the theoretical value calculated from the crystal structure (2.382 S), implying that the compact conformation is retained in solution. The absence of a structural change suggests that the compact ABD conformation observed in the majority of the structures is highly stable and is preserved in solution, even in FSGS mutant ABDs.
Keywords: spectrin family, actin-binding protein, missense mutations, kidney disease
α-Actinin belongs to the spectrin family of actin-binding proteins, generally involved in actin cytoskeleton scaffolding and organization 1; 2. In addition to its classical role as an actin filament cross-linker, α-actinin serves as a general linkage between the actin cytoskeleton and multiple signaling and membrane proteins 3. There are four α-actinin isoforms closely related at the amino acid sequence level: α-actinin-1 is ubiquitously expressed and is most commonly associated with focal adhesions 4, α-actinin-2 and α-actinin-3 are expressed in muscle cells and are involved in actin filament cross-linking in the Z disc 5, α-actinin-4 is widely expressed and is implicated in the maintenance of cell shape and architecture 6.
Like most cytoskeletal proteins, members of the spectrin family are characterized by the presence of discrete protein-protein interaction and regulatory modules. In α-actinin, whose functional unit is an antiparallel dimer, each chain of molecular mass ∼100 kDa presents an N-terminal actin-binding domain (ABD), four spectrin repeats, and a C-terminal pair of EF-hands. The ABD consists of a tandem pair of calponin-homology (CH) domains (CH1 and CH2).
The crystal structures of several ABDs have been determined, including those of human, A. thaliana and S. pombe fimbrin 7; 8, utrophin 9, dystrophin 10, human and mouse plectin 11; 12, and human α-actinin-3 13 and α-actinin-1 14. With the exception of the first ABD of A. thaliana fimbrin 8, which contains unique mutations at the CH1-CH2 interface (discussed below), all the ABD structures display a similar “compact” conformation characterized by extensive interactions between the two CH domains. In two of the structures 10; 15, the compact conformation results from domain swapping between two ABD molecules in the asymmetry unit of the crystals.
Mutations in the ABD of α-actinin-4 (Lys255Glu, Ser262Pro and Thr259Ile) are associated with a common renal lesion in humans known as familial focal segmental glomerulosclerosis (FSGS) 16. The FSGS-linked mutations disrupt the normal localization of α-actinin-4 in actin stress fibers and focal adhesions and promote the formation of actin aggregates in kidney podocytes 17; 18; 19. All the mutations lead to an apparent increase in the actin-binding affinity of α-actinin-4 in vitro 16; 18. One of the mutations in particular, Lys255Glu in CH2 (human α-actinin-4 sequence, O43707), is positioned in the center of the actin-binding interface of the ABD, which has been mapped by mutagenesis, sequence conservation, and NMR studies 14; 20; 21; 22; 23; 24; 25. In a structure-based analysis of sequence conservation in the ABD 14, Lys255 was found to be highly conserved and to form strong hydrophobic, and occasionally π-cation contacts with Trp147 of CH1. The inter-CH interaction between Trp147 and Lys255 was deemed critical for stability of the compact conformation of the ABD 14, since these two amino acids are mutated in the first ABD of A. thaliana fimbrin, which is the only ABD with a non-compact structure 8. The apparent importance of Lys255 for maintenance of the compact ABD conformation and the observation that the Lys255Glu mutant has increased affinity for F-actin 16; 18 suggested that this mutation may facilitate a transition toward the “open” ABD structure, which some EM studies have found is required for F-actin binding 26; 27. However, whether the ABD opens up upon binding to F-actin is an unresolved question. Other EM studies of actin filaments decorated with the ABDs of various members of the spectrin family, α-actinin included, have suggested that the compact conformation is generally preserved in F-actin bound ABDs 28; 29; 30. On the other hand, analysis of electron micrographs of 2-D crystals of F-actin cross-linked with full-length α-actinin suggests that the ABD can adopt either an open or a closed conformation 31; 32. To test whether the FSGS-linked mutations produce an open ABD conformation, we determined the crystal structure of the Lys255Glu mutant ABD of α-actinin-4. We further compared, using analytical ultracentrifugation, the conformations of wild type and mutant ABDs at different temperatures in solution. The results consistently show that wild type and mutant ABDs retain in solution the compact conformation observed in the majority of the structures, including the structure determined here of the ABD of α-actinin-4 Lys255Glu mutant.
Crystal structure of the ABD of α-actinin-4 Lys255Glu mutant
The ABD of human α-actinin-4 (O43707, Ala47-Gly270) shares 94 % and 90 % sequence identity respectively with the ABDs of human α-actinin-1 (P12814, Ala28-Gly251) and α-actinin-3 (Q08043, Ala42-Gly265), and it was expected to share a similar structure with these two ABDs 13; 14. However, it was unknown what would be the effect of the FSGS-linked mutation Lys255Glu on the overall structure of the ABD. To answer this question, we determined the crystal structure of the ABD of α-actinin-4 (Lys255Glu mutant) at 2.2 Å resolution (Figure 1(a) and Table 1).
Figure 1.

Structure of the actin-binding domain (ABD) of α-actinin-4 Lys255Glu mutant. (a) Overall view of the structure of the ABD (CH1, blue; CH2, red). The linker loop between CH1 and CH2 (α-actinin-4 amino acids Asp157 to Ser164) is shown in black. Helices are labeled A to G (this ABD does not contain helix D). Note that although there are two molecules in the asymmetric unit of the crystal, their overall structures are very similar (Cα rms deviation of 0.5 Å) and only one molecule is shown (chain A). In addition to the amino acids of the ABD, chain A includes an extra Phe residue at the N-terminus (Phe46) and a Leu residue at the C-terminus (Leu272) from the expression plasmid. The side chains of Trp147 and the mutated amino acid Glu255 are shown. Trp147 and the wild type residue Lys255 are highly conserved in the spectrin family, making part of the actin-binding interface, and are typically involved in strong inter-CH contacts 14 (b) Superimpositions of the structures of the ABD of α-actinin-4 with those of α-actinin-1 (gold, Cα rms deviation 0.71 Å) and α-actinin-3 (green, Cα rms deviation 0.57 Å). Note that despite the presence of the Lys255Glu mutation in the ABD of α-actinin-4, the overall structures of the three α-actinin ABDs are nearly identical. (c) Close view of the Trp-Lys stacking interaction at the interface of CH1-CH2 in the ABDs of α-actinin-1 and α-actinin-3, and the Lys255Glu mutation in the ABD of α-actinin-4 that brakes this important interaction.
Experimental conditions: Fragments encoding the ABD of human α-actinin-4 (O43707, amino acids Ala47-Ala271) were generated by PCR from wild type and mutant α-actinin-4 cDNAs, subcloned into pGEX-4T1-HTa and verified by sequence analysis. GST-proteins were expressed in E. coli 5-alpha (NEB) and purified using Glutathione-Sepharose (GE Healthcare). Cleavage of the GST tag was carried out using Tobacco-Etch-Virus (TEV) protease, and proteins were purified using Ni Sepharose 6 Fast Flow (GE Healthcare). Protein purity and concentration were verified using SDS-PAGE and Bradford methods. The ABD of α-actinin-4 Lys255Glu mutant was concentrated to ∼8 mg ml−1 using Amicon Ultra centrifugal filters (Amicon) in 50 mM Tris-HCl pH 8.0, 100 mM NaCl and 1 mM DTT. Crystals were obtained using the hanging drop vapor diffusion method at 4°C by mixing 2 μl of the protein solution and 2 μl of a well solution containing 100 mM Imidazole pH 7.1, 50 mM NaCl, 1 mM EDTA, 5 % (v/v) glycerol, and 21 % (w/v) polyethylene glycol 5000 mono-methyl-ether. The percentage of glycerol was raised to ∼20 % for crystal freezing prior to X-ray data collection. An X-ray diffraction dataset was collected to the resolution of 2.2 Å from a crystal frozen in liquid nitrogen, and using the beamline F2 of the Cornell High Energy Synchrotron Source (CHESS). The diffraction data were indexed and scaled with the program HKL2000 (HKL Research, Inc) (Table 1). A molecular replacement solution for the two independent ABD molecules in the asymmetric unit of the crystals was obtained with the program AMoRe 36, and using the 1.7 Å resolution crystal structure of the ABD of α-actinin-1 14 as a search model. Model building and refinement were carried out with the program Coot 37, and the CCP4 program Refmac. The refinement of the structure converged to R-factor and R-free values of 17.1% and 22.4 %, respectively (Table 1). The electron density map reveals all the amino acids of the two ABD molecules in the asymmetric unit of the crystal. The construct crystallized here contained extra amino acids at the N-terminus (GAMDPEF) and the C-terminus (LDELN) resulting from the expression vector. Some of these amino acids are also observed in the electron density map and form part of the refined model (PDB Code 2R0O).
Table 1.
Crystallographic Data and Refinement Statistics
| Diffraction statistics | |
| Space group | P 21 21 21 |
| Cell parameters | |
| a, b, c (Å) | 61.697, 61.533, 174.936 |
| α, β, γ (o) | 90.0, 90.0, 90.0 |
| Data resolution (Å) | 42.37 − 2.20 (2.28 − 2.20) |
| Completeness (%) | 87.5 (69.3) |
| Redundancy | 5.6 (3.6) |
| Unique reflections | 25,757 (1,298) |
| R-mergea (%) | 8.8 (28.3) |
| Average I/σ | 33.8 (6.6) |
| Refinement statistics | |
| Refinement resolution (Å) | 42.37 − 2.20 (2.257 − 2.20) |
| R-factorb (%) | 17.1 (18.3) |
| R-freec (%) | 22.4 (30.0) |
| Rms deviations | |
| Bond length (Å) | 0.014 |
| Bond angles (o) | 1.398 |
| Average B-factor (Å2) | 15.41 |
| PDB Code | 2R0O |
Values in parentheses correspond to highest resolution shell
R-merge=Σ(I-<I>)/ΣI; I and <I> are the intensity and the mean value of all the measurements of an individual reflection
R-factor=Σ|Fo - Fc|/Σ|Fo|; Fo and Fc are the observed and calculated structure factors
R-free; R-factor calculated for a randomly selected subset of the reflections (5%) that were omitted during the refinement
There are two ABD molecules in the asymmetric unit of the crystal (chains A and B). Since these molecules are crystallographically independent, this circumstance allowed for two separate observations of the conformation of the mutant ABD. The two ABD molecules, however, presented very similar structures (Cα rms deviation of 0.5 Å) and both displayed the canonical compact conformation observed in nearly all the ABD structures determined thus far 7; 8; 9; 10; 11; 12; 13; 14. The current structure is most similar to those of α-actinin-1 (Cα rms deviation of 0.71 Å) and α-actinin-3 (Cα rms deviation of 0.57 Å) (Figure 1(b)). The linker loops that connect the CH domains are also very similar among the three α-actinin structures, despite the fact that this region is generally considered to be more flexible. It thus appears that the Lys255Glu mutation in α-actinin-4 does not affect the global structure of the ABD, at least within the context of the crystal structure.
In the compact ABD conformation that characterizes the current structure, the two CH domains are involved in extensive (mainly hydrophobic) contacts with one another (Figure 1). Lys255 in CH2 plays a central role in these interactions. This amino acid is typically involved in strong hydrophobic contacts with the side chain of Trp147 of CH1 14. Both Lys255 and Trp147 are highly conserved among members of the spectrin family, and are predicted to form part of the actin-binding interface 14; 20; 21; 22; 23; 24; 25. Interestingly, when the two CH domains that form the ABD of α-actinin-4 are superimposed upon one another (Cα rms deviation of 1.9 Å), the amino acid structurally equivalent to Lys255 of CH2 is also a lysine in CH1 (Lys140). Lys140 is relatively well conserved within the spectrin family (although less well conserved than Lys255). This conservation is probably significant, since the overall sequence identity between the two CHs is low (17%) and both Lys140 and Lys255 are solvent exposed. Therefore, it appears that there is evolutionary pressure toward the conservation of this particular position in the CH architecture, and the Trp147-Lys255 stacking interaction between CHs, in particular.
In the current structure, the side chain of Glu255, which is well defined in the electron density map (Figure 2(a)), turns away from the side chain of Trp147 (Figure 2(b)). This rotamer orientation of Glu255, which is conserved in both chains of the structure (Figure 2(b)), results in an open cavity on the actin-binding surface of the ABD, at the interface between CH1 and CH2. In both chains, a molecule of glycerol (present in the crystallization buffer) occupies this cavity. However, the exact position of glycerol is not conserved between chains (Figure 2(b)). Despite this difference, the amino acids that surround the mutated residue Glu255 occupy similar positions in the two chains of the asymmetric unit (Figure 2(b)), as well as in the structures of the ABDs of α-actinin-114 and α-actinin-3 13 (Figure 1(c)).
Figure 2.

Close view of the structure around residue Glu255. (a) 2Fo-Fc electron density map contoured at 1.8 σ around (Chain B). Note that a molecule of glycerol occupies the space that is typically engaged by the side chain of Lys255. (b) Superimposition of Chains A (gray Cα atoms) and B (yellow Cα atoms). Note that chain A also contains a molecule of glycerol in the space near the mutation, but its location is somewhat different from that of Chain B.
As established above, the presence of the mutation Lys255Glu does not affect the overall structure of the ABD, at least in isolation. However, a very different picture emerges if the electrostatic characteristics of the ABD are taken into account. By restoring the endogenous Lys255, according to its position in the structure of the ABD of α-actinin-1 14, a comparison of the electrostatic surfaces of the wild type and Lys255Glu mutant ABDs reveals major differences (Figure 3(a,b)). These differences, which coincide with the location of the predicted actin-binding interface, may possibly explain the different actin-binding properties of Lys255Glu α-actinin-4 16; 18. However, this explanation cannot be extended to the other two FSGS-linked mutations (Ser262Pro and Thr259Ile), which occur at the interface between CH1 and CH2 and are buried in the structure (Figure 3(c,d)). To test whether the conformation of mutant ABD constructs changes in solution, we used analytical ultracentrifugation.
Figure 3.
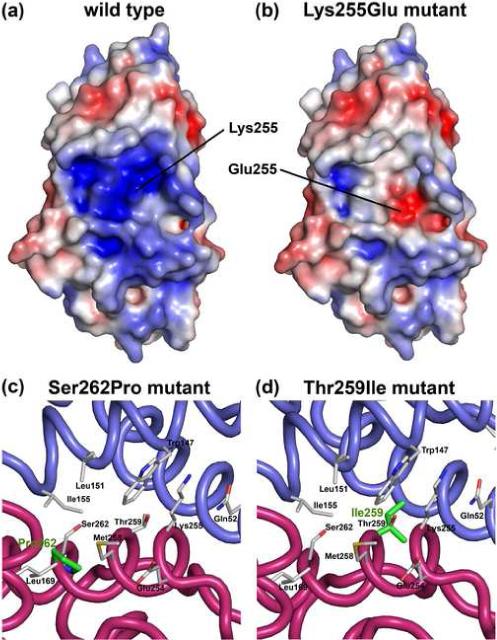
Different effects of FSGS-linked mutations on the structure of the ABD of α-actinin-4. (a, b) Electrostatic surface representation of the ABD of α-actinin-4 wild type and Lys255Glu mutant calculated with the program APBS 38 and displayed with the program PyMOL (http://www.pymol.org). The surface is colored according to charge, using a linear color ramp (−5 kT, red to 5 kT, blue). For this figure, the wild type structure was generated by restoring the endogenous residue Lys255 (indicated) according to its position in the structure of the ABD of α-actinin-1 14. (c,d) Mutations Ser262Pro and Thr259Ile (shown in green) are buried in the compact structure of the ABD and do not affect the electrostatic surface. However, these two mutations occur in helix G, which is at the interface between CH1 (blue) and CH2 (red) and are likely to affect inter-CH contacts.
Conformation of the wild type and FSGS mutant ABDs in solution
The sedimentation coefficient of macromolecules is sensitive to conformational changes in solution. For example, the myosin light chain Mlc1p adopts two radically different conformations, compact and extended, depending on the specific sequence of the bound myosin IQ domain 33. The sedimentation coefficient of the compact Mlc1p-IQ complex (1.95 S) is 8.2% greater (i.e. sediments faster) than that of the extended complex (1.79 S), while their molecular masses are nearly identical (19,300 Da). A similar change in sedimentation coefficient may be expected if the ABD of α-actinin-4 adopts an open conformation in solution as a result of the FSGS mutations, in particular since the molecular mass of the ABD (25,692 Da) is higher than that of Mlc1p-IQ complexes. Therefore, sedimentation velocity analyses were performed on the wild type and mutant ABDs of α-actinin-4 to assess whether their free hydrodynamic conformations are measurably different (Figure 4 and Table 2).
Figure 4.
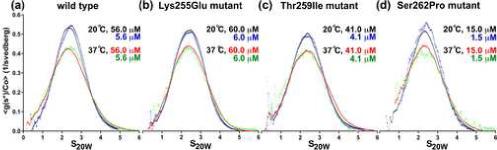
Conformation of wild type and mutant ABDs in solution by analytical ultracentrifugation. g(s) plots produced with the program SEDANAL 39 showing two concentrations at two different temperatures (20°C and 37°C) of (a) wild type, (b) Lys255Glu, (c) Thr259Ile and (d) Ser262Pro mutant ABDs. The x-axis corresponds to the sedimentation coefficient in Svedberg units converted according to the general convention to S20W values (S value at 20°C in water). The y-axis represents the sedimentation distribution function normalized to the loading concentration. The entire global fit of wild type and mutant ABDs consisted of 900 scans from experiments at two different concentrations and temperatures, resulting in an average S value of 2.50 ± 0.03 S (see Table 2 for details).
Experimental conditions: Sedimentation velocity experiments were conducted in a Beckman Optima XL-I ultracentrifuge (Beckman, Palo Alto, CA), using either an An60 Ti four-hole rotor or an An50 Ti eight-hole rotor. Data were acquired with the interference optics system using sapphire windows and meniscus-matching 12-mm aluminum-filled Epon centerpieces with interference slit window holders on the top window (Biomolecular Interaction Technology Center, Durham, NH). The wild type and mutant ABD constructs were dialyzed exhaustively against 100 mM NaCl and 20 mM Tris-HCl pH 8.0. Velocity experiments were conducted at 50,000 rpm and 20°C. Sedimentation velocity data were analyzed using the program SEDANAL 39. Curve fits were deemed acceptable only if the standard deviation of the fit was comparable to the previously measured optical noise of the system, 1 × 10−2 fringes. The fit S values were converted to standard S20W values (sedimentation coefficient of the molecule if it were sedimented at 20°C in water) by the program SEDNTERP 40.
Table 2.
Sedimentation velocities of wild type and mutant ABDs of α-actinin-4
| ABD Construct | T (°C) | Standard deviation of fit (mg/ml) | S20w | % deviation from WT |
|---|---|---|---|---|
| WT (1) | 20 | 0.00284 | 2.46 +/− 0.01S | −1.6% |
| WT (2) | 20 | 0.00491 | 2.50 +/− 0.01S | 0.0% |
| WT | 37 | 0.00407 | 2.52 +/− 0.01S | 0.8% |
| Lys255Glu (1) | 20 | 0.00304 | 2.50 +/− 0.01S | 0.0% |
| Lys255Glu (2) | 20 | 0.00384 | 2.49 +/− 0.01S | −0.4% |
| Lys255Glu | 37 | 0.00338 | 2.53 +/− 0.01S | 1.2% |
| Ser262Pro | 20 | 0.00256 | 2.49 +/− 0.01S | −0.4% |
| Ser262Pro | 37 | 0.00312 | 2.49 +/− 0.01S | −0.4% |
| Thr259Ile | 20 | 0.00350 | 2.46 +/− 0.01S | −1.6% |
| Thr259Ile | 37 | 0.00437 | 2.54 +/− 0.01S | 1.6% |
| Average | 0.00356 | 2.50 |
For each construct, two or more concentrations were analyzed at two different temperatures (20°C and 37°C). Global fits to single species models resulted in very similar S values for all the constructs (2.50 ± 0.03 S), independent of concentration and temperature (Figure 4 and Table 2). These results strongly suggest that all the ABD constructs have very similar conformations in solution.
In an attempt to assess the relationship between the conformation of the ABD in solution and that of the crystal structure, the program HYDROPRO 34 was used to calculate a theoretical sedimentation coefficient from the atomic coordinates of the Lys255Glu mutant ABD. The calculated value, 2.382 S, is slightly smaller than the measured value. A smaller S value indicates a slower sedimenting species and a more extended conformation at constant mass. In other words, the ABD constructs (wild type and mutants) sediment faster than predicted from the crystal structure, implying that their solution conformations are as compact (or more) than that of the structure. More likely, however, the crystal structure accurately represents the conformation of the ABD in solution and a smaller theoretical sedimentation coefficient probably reflects underestimation of hydration by the program HYDROPRO.
In summary, the Lys255Glu mutation within the ABD of α-actinin-4 has been linked to FSGS and increases the actin-binding affinity of α-actinin-4 (Figure 5) 16; 18. The mutation falls in the middle of what has been predicted to be the actin-binding interface of the ABD and affects an important and highly conserved inter-CH contact with Trp147 14. Therefore, this mutation was predicted to weaken inter-CH interactions, facilitating a transition toward the open ABD conformation, which some EM studies have suggested is required for F-actin binding 26; 27. However, the crystal structure of the mutated ABD (Figure 1) reveals the canonical compact conformation. The compact conformation appears to be conserved in solution since the sedimentation coefficient of the mutated and wild type ABDs determined by analytical ultracentrifugation at different temperatures are nearly identical (Figure 4), and are in good agreement with the theoretical value calculated from the crystal structure. Similarly, the sedimentation coefficients of mutants Ser262Pro and Thr259Ile are undistinguishable from that of the wild type protein, suggesting that their solution conformations are also compact.
Figure 5.

Binding curves of wild type, Lys255Glu, Thr259Ile and Ser262Pro ABDs of α-actinin-4 show increased actin-binding affinity of the mutants. The dissociation constant (KD) of the wild type ABD could not be determined due to its low binding affinity. Mutants Lys255Glu, Thr259Ile and Ser262Pro were found to have increased binding affinities (Kd 25−35 μM).
Experimental conditions: Recombinant ABDs were mixed with 5 μM G-actin in a solution containing 20 mM Tris-HCl pH 7.4, 0.5 mM ATP, 2 mM MgCl2, 120 mM NaCl and 0.5 mM EGTA for 1h at room temperature. To determine the KD, 5 μM G-actin was incubated with increasing amounts of purified WT and mutant ABD (5 to 140 μM). F-actin was sedimented at 100,000 × g for 30 min at 22°C. Proteins in the supernatant and pellet were solubilized in equal amounts of SDS sample buffer, boiled and subjected to SDS-PAGE. Protein bands were visualized by Coomassie Blue staining, and band intensities were quantified using the program ImageJ (http://rsb.info.nih.gov/ij/). One-site binding hyperbola nonlinear regression analysis, Scatchard analysis, KD calculation and 95% confidence intervals were performed with the program GraphPad Prism (version 4.00).
The absence of a conformational change suggests that the compact ABD conformation observed in the majority of the structures 7; 8; 9; 10; 11; 12; 13; 14 is highly stable and that it is preserved in solution even in FSGS mutant ABDs. It also appears that the increased actin-binding affinities of α-actinin-4 mutants (Figure 5) 16; 18 cannot be explained by a single structural mechanism. Thus, the Lys255Glu mutation occurs on the surface of the ABD and has a very limited effect on its overall structure (Figure 1). Yet, this mutation probably weakens inter-CH contacts and has a major impact on the electrostatic charge distribution at the actin-binding interface (Figure 3(a,b)). On the other hand, mutations Ser262Pro and Thr259Ile are buried in the compact structure and do not affect the electrostatic surface of the ABD. However, these two mutations occur in helix G, which is at the interface between CH1 and CH2 (Figure 3(c,d)), and probably affect inter-CH contacts. The incorporation of a Pro residue at position 262 may produce a kink in helix G, whereas a larger Ile side chain at position 259 could result in steric hindrance, both of which would weaken inter-CH contacts. The analytical ultracentrifugation results (Figure 4 and Table 2) indicate that these changes are insufficient to produce an open ABD conformation in solution. It thus appears that the exposure of hydrophobic surfaces in CH1 and CH2, which face each other in the compact conformation, is energetically unfavorable. However, this study did not address the effect of F-actin on the conformation of the ABD. Weakening of inter-CH contacts by FSGS-linked mutations may facilitate opening of the ABD in the presence of F-actin, as previously suggested 35.
ACKNOWLEDGMENTS
This work was supported by NIH grants HL086655 and DK59588, and a National Kidney Foundation postdoctoral fellowship to A.W. Use of the Cornell High Energy Synchrotron Source (CHESS) and Macromolecular Crystallography Facility (MacCHESS) was supported by NSF grant DMR-0225180 and NIH grant RR-01646.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Gimona M, Djinovic-Carugo K, Kranewitter WJ, Winder SJ. Functional plasticity of CH domains. FEBS Lett. 2002;513:98–106. doi: 10.1016/s0014-5793(01)03240-9. [DOI] [PubMed] [Google Scholar]
- 2.Broderick MJ, Winder SJ. Spectrin, alpha-actinin, and dystrophin. Adv Protein Chem. 2005;70:203–46. doi: 10.1016/S0065-3233(05)70007-3. [DOI] [PubMed] [Google Scholar]
- 3.Otey CA, Carpen O. Alpha-actinin revisited: a fresh look at an old player. Cell Motil Cytoskeleton. 2004;58:104–11. doi: 10.1002/cm.20007. [DOI] [PubMed] [Google Scholar]
- 4.Pavalko FM, Burridge K. Disruption of the actin cytoskeleton after microinjection of proteolytic fragments of alpha-actinin. J Cell Biol. 1991;114:481–91. doi: 10.1083/jcb.114.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beggs AH, Byers TJ, Knoll JH, Boyce FM, Bruns GA, Kunkel LM. Cloning and characterization of two human skeletal muscle alpha-actinin genes located on chromosomes 1 and 11. J Biol Chem. 1992;267:9281–8. [PubMed] [Google Scholar]
- 6.Lachapelle M, Bendayan M. Contractile proteins in podocytes: immunocytochemical localization of actin and alpha-actinin in normal and nephrotic rat kidneys. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;60:105–11. doi: 10.1007/BF02899534. [DOI] [PubMed] [Google Scholar]
- 7.Goldsmith SC, Pokala N, Shen W, Fedorov AA, Matsudaira P, Almo SC. The structure of an actin-crosslinking domain from human fimbrin. Nat Struct Biol. 1997;4:708–12. doi: 10.1038/nsb0997-708. [DOI] [PubMed] [Google Scholar]
- 8.Klein MG, Shi W, Ramagopal U, Tseng Y, Wirtz D, Kovar DR, Staiger CJ, Almo SC. Structure of the actin crosslinking core of fimbrin. Structure (Camb) 2004;12:999–1013. doi: 10.1016/j.str.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Keep NH, Winder SJ, Moores CA, Walke S, Norwood FL, Kendrick-Jones J. Crystal structure of the actin-binding region of utrophin reveals a head-to-tail dimer. Structure Fold Des. 1999;7:1539–46. doi: 10.1016/s0969-2126(00)88344-6. [DOI] [PubMed] [Google Scholar]
- 10.Norwood FL, Sutherland-Smith AJ, Keep NH, Kendrick-Jones J. The structure of the N-terminal actin-binding domain of human dystrophin and how mutations in this domain may cause Duchenne or Becker muscular dystrophy. Structure Fold Des. 2000;8:481–91. doi: 10.1016/s0969-2126(00)00132-5. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Alvarez B, Bobkov A, Sonnenberg A, de Pereda JM. Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin beta4. Structure (Camb) 2003;11:615–25. doi: 10.1016/s0969-2126(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 12.Sevcik J, Urbanikova L, Kost'an J, Janda L, Wiche G. Actin-binding domain of mouse plectin. Crystal structure and binding to vimentin. Eur J Biochem. 2004;271:1873–84. doi: 10.1111/j.1432-1033.2004.04095.x. [DOI] [PubMed] [Google Scholar]
- 13.Franzot G, Sjoblom B, Gautel M, Djinovic Carugo K. The crystal structure of the actin binding domain from alpha-actinin in its closed conformation: structural insight into phospholipid regulation of alpha-actinin. J Mol Biol. 2005;348:151–65. doi: 10.1016/j.jmb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Borrego-Diaz E, Kerff F, Lee SH, Ferron F, Li Y, Dominguez R. Crystal structure of the actin-binding domain of alpha-actinin 1: Evaluating two competing actin-binding models. J Struct Biol. 2006;155:230–8. doi: 10.1016/j.jsb.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Keep NH, Norwood FL, Moores CA, Winder SJ, Kendrick-Jones J. The 2.0 A structure of the second calponin homology domain from the actin-binding region of the dystrophin homologue utrophin. J Mol Biol. 1999;285:1257–64. doi: 10.1006/jmbi.1998.2406. [DOI] [PubMed] [Google Scholar]
- 16.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–6. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 17.Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, Pollak MR. Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol. 2004;2:e167. doi: 10.1371/journal.pbio.0020167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weins A, Kenlan P, Herbert S, Le TC, Villegas I, Kaplan BS, Appel GB, Pollak MR. Mutational and Biological Analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2005;16:3694–701. doi: 10.1681/ASN.2005070706. [DOI] [PubMed] [Google Scholar]
- 19.Michaud JL, Chaisson KM, Parks RJ, Kennedy CR. FSGS-associated alpha-actinin-4 (K256E) impairs cytoskeletal dynamics in podocytes. Kidney Int. 2006;70:1054–61. doi: 10.1038/sj.ki.5001665. [DOI] [PubMed] [Google Scholar]
- 20.Bresnick AR, Janmey PA, Condeelis J. Evidence that a 27-residue sequence is the actin-binding site of ABP-120. J Biol Chem. 1991;266:12989–93. [PubMed] [Google Scholar]
- 21.Corrado K, Mills PL, Chamberlain JS. Deletion analysis of the dystrophin-actin binding domain. FEBS Lett. 1994;344:255–60. doi: 10.1016/0014-5793(94)00397-1. [DOI] [PubMed] [Google Scholar]
- 22.Levine BA, Moir AJ, Patchell VB, Perry SV. The interaction of actin with dystrophin. FEBS Lett. 1990;263:159–62. doi: 10.1016/0014-5793(90)80728-2. [DOI] [PubMed] [Google Scholar]
- 23.Levine BA, Moir AJ, Patchell VB, Perry SV. Binding sites involved in the interaction of actin with the N-terminal region of dystrophin. FEBS Lett. 1992;298:44–8. doi: 10.1016/0014-5793(92)80019-d. [DOI] [PubMed] [Google Scholar]
- 24.Fabbrizio E, Bonet-Kerrache A, Leger JJ, Mornet D. Actin-dystrophin interface. Biochemistry. 1993;32:10457–63. doi: 10.1021/bi00090a023. [DOI] [PubMed] [Google Scholar]
- 25.Kuhlman PA, Hemmings L, Critchley DR. The identification and characterisation of an actin-binding site in alpha-actinin by mutagenesis. FEBS Lett. 1992;304:201–6. doi: 10.1016/0014-5793(92)80619-r. [DOI] [PubMed] [Google Scholar]
- 26.Galkin VE, Orlova A, VanLoock MS, Rybakova IN, Ervasti JM, Egelman EH. The utrophin actin-binding domain binds F-actin in two different modes: implications for the spectrin superfamily of proteins. J Cell Biol. 2002;157:243–51. doi: 10.1083/jcb.200111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moores CA, Keep NH, Kendrick-Jones J. Structure of the utrophin actin-binding domain bound to F-actin reveals binding by an induced fit mechanism. J Mol Biol. 2000;297:465–80. doi: 10.1006/jmbi.2000.3583. [DOI] [PubMed] [Google Scholar]
- 28.McGough A, Way M, DeRosier D. Determination of the alpha-actinin-binding site on actin filaments by cryoelectron microscopy and image analysis. J Cell Biol. 1994;126:433–43. doi: 10.1083/jcb.126.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanein D, Volkmann N, Goldsmith S, Michon AM, Lehman W, Craig R, DeRosier D, Almo S, Matsudaira P. An atomic model of fimbrin binding to F-actin and its implications for filament crosslinking and regulation. Nat Struct Biol. 1998;5:787–92. doi: 10.1038/1828. [DOI] [PubMed] [Google Scholar]
- 30.Sutherland-Smith AJ, Moores CA, Norwood FL, Hatch V, Craig R, Kendrick-Jones J, Lehman W. An atomic model for actin binding by the CH domains and spectrin-repeat modules of utrophin and dystrophin. J Mol Biol. 2003;329:15–33. doi: 10.1016/s0022-2836(03)00422-4. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Taylor DW, Taylor KA. A 3-D reconstruction of smooth muscle alpha-actinin by CryoEm reveals two different conformations at the actin-binding region. J Mol Biol. 2004;338:115–25. doi: 10.1016/j.jmb.2004.02.034. [DOI] [PubMed] [Google Scholar]
- 32.Hampton CM, Taylor DW, Taylor KA. Novel structures for alpha-actinin:F-actin interactions and their implications for actin-membrane attachment and tension sensing in the cytoskeleton. J Mol Biol. 2007;368:92–104. doi: 10.1016/j.jmb.2007.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terrak M, Wu G, Stafford WF, Lu RC, Dominguez R. Two distinct myosin light chain structures are induced by specific variations within the bound IQ motifs-functional implications. Embo J. 2003;22:362–71. doi: 10.1093/emboj/cdg058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–30. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weins A, Schlondorff JS, Nakamura F, Denker BM, Hartwig JH, Stossel TP, Pollak MR. Disease-associated mutant alpha-actinin-4 reveals a mechanism for regulating its F-actin-binding affinity. Proc Natl Acad Sci U S A. 2007;104:16080–5. doi: 10.1073/pnas.0702451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navaza J. AMoRe: an automated package for molecular replacemenet. Acta Cryst. 1994;A50:157–163. [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stafford WF, Sherwood PJ. Analysis of heterologous interacting systems by sedimentation velocity: curve fitting algorithms for estimation of sedimentation coefficients, equilibrium and kinetic constants. Biophys Chem. 2004;108:231–43. doi: 10.1016/j.bpc.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 40.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. In Analytical Ultracentrifugation in Biochemistry and Polymer Sciences. Royal Society of Chemistry; Cambridge, UK: 1992. pp. 90–125. [Google Scholar]