

Abstract
Src family kinases (SFKs) interact with a number of cellular receptors. They participate in diverse signaling pathways and cellular functions. Most of the receptors involved in SFK signaling are characterized by similar modes of regulation. This computational study discusses a general kinetic model of SFK-receptor interaction. The analysis of the model reveals three major ways of SFK activation: release of inhibition by C-terminal Src kinase, weakening of the inhibitory intramolecular phosphotyrosine-SH2 interaction, and amplification of a stimulating kinase activity. The SFK model was then extended to simulate interaction with growth factor and T-cell receptors. The modular SFK signaling system was shown to adapt to the requirements of specific signaling contexts and yield qualitatively different responses in the different simulated environments. The model also provides a systematic overview of the major interactions between SFKs and various cellular signaling systems and identifies their common properties.
INTRODUCTION
Many cell surface receptors share a common mode of activation. Binding of a signaling molecule to the extracellular, ligand-binding domain activates the specific receptor and triggers subsequent intracellular events. This typically involves dimerization of activated receptor molecules and, due to the intrinsic tyrosine kinase activity of many receptors, phosphorylation of tyrosine residues in the intracellular receptor domains, as well as phosphorylation of other target proteins. A large variety of signaling proteins can attach to these phosphorylated tyrosine residues through interaction between the phosphotyrosines in the receptor and Src homology 2 (SH2) domains in the binding protein. In some cases, this causes the activation of the binding protein, which then performs subsequent cellular functions. In other cases, the transportation of an enzyme near the membrane directs its activity to other membrane-located proteins, having a similar, activating effect.
One important class of receptor binding protein is the family of Src kinases. The nine members of this family play roles in the cellular events mediated by receptors such as the epidermal growth factor receptor (EGFR), the platelet-derived growth factor receptor (PDGFR), or the T-cell receptor (TCR) (1,2). Some of the Src family kinases (SFKs) are expressed ubiquitously, like Src, which also contributes to cell-cycle-specific events. Other SFKs, such as Fyn and Lck, are only found in specific cell types (T-lymphocytes, in this case). All SFKs are membrane-anchored and share a common domain architecture.
In recent years, the modes of SFK regulation have been elucidated to such a degree that we now understand the effects of the many kinases and phosphatases the SFKs interact with (see also Fig. 1 B). SFK activity is regulated by a multitude of factors. Most importantly, Tyr-418 (residue numbers refer to human Src) is phosphorylated in a trans-autocatalytic reaction and significantly enhances kinase activity. The C-terminal Src kinase (Csk) is an important negative regulator of SFK activity (3,4). It phosphorylates tyrosine residue 530 of Src (3), a residue which allows the C-terminal tail to interact with the SH2 region within the same molecule. This “tail-bite” conformation is catalytically inactive (5,6). Csk itself is regulated by translocation: the so-called Csk binding protein (Cbp, also known as PAG) is a membrane-anchored adaptor protein that recruits Csk to the membrane, when phosphorylated on tyrosine 317 (7,8). Cbp/PAG and Csk interact through an SH2 domain in Csk that binds to this phosphotyrosine. Src family kinases can phosphorylate this residue, and thus create a negative feedback loop. This mechanism of SFK suppression plays a key role in the signaling contexts of the TCR (9–11), EGFR (12,13), PDGFR (14), and others (15).
FIGURE 1.

(A) Common reaction scheme of interactions between receptor (R), kinase (Src), and SH2-domain containing proteins (SH2). (B) Phosphorylation sites of Src and enzymes responsible for phosphorylation and dephosphorylation.
The fact that Src family kinases interact with a variety of receptors and are also involved in other signaling pathways calls for a general model to explain more about the mechanism of interaction with and the physiological benefit gained from involvement of SFKs. Such a general model would provide a useful basis for reasoning about any of the specific signaling systems—for example, for drawing parallels between them and elucidating unknown molecular mechanisms. It would also help to answer the question about the precise physiological role played by SFKs.
However, there are a few uncertainties and even conflicting reports that require closer investigation. For example, it is currently not known which phosphatase is involved in the dephosphorylation of Cbp/PAG, which would release Csk and allow for the activation of SFKs. Matsuoka et al. (12) have observed that upon stimulation of the EGF receptor, Src is activated and causes an increase in Cbp/PAG phosphorylation, which is consistent with the interactions described above. On the other hand, various groups have shown that in unstimulated T-cells, Cbp/PAG phosphorylation levels are high and stimulation of the T-cell receptor causes Cbp/PAG dephosphorylation, allowing SFK activation (8,16). In these two cases, it seems that two different mechanisms underlie the receptor-mediated activation of SFKs.
If the Src family of kinases and their main regulators constitute a reusable module within diverse signaling contexts, several questions arise:
How are SFKs activated by each receptor?
What are the requirements for successful employment of SFKs?
How can the SFK module adapt to the requirements of the various signaling contexts?
To investigate the mechanisms that lead to the activation of SFKs upon receptor stimulation, we constructed a kinetic model of the common mechanisms of SFK interaction that are shared by most receptor tyrosine kinases and investigated the model's dynamics. For this, we made use of a previously published mathematical model for Src activation at mitosis (17). This model was extended to include receptor interactions and updated with some quantitative and semiquantitative information. An important goal of this study was to elicit the properties and mechanisms which are common among signaling systems involving SFKs. Establishing these commonalities will facilitate the formulation of general principles governing such systems. Using numerical analysis methods, we identified and characterized three mechanisms that lead to SFK activation: dephosphorylation of Cbp/PAG, weakening of the intramolecular SH2-pTyr (“tail-bite”) interaction, and activation of a small pool of stimulating SFK molecules. The simulations show how these mechanisms function in the context of the EGFR and TCR.
METHODS
Protein interactions
The major protein interactions in SFK-receptor signaling were identified from published literature. From these, a common set of reactions was derived that describes the mode of regulation between the receptor systems under investigation. Only well-characterized protein interactions were taken into account. The simplified reaction scheme on which the model is based is given in Fig. 1 (explained in the text below).
Model definition
The concentrations of each molecular species in the network were represented as a dimensionless variable in the model, i.e., the variables are not calibrated and assume values from the unit interval ([0…1]). This was due to two reasons:
The intention was to construct a general model, which would not describe quantitative concentration changes in a particular system, but rather maintain validity across a wide range of contexts; different members of the Src family in different cellular environments are likely to differ in their kinetic rate constants.
The published information available on the relevant reactions (particularly the SFK subsystem) does not permit an accurate, quantitative description at this time. However, where available, quantitative information was taken into account.
The reactions between molecular species were modeled using ordinary differential equations. In this approach, the dynamic behavior of the system is defined by a set of parameterized equations that determine how the concentration of each species changes over time. These equations are derived from elementary chemical kinetic laws, such as the law of mass action. The result is a model system that reproduces the temporal behavior of the biological system.
The full kinetic specification of the model is given in Table 1. Reaction 1 defines ligand degradation, Reactions 2–9 define reactions involving the receptor, and Reactions 10–16 represent SFK regulation. A set of auxiliary algebraic equations define the relationships between concentrations and enzymatic activities of the molecular species, conservation relationships, and others. Activity in this context refers to an effective concentration that determines the velocity of the enzyme-catalyzed reaction.
TABLE 1.
List of reactions and kinetic equations
| Number | Reaction | Kinetics |
|---|---|---|
| Ligand degradation | ||
| 1 | lig → Ø | v1 = c1 × lig |
| Ligand binding and receptor phosphorylation | ||
| 2 | 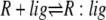 |
v2 = c2 × R × lig − c–2 × R:lig |
| 3 | 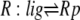 |
v3 = c3 × R:lig − c–3 × Rp |
| 4 |  |
v4 = c4 × aSrc × Rp − c–4 × Rppp |
| Src binding to phosphorylated receptor | ||
| 5a | 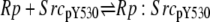 |
v5a = c5 × Rp × SrcpY530 − c–5 × Rp:SrcpY530 |
| 5b | 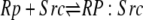 |
v5b = c5 × Rp × Src − c–5 × Rp:Src |
| 5c | 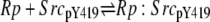 |
v5c = c5 × Rp × SrcpY419 − c–5 × Rp:SrcpY419 |
| 5d | 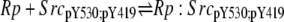 |
v5d = c5 × Rp × SrcpY530;pY419 − c–5 × Rp:SrcpY530;pY419 |
| 5e | 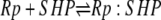 |
v5e = c5 × Rp × SHP − c–5 × Rp:SHP |
| Src binding to hyperphosphorylated receptor | ||
| 6a | 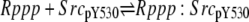 |
v6a = c6 × Rppp × SrcpY530 − c–6 × Rppp:SrcpY530 |
| 6b | 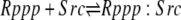 |
v6b = c6 × Rppp × Src − c–6 × Rppp:Src |
| 6c |  |
v6c = c6 × Rppp × SrcpY419 − c–6 × Rppp:SrcpY419 |
| 6d | 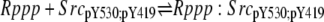 |
v6d = c6 × Rppp × SrcpY530;pY419 − c–6 × Rppp:SrcpY530;pY419 |
| 6e | 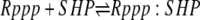 |
v6e = c6 × Rppp × SHP − c–6 × Rppp:SHP |
| Autophosphorylation of receptor-bound Src | ||
| 7a |  |
v7a = c7 × aSrc × Rp:SrcpY530 − c–7 × Rp:SrcpY530;pY419 |
| 7b | 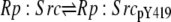 |
v7b = c7 × aSrc × Rp:Src − c–7 × Rp:SrcpY419 |
| 8a |  |
v8a = c8 × aSrc × Rppp:SrcpY530 − c–8 × Rppp:SrcpY530;pY419 |
| 8b |  |
v8b = c8 × aSrc × Rppp:Src − c–8 × Rppp:SrcpY419 |
| N-terminal Src phosphorylation (independent of pY530 and pY419) | ||
| 9 |  |
v9 = c9 × aR × (1 − Srcp(SH3)) − c–9 × Srcp(SH3) |
| Src family kinase regulation | ||
| 10 | 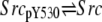 |
v10 = k′2 × aPTPα × SrcpY530 − k1 × aCsk × Src |
| 11 |  |
v11 = k3 × aSrc × Src − p1 × SrcpY419 |
| 12 |  |
v12 = k1 × aCsk × SrcpY419 − k′2 × aPTPα × SrcpY530;pY419 |
| 13 | SrcpY530;pY419 → SrcpY530 | v13 = p1 × SrcpY530;pY419 |
| Csk regulation | ||
| 14 | 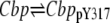 |
v14 = (k+Cbp + kCbp × aSrc) × Cbp − (k–Cbp + kSHP × aSHP) × CbppY317 |
| 15 |  |
v15 = kCbp;on × CbppY317 × Csk − kCbp;off × CbppY317:Csk |
| PTPα regulation | ||
| 16 |  |
v16 = (p3 + aSrc) × PTPα − p2 × PTPαpY798 |
| Receptor complex dissociation | ||
| 17a–c | Rx → R + lig | v17x = kp × Rx |
| 17d–m | R:y → R + lig + y | v17x = kp × Rx:y |
| Auxiliary equations | ||
| Kinase activities of Src, Csk, and receptor kinase | ||
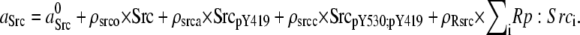
| ||
| aCsk = CbppY317:Csk. | ||
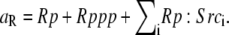
| ||
| Phosphatase activities of PTPα and SHP | ||

| ||
| aSHP = Rp:SHP + Rppp:SHP. | ||
| Conservation of mass | ||
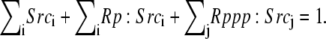
| ||
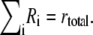
| ||
| Cbp + CbppY317 + CbppY317:Csk = Cbptotal. | ||
| Csk + CbppY317:Csk = Csktotal. | ||
| PTPα + PTPαpY798 = PTPαtotal. | ||
| Phosphorylation-mediated weakening of the Src tail-bite | ||
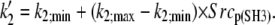
| ||
Parameter values
The parameter values were derived through mainly two principal methods: first, directly or indirectly from published data, and second, from prior knowledge. Useful published data is sometimes available in the form of quantitative equilibrium measurements. For example, the basal level of PTPα phosphorylation in T-cells was determined as ∼20% (18). From this value, the ratio of the parameters p2 and p3 (which control phosphorylation of PTPα Tyr-798) could be derived as p2/(p2 + p3) ≈ 0.20. Similarly, the known basal levels of Csk activity for the two systems considered have been taken into account for the derivation of k–Cbp and k+Cbp. Also, sometimes relative total concentrations are available, as in the case of Csk and Cbp/PAG: Csk is present in excess and only 5% of Csk is recruited to the membrane via phosphorylated Cbp/PAG (11). This is reflected by the values of Cbptotal = 1 and Csktotal = 20.
So-called prior knowledge is typically available in the form of qualitative features of the system in question and plausible assumptions on its behavior. The nature of these assumptions is behavioral, as opposed to mechanistic. For example, it is known that Fyn and Lck activity show a single transient peak after TCR activation. Any parameter set that produces no response, sustained response, or multiple peaks can therefore be disregarded. Parameter values were adjusted so that the obtained behavior is compatible with known behavioral properties of the system.
To cope with residual uncertainty we previously carried out a robustness study on the SFK subsystem (19). The results showed that the major systems-level characteristics of the SFK model, namely the bistable switch and the excitable behavior that produced transient responses to stimuli, were predominantly determined by the topology rather than by particular parameter values.
Since the receptor subsystem model developed here is a rather abstract representation of the molecular events involved in receptor activation, many of the parameter values do not correspond to actual rate constants of elementary reactions. They are therefore not an exact mechanistic representation, but were devised to ensure qualitative consistency with previously published models, such as in the literature (20,21). Details are given in Results below.
SFK subsystem
The dynamics of the protein interaction network involving Src, Cbp/PAG, Csk, and PTPα (or, as an alternative interpretation, CD45) have been modeled in Fuß et al. (17). Some minor changes to the model parameters (see Methods) were introduced to better reflect the situation in the contexts under investigation.
The simulations of the SFK subsystem (Figs. 2 and 3) were performed with a total receptor concentration of Rtotal = 0. Therefore, only Reactions 10–16 effectively contributed to the dynamic behavior of the model.
FIGURE 2.

Activation of the SFK subsystem. Three bifurcation diagrams indicating the relative enzyme activities at steady-state (continuous line, stable; dashed line, unstable). (A) Excitation of SFK activity. Dotted line represents a possible excitation trajectory. (B) Bifurcation curves for different values of k2, representing allosteric effects of Ser/Thr-phosphorylation in the N-terminal SFK region. (C) Kinase activity required for overcoming Csk inhibition and triggering the SFK autophosphorylation reaction. Graphs are based on parameters in Table 2.
FIGURE 3.

Bifurcation diagrams illustrating the effect of a Cbp-pY317 targeting phosphatase. The system has a Hopf bifurcation (H) and an infinite period bifurcation (IP). (Heavy lines) stable steady state; (dashed lines) unstable steady-state; and (dots) maxima and minima of stable periodic solutions (limit cycles). Unstable limit cycles not shown.
Receptor interaction
A simplified model of receptor activation was used to provide a framework for the simulation of SFK-receptor interactions. The reactions considered were similar to the EGFR activation model published by Kholodenko et al. (20), which describes the immediate events after activation of the EGFR. In addition to the reactions modeled there, this study also takes dissociation of the ligand from the activated receptor complex into account. With respect to this, the model resembles the scheme proposed by McKeithan (22).
The numerous tyrosine phosphorylation sites of the various receptors were classified into two groups: receptor autophosphorylation sites and hyperphosphorylation sites, which are targeted by SFKs (23). The reactions for receptor activation modeled in this study are shown in Fig. 1. Reactions 5 and 6 represent the binding of an SH2 domain containing protein to the receptor. Src family kinases bound to the receptor were assumed to obey the same kinetics as the free SFK except that the inhibitory phosphorylation of the receptor-bound SFK was assumed to be ineffective.
As before, the rate constants chosen do not reflect concrete, quantitative kinetic information, but provide plausible approximations that allow generalization of the receptor model to a wide range of signaling contexts. Most reactions in the model do not correspond to actual elementary biochemical reactions, since there is a considerable amount of uncertainty and also variation between different receptors. For this reason, the kinetic equations have the form of pseudo first-and second-order mass action kinetics rather than, for example, Michaelis-Menten kinetics. This approximation is valid for small substrate concentrations.
The application of ligand was simulated by increasing the variable lig, which corresponds to the concentration of free ligand. Removal of ligand was controlled by adjusting parameter c1 (Reaction 1).
Numerical analysis
Simulations were carried out with XPPAut 5.96 (24). Integration was performed with the fourth-order Runge-Kutta method and a step size of ΔT = 0.05 or less. Bifurcation diagrams were created with the integral XPPAut interface to AUTO (25).
RESULTS
Characterization of the SFK subsystem
To understand the SFK-specific events after receptor stimulation, we first illustrated the dynamic properties of the SFK subsystem (17). The subsystem model represents the dynamics of the protein interactions among an Src family kinase, Cbp/PAG, and Csk, and a phosphatase such as PTPα or CD45 that interacts with the SFK in a positive feedback loop. Using numerical analysis methods, this system was shown to allow for bistability, a common dynamic motif that is often encountered in switchlike biochemical systems (e.g., genetic switches (26) or cell-cycle phase transitions (27)). In a bistable system, a given set of parameters permits two distinct stable equilibrium solutions. The state of a bistable system can usually be toggled by crossing a bifurcation point, beyond which one of the stable states disappears, rendering the system monostable. Bistable systems also normally display hysteresis: the system state depends on its immediate history. Switching mainly occurs at two distinct points that enclose the region of bistability.
In the SFK system, switching can be achieved by altering the activity of Csk. The bifurcation diagram in Fig. 2 A illustrates this. Two stable equilibrium states (thick lines) exist. The state of low SFK activity is only stable for Csk activities above a certain threshold (bifurcation point 1). If Csk activity falls below this threshold (for example, due to Cbp dephosphorylation and Csk release), the system is attracted by the high SFK activity state. For default parameters, the threshold value was 0.434, which corresponds to 43.4% of the total Cbp/PAG in the cell being phosphorylated and bound to Csk. The increasing SFK activity in turn causes rephosphorylation of Cbp/PAG and subsequent suppression of SFK activity by Csk. The course of these events is illustrated by the trajectory in Fig. 2 A (dotted line).
Requirements for SFK activation
Cbp/PAG dephosphorylation
As explained above, the level of Cbp/PAG phosphorylation determines the stability of the two steady states and is therefore an important property for SFK regulation. For the system to remain in the low SFK state, the unstimulated cell must maintain a certain level of Cbp/PAG phosphorylation, which is at least above bifurcation point 1 (see Fig. 2 A).
Based on this conceptual model, early events after receptor stimulation must lead to an increased tyrosine phosphatase activity targeting Cbp/PAG (or alternatively, in case of constitutive tyrosine phosphatases, a decrease in Cbp/PAG-directed tyrosine kinase activity). We explored the effects of varying the rate of Cbp/PAG dephosphorylation by incorporating a dephosphorylation reaction into our model. Dephosphorylation was assumed to follow first-order kinetics with the rate constant k–Cbp.
We performed bifurcation analysis on the extended model using k–Cbp as a bifurcation parameter. The parameter was varied between 0 and 0.1, which corresponds to a dephosphorylation rate equivalent to the maximally achievable rate at which Src can phosphorylate Cbp (assuming default parameters). The two bifurcation diagrams in Fig. 3 illustrate the system's behavior in response to different phosphatase activities. The two bifurcation points (IP) divide the graph into three regions, which are discussed in the following.
Low phosphatase activity creates a stable equilibrium state of low SFK activity and medium to high Csk activity (region I). The latter depends on the value of k–Cbp. However, phosphatase activities within this region are too weak to cause activation of significant amounts of SFK. Rather, kinase and phosphatase activities center the system on a position close to the bifurcation point.
If phosphatase activity further exceeds a threshold (kCbp = 0.0092), SFK activity can be excited, after a similar trajectory as shown in Fig. 2 A. As described above, this activation of SFKs is followed by recruitment of Csk to the membrane and downregulation of the SFK. Because, in our simple model, the phosphatase activity is constitutive and persists even after SFK activity is suppressed, the cycle continues and another round of Cbp/PAG dephosphorylation begins. Region II in the bifurcation diagram therefore shows oscillating behavior. The transition from stable low SFK activity to oscillating behavior is marked by a Hopf bifurcation point (H). Finally, under the influence of strong phosphatase activity (>k–Cbp = 0.05), SFK activation becomes constitutive, and the oscillations disappear (region 3). This bifurcation was identified as an infinite period bifurcation (IP). In summary, there is a low phosphatase activity region (I), which corresponds to SFK repression, and two high phosphatase activity regions, which create oscillatory (II) or stably sustained SFK activity (III).
There is currently no experimental evidence for the sustained oscillations that the model predicts. Normally, a single stimulus yields a transient period of SFK activation, which is then followed by stable suppression of SFKs (16). This could mean that the system does not stay in the oscillatory region for a sustained time (possibly even shorter than the oscillation period), or that only the two steady-state regions play biological roles. In either case, to maintain the validity of this phosphatase model and to accommodate for transient activation, we would need to postulate a mechanism by which the Cbp/PAG targeting phosphatase is deactivated during the excitation of the SFK.
Weakening of the “tail-bite”
It is certainly easy to imagine several possible implementations of such a mechanism. One scenario is described below in the context of the T-cell receptor. However, our results suggest an alternative that does not require the postulation of hypothetical enzymatic interactions. There have been reports about phosphorylation sites in the N-terminal region and the SH3 domain of SFKs that weaken the intramolecular association between the C-terminal phosphotyrosine and the SH2 domain: the PDGF receptor phosphorylates tyrosine residues 138 (28) and 213 (29) of Src (reviewed in (1)). The cell-cycle-specific kinase cdk1 phosphorylates different serines and threonines during mitosis, which has the same effect on the tail-bite interaction (30). It has been found that in this state access of phosphatases that dephosphorylate the inhibitory tyrosine is enhanced (30). A recent study has shown that an artificially strengthened Lck tail-bite interaction prevents normal TCR signaling (31).
In the model investigated here, the state of these phosphorylation sites is collectively represented in the parameter k2 (see also (17)), a pseudo second-order rate constant for the dephosphorylation of the C-terminal phosphotyrosine by PTPα or CD45. Fig. 2 B contains variations of the bifurcation diagram of Fig. 2 A. The curves shown correspond to 20%, 60%, and 100% of the normal dephosphorylation rate (default parameters), on which Fig. 2 A is based. The shape of the steady-state branches and the position of the bifurcation points were dependent on k2. An increase of k2 could move the system from the bistable region into the monostable region (left in the graphs), which is characterized by high SFK activity. It is therefore conceivable that activation of SFKs is triggered not (or not only) by an increase of C-terminal phosphatase activity, but by an increased kinase activity that targets N-terminal serine, threonine, and tyrosine residues in SFKs.
Small stimulating kinase activity
Fig. 2 A also indicates another mechanism which could lead to SFK activation. When the system state is within the bistable region, an addition of a certain amount of active SFK could trigger the autophosphorylation reaction and make the system jump to the state of high SFK activity (upper stable branch). The triggering kinase activity must be large enough to allow the system to cross the unstable steady state (dashed line in Fig. 2 A). This threshold increases with the level of Cbp/PAG phosphorylation.
This possibility is interesting because a number of receptors are known to bind SFKs via their SH2 domain to phosphotyrosines in the juxtamembrane region of the receptor. Since affinities of the SFK SH2 domains for receptor phosphotyrosines are higher than for the C-terminal phosphotyrosine, this binding event releases the intramolecular block (32). It is commonly assumed that small numbers of SFK molecules activated in such a way could trigger autophosphorylation of the remaining SFK population.
Using the SFK model we determined the minimal kinase activity needed for triggering autophosphorylation under absence of any other activating influences. To simulate a triggering kinase activity the model parameter 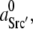 which represents background SFK activity, was increased in small steps. This had an impact on the positions of the bifurcation points described above. Higher values of
which represents background SFK activity, was increased in small steps. This had an impact on the positions of the bifurcation points described above. Higher values of 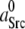 demanded a higher Csk activity for maintaining SFKs in an inactive state. The correlation is shown in Fig. 2 C. At Csk activities below point a in Fig. 2 C, SFK was constitutively active and no additional kinase activity was required to trigger SFK activation. Between points a and b, the threshold increased up to a maximum (point b). For default parameters (Table 2), this maximum required activity corresponded to 1.3% of the SFK population bound to the receptor and activated. Of course, this value is highly sensitive to a number of model parameters, such as the ratio of Src/Cbp. (A 1:1 ratio was assumed here.) However, these findings confirm that a very small amount of activated SFK, as can be caused by receptor binding, may be sufficient to trigger a response. On the other hand, to the right of point b high Csk activity suppressed all net autophosphorylation activity, regardless of the amount of stimulating kinase activity. This point corresponded to 0.95 units of active Csk per unit of SFK.
demanded a higher Csk activity for maintaining SFKs in an inactive state. The correlation is shown in Fig. 2 C. At Csk activities below point a in Fig. 2 C, SFK was constitutively active and no additional kinase activity was required to trigger SFK activation. Between points a and b, the threshold increased up to a maximum (point b). For default parameters (Table 2), this maximum required activity corresponded to 1.3% of the SFK population bound to the receptor and activated. Of course, this value is highly sensitive to a number of model parameters, such as the ratio of Src/Cbp. (A 1:1 ratio was assumed here.) However, these findings confirm that a very small amount of activated SFK, as can be caused by receptor binding, may be sufficient to trigger a response. On the other hand, to the right of point b high Csk activity suppressed all net autophosphorylation activity, regardless of the amount of stimulating kinase activity. This point corresponded to 0.95 units of active Csk per unit of SFK.
TABLE 2.
Parameters of the SFK subsystem
| Parameter | Value |
|---|---|
| k1 | 1 |
| k2 | 0.6 |
| k3 | 1 |
| k4 | 1 |
| p1 | 0.2 |
| p2 | 0.15 |
| p3 | 0.035 |
| kCbp | 0.1 |
| kCSK;off | 0.1 |
| kCSK;on | 0.1 |
| Cbptotal | 1 |
| Csktotal | 20 |
| PTPαtotal | 1 |
| ρsrco | 0 |
| ρsrca | 1 |
| ρsrcc | 1 |
 |
0 |
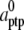 |
0 |
Parameters are in arbitrary, dimensionless units (see Methods).
Note that point b is not equivalent to bifurcation point 2. Even within parts of the monostable region of Fig. 2 A it was possible to trigger transient autophosphorylation, provided the stimulating SFK activity was high enough.
Receptor dynamics
To characterize the three activating mechanisms (Cbp/PAG dephosphorylation, weakening of the tail-bite, and small stimulating kinase activity) in the context of receptor dynamics, a simplified, general kinetic model of receptor activation was constructed. The dynamics of receptor-ligand interaction and subsequent activation have been studied extensively in kinetic models (for an overview, see (21)). Our model was constructed with the specific aim of characterizing receptor-SFK interaction in a general approach, which could explain observations in various receptor signaling contexts.
Fig. 1 A shows the reaction scheme underlying the receptor activation part of our model. The scheme is similar to the one modeled by Kholodenko et al. (20), but dissociation of the dimerized and activated receptor-ligand complexes (Reaction 17) was also taken into account (in a similar way to the model of McKeithan (22)). Since little quantitative data are available on the relative abundance of the relevant proteins of the SFK subsystem, the entire model was kept dimensionless.
Receptor activation was modeled in three steps: ligand binding (Reaction 2), receptor phosphorylation (3), and hyperphosphorylation (4). Reactions 5 and 6 represent binding of SH2 domain containing proteins, such as SFKs, to the receptor. (These reactions appear in several instances in Table 1, one for each species of SH2 containing protein.)
The receptor also participates in SFK regulation (Fig. 1 B). Reaction 9 models the weakening of the tail-bite through phosphorylation within the N-terminal and SH3 regions of SFKs. The amount of phosphorylation influences the parameter k′2 (see auxiliary equations in Table 1), which controls dephosphorylation of the SFK negative regulatory phosphorylation site. Receptor-bound SFK proteins were assumed to allow autophosphorylation (Reactions 7 and 8).
Growth factor receptors
The reaction scheme in Fig. 1 directly corresponds to the immediate events after EGFR stimulation. The binding of growth factors to the receptor (Reaction 2) causes homodimerization of two receptor molecules (not explicitly represented in the model). The EGF receptor proteins are phosphorylated on several tyrosine residues by trans-autophosphorylation (Reaction 3) (33). Src also phosphorylates tyrosines 845 and 1101 (23) in the receptor (Reaction 4). The phosphotyrosines then serve as binding sites for Src (Reactions 5 and 6).
Due to the abstract representation of these reactions, parameter values for the receptor subsystem were kept at unity or decimal powers. The dissociation rates and equilibrium constants are in approximate accordance with the model of Kholodenko et al. (20). For initial simulations, a constitutive phosphatase activity of k–Cbp = 0.02 was assumed. This phosphatase activity created an equilibrium level of Cbp/PAG phosphorylation close to bifurcation point 1 in Fig. 2 A at ∼0.181 (corresponding to 18.1% of the total Cbp/PAG being phosphorylated on Tyr-317). Simulation was started from this equilibrium point. Stimulation of the receptor was simulated by adding an amount of ligand at time t = 0. The slow degradation of free ligand (c1 = 0.05) provides for transient rather than continuous stimulation.
The simulation (Fig. 4 A) produced a short, transient response of SFK activity, which rapidly brought Cbp/PAG phosphorylation to a maximum. The constitutive phosphatase activity then caused Cbp/PAG phosphorylation to slowly return to its original equilibrium state. For certain parameters, the system returned to its equilibrium in damped oscillations. Fig. 4 A shows the nonoscillating case.
FIGURE 4.

Simulations of the general receptor model with the growth factor parameter set. At the times indicated by triangle symbols, one unit of ligand was added. (A) single stimulation; and (B) subsequent stimulation demonstrating the refractory period. (Dashed lines) Cbp/PAG phosphorylation; (dash-dotted lines) receptor phosphorylation; and (continuous lines) SFK activation (phosphorylation of the autocatalytic site).
In conjunction with constitutive phosphatases, the feedback between SFK activity and Cbp/PAG phosphorylation positioned the system on an excitable spot, which allowed a sensitive response to an appropriate stimulus. These results indicate that neither direct nor indirect upregulation of a Cbp/PAG-specific phosphatase activity by the activated receptor is required. The weakening of the inhibitory, intramolecular interaction through receptor-mediated phosphorylation was enough to trigger SFK activation.
To test how the model system responds to subsequent stimulations of the receptor, another quantity of ligand was added at different times after the initial stimulation. We found that the system showed refractory behavior within a certain period after response to primary stimulation. SFK and receptor activity was low during that period, but excess Cbp/PAG phosphorylation prevented a response of SFKs to subsequent stimulation. Fig. 4 B shows a typical example of unsuccessful secondary stimulation within the refractory period. While there were significant amounts of receptor phosphorylation in response to the stimulus, SFK activity remained low. Since SFK activity also contributes to receptor phosphorylation (and therefore activation), the receptor kinase itself exhibits an increased activation threshold during the refractory period. The length of the refractory period was mainly dependent on the activity of the constitutive phosphatase and the rates of association and dissociation between Cbp/PAG and Csk (kCSK;on and kCSK;off).
T-cell receptor
The results presented in the previous section could explain SFK activation in the context of the EGF and PDGF receptors, and how unstimulated cells could maintain a low level of Cbp/PAG phosphorylation. However, as mentioned in the introduction, a different situation seems to exist in other signaling contexts, such as in T-lymphocytes. These cells maintain a high level of Cbp/PAG phosphorylation, which is rapidly reduced upon ligand binding, followed by activation of the Src family member Fyn.
Fig. 1 can be reinterpreted to accurately describe the interaction between SFKs and the T-cell receptor (TCR) complex. The TCR consists of a number of proteins, the α-, β-, γ-, and ζ-chains, which form the receptor complex. The coreceptors CD4 and CD8, which bind Lck, are of importance for SFK regulation in T-lymphocytes. Reaction 2 corresponds to the binding of an antigen, contained in a major histocompatibility complex molecule on the surface on an antigen-presenting cell. This binding event recruits CD4/8 molecules to the receptor complex and brings Lck close to the receptor. Lck then phosphorylates tyrosine phosphorylation motifs (called immunoreceptor tyrosine-based activation motifs or ITAMs) in the TCR ζ-chains (Reaction 3) and thereby facilitates binding of proteins to these ITAMs. The activity of Lck causes recruitment of Fyn to the TCR complex (34,35), presumably by binding to phosphorylated tyrosines (Reaction 5). Whether a similar hyperphosphorylation mechanism as in Reactions 4 and 6 exists for the TCR is not clear. For example, Fyn might target tyrosines in the receptor complex other than those phosphorylated by Lck. The rate constants for these equations (c4 and c6) were set to zero in the default parameter set (simulations shown in Fig. 5). However, we found that the qualitative behavior of the system as documented below was not altered if these rate constants were kept at the same levels as in the growth factor parameter set.
FIGURE 5.

Simulations of the general receptor model with the T-cell receptor parameter set. The model receptor was subjected to three different stimuli, indicated by the bars at the top: (A) exposure to a low-affinity ligand; (B) short exposure to a high-affinity ligand; and (C) long exposure to the same high-affinity ligand. (Dashed lines) Cbp/PAG phosphorylation; (dash-dotted lines) receptor phosphorylation; and (continuous lines) SFK activation (phosphorylation of the autocatalytic site).
It is currently unknown how Cbp/PAG is dephosphorylated upon TCR stimulation. Whatever mechanisms cause Cbp/PAG dephosphorylation, net phosphatase activity must increase significantly within a minute of receptor stimulation. Since a number of signaling molecules have been reported to bind to the phosphorylated ITAMs of the T-cell receptor, we assumed as a working hypothesis, that one of the early binding proteins is an SH2 domain containing phosphatase that targets Cbp/PAG (see also Discussion). We also assumed that binding of that phosphatase to the ITAMs significantly increased its activity, as is the case with many SH2-domain-containing proteins besides the SFKs, such as the phosphatase SHP-2 (36). While an indirect activation of phosphatases is conceivable, these assumptions represented the minimal hypotheses necessary to implement the implied phosphatase activity. An interaction between Lck and SHP-1 that is involved in TCR ligand discrimination has been previously described (37) and could be responsible for triggering Cbp/PAG dephosphorylation.
The TCR model was simulated with the parameters in Table 3 (column headed TCR). Only minimal changes were made to the parameter set used in the growth factor simulations. A constitutive kinase activity k+Cbp created the high default level of Cbp/PAG phosphorylation. The simulations in Fig. 5 show the model's responses to different stimuli (indicated by bars above the graphs). In Fig. 5 A, a ligand of weak affinity, represented by a dissociation constant of kp = 0.6, was applied at time t = 0 and not removed by degradation (c1 = 0). Due to the high dissociation rate the ligand was able to activate only small amounts of phosphatase, resulting in a small reduction of Cbp/PAG phosphorylation without subsequent SFK activation. In Fig. 5 B, a ligand of high affinity (kp = 0.1) was used, but was removed after a short time of t = 25. Although this stimulus was significantly stronger than the ones used in the above simulations of the growth factor receptor, it was not sufficient to produce SFK activation in the T-cell receptor model. A significant amount of receptor molecules (80.0% at maximum) became phosphorylated, followed by a decrease in Cbp/PAG phosphorylation from 91.5% to 71.9%. However, after ligand depletion these effects were reversed and no SFK activation occurred. Finally, in Fig. 5 C the receptor was stimulated with the same high affinity ligand for 100 units of time. The result was sufficient Cbp/PAG dephosphorylation to trigger SFK activation. The period of active SFK lasted ∼2.4 times as long as the initial stimulus. A refractory period was not observed with this parameter set. Instead, subsequent stimulations prolonged the period of SFK activity (data not shown).
TABLE 3.
Parameters and initial conditions of the receptor subsystem for simulation of growth-factor receptor (GFR) and T-cell receptor (TCR) behavior
| GFR | TCR | |
|---|---|---|
| Parameter | ||
| rtotal | 0.2 | 0.2 |
| c1 | 0.05 | 0/0.05 |
| c2 | 1 | 1 |
| c–2 | 0.1 | 0.1 |
| c3 | 1 | 1 |
| c–3 | 0.01 | 0.01 |
| c4 | 1 | 0 |
| c–4 | 0.01 | 0 |
| c5 | 1 | 1 |
| c–5 | 1 | 1 |
| c6 | 1 | 0 |
| c–6 | 1 | 0 |
| c7 | = k3 | = k3 |
| c–7 | = p1 | = p1 |
| c8 | = k3 | = k3 |
| c–8 | = p1 | = p1 |
| c9 | 1 | 1 |
| c–9 | 0.01 | 0.01 |
| kp | 0.1 | 0.1/0.6 |
| k–Cbp | 0.02 | 0.01 |
| k+Cbp | 0 | 0.005 |
| kSHP | NA | 2 |
| k2;min | 0.3 | 0.3 |
| k2;max | 0.6 | 0.6 |
| ρRsrc | 1 | 1 |
| Initial conditions | ||
| Variable* | ||
| SHP | 0 | 1 |
| Cbp/PAGpY317 | 0.009 | 0.045 |
| Cbp/PAG:Csk | 0.172 | 0.869 |
| PTPαpY798 | 0.198 | 0.198 |
Parameters are in arbitrary, dimensionless units (see Methods).
Zero for all variables other than those noted in Table 3.
If hyperphosphorylation of receptor proteins by Src-family kinases was enabled (parameters c4 and c6 set to GFR values), a prolonged period of SFK activation, with otherwise unaltered behavior, was observed (data not shown).
DISCUSSION
SFK activation in context
SFK activation is a complex issue. Various ways of releasing the different autoinhibitory mechanisms have been suggested (38). In this study, mathematical modeling was used to characterize three such mechanisms:
Mechanism 1. Release of inhibition by Csk through Cbp/PAG dephosphorylation.
Mechanism 2. Weakening of the inhibitory Src tail-bite.
Mechanism 3. Autocatalytic amplification of a triggering kinase activity.
The contribution of this model is a mechanistic representation that integrates these three mechanisms. The three mechanisms quantitatively depend on each other, and none of them is, in general, on its own sufficient to activate the SFK. However, depending on the physiological values of k2 for the different states of the phosphorylation sites that influence Mechanism 2, either Mechanism 1 or 2 on their own may provide a large enough stimulus to trigger SFK autophosphorylation and activation. Mechanism 3 was found to be sufficient below a threshold activity of Csk (point b in Fig. 2 C).
In different signaling contexts these mechanisms are implemented by different means. Table 4 lists the known implementations in the systems investigated here. In many cases these mechanisms are not known. This especially applies to the release of Csk activity. So far no receptor-activated phosphatase has been identified that specifically dephosphorylates Cbp/PAG. SHP-2 is a likely candidate for such a phosphatase. This phosphatase is known to interact with the T-cell receptor and is activated by association of its SH2 domains with phosphotyrosines (36,39). While the experiments performed by Davidson et al. (40) indicate that its relative, SHP-1, is not involved in Cbp/PAG dephosphorylation, it is still unclear whether SHP-2 is.
TABLE 4.
Tabular overview of the main mechanisms involved in SFK interaction in various signaling contexts
| Signaling context | Csk deactivation | Initial kinase activity | Tail-bite weakening |
|---|---|---|---|
| Cell cycle | ? | ? | cdc2 phosphorylating Ser-72, Thr-46, and Thr-72 |
| T-cell activation | ? | Complex CD4/8:Lck | ? |
| PDGFR stimulation | ? | Complex PDGFR:Src | PDGFR phosphorylating Tyr-138 |
| EGFR stimulation | Low default Cbp/PAG phosphorylation | Complex EGFR:Src | ? |
Recent evidence points to a possible role for the Src family member FynT. Davidson et al. (40) have identified an interaction between Cbp/PAG and FynT in resting T-cells. Upon stimulation of the TCR, they observed dissociation of FynT, which preceded Cbp/PAG dephosphorylation (40). FynT could therefore function as the kinase that creates the high level of Cbp/PAG phosphorylation in unstimulated cells. It is conceivable that the observed dissociation of FynT is caused by FynT binding to another phosphorylation site with higher affinity. The dissociation of FynT from the complex with Cbp/PAG might make the latter accessible to (possibly constitutive) phosphatases.
However, the simulations of the growth factor receptor system presented here (Fig. 4) have shown that a specific, receptor-activated phosphatase is not required for SFK activation. A constitutively active, and possibly nonspecific phosphatase was sufficient due to the combination of several activating mechanisms employed.
Requirements of individual system contexts
The Cbp/PAG dephosphorylation paradox and the results presented here raise an important question: why do different receptors require qualitatively different interactions with the SFK system? To answer this question one has to consider the kinds of ligands encountered by the receptor and the nature of the biological responses. The ligands of the T-cell receptor are antigens bound to major histocompatibility complex molecules on the surface of an antigen-presenting cell. The triggering of an appropriate response is complicated by the presence of self-antigens (from the organism's own proteins) with very similar affinities and the short lengths of antigen peptides. Specific foreign antigen recognition is, however, required for the correct function of the immune system.
McKeithan proposed a solution to this problem, called kinetic proofreading (22). Kinetic proofreading is a mechanism employed by receptors (and other biochemical systems, such as DNA replication) for distinguishing between rather similar levels of affinities. In T-lymphocytes, this concept is important for the correct training of the immune system. Since the T-cell receptor undergoes many different modifications before it triggers the final biological response, a long-lasting and strong interaction between the antigen and the receptor is required. A self-antigen will dissociate before the receptor is fully activated and therefore only trigger the first few steps of receptor activation. These initial steps would be rapidly reversed after ligand dissociation (22). The simulations of T-cell receptor dynamics presented here indicate that the activation of SFKs required both a certain strength of interaction (represented by a low dissociation constant Kd), as well as a minimum duration of ligand binding. The results indicate that SFK regulation could possibly play a role in kinetic proofreading in the context of the T-cell receptor.
Growth factor receptors, on the other hand, are activated by free protein ligands rather than binding of cell surface antigens. While T-cells respond to only one or few antigen-presentation events per cell, growth factors such as EGF and PDGF transmit signals to a whole population of receptors on the membrane surface. These receptors guarantee high ligand specificity by integrating the signals of a number of receptors and can therefore provide both a highly sensitive and selective response. In the case of the TCR, selectivity requires additional mechanisms, such as the above-mentioned kinetic proofreading, or the dephosphorylation mechanism tested in this study.
To our knowledge, a specific phosphatase that dephosphorylates Cbp/PAG in response to a receptor signal has not been found. The model does, however, assume that such a phosphatase is recruited by the T-cell receptor. It should be emphasized that this assumption was based on the observation that Cbp/PAG is rapidly dephosphorylated upon TCR stimulation (9) and represents just one plausible mechanistic hypothesis. It is quite likely that several constitutive and TCR activated kinases and phosphatases, Fyn and Lck included, control the phosphorylation status of Cbp/PAG. Both spatial regulation mechanisms, such as transportation of enzymes inside lipid rafts or recruitment to the receptor complex, as well as allosteric regulation of activity may play a role. Many phosphotyrosine binding sites may compete with each other, making the network of protein interactions rather complex. More generally, the exact role that Fyn and Lck play in kinetic proofreading is currently not clear.
A recent kinetic model by Wylie et al. (41) investigated an altogether different mechanism for ligand discrimination that does not rely on Fyn interaction or Cbp/PAG dephosphorylation, but on a mechanism that enhances Lck activity for endogenous ligands. It is difficult to compare their findings with our study, as different subsets of molecular components were taken into consideration. It is, however, conceivable that both of the proposed mechanisms for ligand discrimination independently play a role in TCR signaling. The integration of a more complete account of SFK signaling, such as presented in this study, with detailed kinetics of the TCR subsystem, as presented by Wylie et al. and others, would likely provide useful insights into the mechanisms of kinetic proofreading in T-cells.
Another interesting observation in this context is that the signaling through antigen receptors other than the TCR have also been correlated with kinetic proofreading, but do not rely on high basal Cbp/PAG phosphorylation. For example, activation of the IgE high affinity receptor FcεRI causes an increase in Cbp/PAG phosphorylation (42). As such, the behavior of these cells rather resembles our simulations of the growth factor receptor. This indicates that the mechanism of kinetic proofreading associated with the FcεRI is different than the one proposed in our study. Informal models explaining the kinetic proofreading mechanisms in this context have been proposed (43). Importantly, however, these observations suggest that signaling molecules such as Cbp/PAG may be employed in substantially different ways in different contexts, even within the same cell type. To clarify this issue, the responses of Cbp/PAG and other kinases in the system need to be experimentally correlated to the various receptors. Investigating the differences between them will likely provide useful insights into T-cell physiology.
Testable model predictions
The model makes a number of theoretical predictions that are verifiable experimentally. We already mentioned that the model predicts what the required and sufficient conditions for SFK activation are with regard to the three mechanisms investigated in this study. The respective activation thresholds, i.e., the bifurcation points, will need to be determined experimentally under experimental conditions that are linked to particular sets of parameters. For example, determining the effect of a weakened SFK tail-bite on these activation thresholds could demonstrate whether the relationship between those three mechanisms are consistent with the model's predictions.
Our results suggest that these mechanisms exist in many or all of the different signaling networks, in which SFKs are involved. Table 4 identifies some of the known implementations and lists those that have not been described so far. This table should therefore provide an agenda for further research.
A novel prediction of the receptor model is that Src response to EGF stimulation may exhibit refractory behavior. Furthermore, the model predicts that at least two classes of receptor-SFK coupling mechanisms exist: one that relies on a relatively low basal Csk activity, and one that requires high basal Csk activity. According to the model, refractory behavior is only possible in the former class. Also, the two classes should react differently to perturbations. For example, basal-low Csk systems require the action of phosphorylation sites that allow the weakening of the autoinhibitory tail-bite, and should react sensitively to the phosphorylation of these sites. Basal-high Csk systems, in contrast, require substantial Cbp/PAG dephosphorylation in addition to (or instead of) the former.
The essential difference between the two classes is the balance of kinases and phosphatases that act on Tyr-317 of Cbp/PAG. This balance is captured in the two parameters k–Cbp and k+Cbp. The measurement of these for different systems could provide an experimental discrimination between the two classes.
CONCLUSIONS
We have proposed a general kinetic model to simulate the interaction between Src family kinases and various cellular receptors. The model is simple at present, and due to an emphasis on generality does not provide quantitative predictions of protein concentrations. The receptor systems investigated are very diverse; therefore the parameter sets used are very rough estimates and may not quantitatively correspond to the actual receptor dynamics.
However, the focus of this study has been on the interaction of receptor systems with SFKs and the computational analysis provided useful insights into their regulation. The three modes of activation (Csk release, weakening of the tail-bite, and amplification of a stimulation kinase activity) were characterized using bifurcation theory techniques (Fig. 2). The results give theoretical evidence that binding and phosphorylation of SFKs by receptor tyrosine kinases can contribute to SFK activation in the system context. The analysis explained the Cbp/PAG dephosphorylation paradox described in the Introduction, predicted refractory behavior in the EGFR system and a signal-response characteristic of the TCR system that is compatible with the theory of kinetic proofreading.
On the one hand, the promiscuity of the Src family of kinases has made it difficult to attribute a specific role to them since their discovery. On the other hand, their involvement in various signaling contexts allows the drawing of parallels between them. The kinetic modeling approach permits a systematic way of hypothesis testing. The model thus provides a useful framework, on which hypotheses about SFK-receptor signaling can be tested.
Acknowledgments
We thank Bard Ermentrout for helpful advice on the use of XPPAut.
Editor: Byron Goldstein.
References
- 1.Abram, C. L., and S. A. Courtneidge. 2000. Src family tyrosine kinases and growth factor signaling. Exp. Cell Res. 254:1–13. [DOI] [PubMed] [Google Scholar]
- 2.Palacios, E. H., and A. Weiss. 2004. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 23:7990–8000. [DOI] [PubMed] [Google Scholar]
- 3.Nada, S., T. Yagi, H. Takeda, T. Tokunaga, H. Nakagawa, Y. Ikawa, M. Okada, and S. Aizawa. 1993. Constitutive activation of Src family kinases in mouse embryos that lack CSK. Cell. 73:1125–1135. [DOI] [PubMed] [Google Scholar]
- 4.Ogawa, A., Y. Takayama, H. Sakai, K. Chong, S. Takeuchi, A. Nakagawa, S. Nada, M. Okada, and T. Tsukihara. 2002. Structure of the carboxyl-terminal Src kinase, Csk. J. Biol. Chem. 277:14351–14354. [DOI] [PubMed] [Google Scholar]
- 5.Kmiecik, T. E, and D. Shalloway. 1987. Activation and suppression of pp60/c-Src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 49:65–73. [DOI] [PubMed] [Google Scholar]
- 6.Xu, W., A. Doshi, M. Lei, M. Eck, and S. Harrison. 1999. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell. 3:629–638. [DOI] [PubMed] [Google Scholar]
- 7.Kawabuchi, M., Y. Satomi, T. Takao, Y. Shimonishi, S. Nada, K. Nagai, A. Tarakhovsky, and M. Okada. 2000. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 404:999–1003. [DOI] [PubMed] [Google Scholar]
- 8.Brdička, T., D. Pavlištova, A. Leo, E. Bruyns, V. Kořínek, P. Angelisová, J. Scherer, A. Shevchenko, A. Shevchenko, I. Hilgert, J. Černy, K. Drbal, Y. Kuramitsu, B. Kornacker, V. Hořejši, and B. Schraven. 2000. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase CSK and is involved in regulation of T-cell activation. J. Exp. Med. 191:1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson, D., M. Bakinowski, M. L. Thomas, V. Hořejši, and A. Veillette. 2003. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol. Cell. Biol. 23:2017–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vang, T., H. Abrahamsen, S. Myklebust, V. Hořejši, and K. Taskén. 2003. Combined spatial and enzymatic regulation of Csk by cAMP and protein kinase A inhibits T-cell receptor signaling. J. Biol. Chem. 278:17597–17600. [DOI] [PubMed] [Google Scholar]
- 11.Vang, T., K. Torgersen, V. Sundvold, M. Saxena, F. Levy, B. Skålhegg, V. Hansson, T. Mustelin, and K. Taskén. 2001. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T-cell receptor. J. Exp. Med. 193:497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuoka, H., S. Nada, and M. Okada. 2004. Mechanism of Csk-mediated down-regulation of Src family tyrosine kinases in epidermal growth factor signaling. J. Biol. Chem. 279:5975–5983. [DOI] [PubMed] [Google Scholar]
- 13.Jiang, L. Q., X. Feng, W. Zhou, P. G. Knyazev, A. Ullrich, and Z. Chen. 2006. Csk-binding protein (Cbp) negatively regulates epidermal growth factor-induced cell transformation by controlling Src activation. Oncogene. 25:5495–5506. [DOI] [PubMed] [Google Scholar]
- 14.Gelderloos, J. A., S. Rosenkranz, C. Bazenet, and A. Kazlauskas. 1998. A role for Src in signal relay by the platelet-derived growth factor-α receptor. J. Biol. Chem. 273:5908–5915. [DOI] [PubMed] [Google Scholar]
- 15.Dey, N., B. W. Howell, P. K. De, and D. L. Durden. 2005. CSK negatively regulates nerve growth factor induced neural differentiation and augments AKT kinase activity. Exp. Cell Res. 307:1–14. [DOI] [PubMed] [Google Scholar]
- 16.Torgersen, K. M., T. Vang, H. Abrahamsen, S. Yaqub, V. Hořejši, B. Schraven, B. Rolstad, T. Mustelin, and K. Taskén. 2001. Release from tonic inhibition of T-cell activation through transient displacement of C-terminal Src kinase (Csk) from lipid rafts. J. Biol. Chem. 276:29313–29318. [DOI] [PubMed] [Google Scholar]
- 17.Fuß, H., W. Dubitzky, C. S. Downes, and M. J. Kurth. 2006. Bistable switching and excitable behavior in the activation of Src at mitosis. Bioinformatics. 22:e158–e165. [DOI] [PubMed] [Google Scholar]
- 18.den Hertog, J., S. Tracy, and T. Hunter. 1994. Phosphorylation of receptor protein-tyrosine phosphatase alpha on Tyr789, a binding site for the SH3–SH2-SH3 adaptor protein GRB-2 in vivo. EMBO J. 13:3020–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuß, H., W. Dubitzky, C. S. Downes, and M. J. Kurth. 2007. Deactivation of Src family kinases: hypothesis testing using a Monte Carlo sensitivity analysis of systems-level properties. J. Comput. Biol. 14:1185–1200. [DOI] [PubMed] [Google Scholar]
- 20.Kholodenko, B. N., O. V. Demin, G. Moehren, and J. B. Hoek. 1999. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 274:30169–30181. [DOI] [PubMed] [Google Scholar]
- 21.Lauffenburger, D. A., and J. J. Linderman. 1993. Receptors: Models for Binding, Trafficking, and Signaling. Oxford University Press, New York.
- 22.McKeithan, T. W. 1995. Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. USA. 92:5042–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biscardi, J. S., M. C. Maa, D. A. Tice, M. E. Cox, T. H. Leu, and S. J. Parsons. 1999. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 274:8335–8343. [DOI] [PubMed] [Google Scholar]
- 24.Ermentrout, G. B. 2006. XPPAUT 5.96. http://www.math.pitt.edu/∼bard/xpp/xpp.html.
- 25.Doedel, E. J. 1981. AUTO: a program for the automatic bifurcation analysis of autonomous systems. In Proceedings of the 10th Manitoba Conference on Numerical Mathematics and Computing. University of Manitoba, Winnipeg, Canada. 30:265–284. http://indy.cs.concordia.ca/auto.
- 26.Griffith, J. 1971. Mathematical Neurobiology: An Introduction to the Mathematics of the Nervous System. Academic Press, New York.
- 27.Tyson, J. J., A. Csikász-Nagy, and B. Novák. 2002. The dynamics of cell cycle regulation. Bioessays. 24:1095–1109. [DOI] [PubMed] [Google Scholar]
- 28.Broome, M. A., and T. Hunter. 1997. The PDGF receptor phosphorylates Tyr-138 in the c-Src SH 3 domain in vivo reducing peptide ligand binding. Oncogene. 14:17–34. [DOI] [PubMed] [Google Scholar]
- 29.Stover, D. R., P. Furet, and N. B. Lydon. 1996. Modulation of the SH2 binding specificity and kinase activity of Src by tyrosine phosphorylation within Its SH2 domain. J. Biol. Chem. 271:12481–12487. [DOI] [PubMed] [Google Scholar]
- 30.Stover, D. R., J. Liebetanz, and N. B. Lydon. 1994. Cdc2-mediated modulation of pp60/c-Src activity. J. Biol. Chem. 269:26885–26889. [PubMed] [Google Scholar]
- 31.Nika, K., L. Tautz, Y. Arimura, T. Vang, S. Williams, and T. Mustelin. 2007. A weak Lck tail-bite is necessary for Lck function in T-cell antigen receptor signaling. J. Biol. Chem. 282:36000–36009. [DOI] [PubMed] [Google Scholar]
- 32.Boggon, T. J., and M. J. Eck. 2004. Structure and regulation of Src family kinases. Oncogene. 23:7918–7927. [DOI] [PubMed] [Google Scholar]
- 33.Downward, J., P. Parker, and M. D. Waterfield. 1984. Autophosphorylation sites on the epidermal growth factor receptor. Nature. 311:483–485. [DOI] [PubMed] [Google Scholar]
- 34.Filipp, D., J. Zhang, B. Leung, A. Shaw, S. Levin, A. Veillette, and M. Julius. 2003. Regulation of Fyn through translocation of activated Lck into lipid rafts. J. Exp. Med. 197:1221–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samelson, L. E., A. F. Phillips, E. T. Luong, and R. D. Klausner. 1990. Association of the Fyn protein-tyrosine kinase with the T-cell antigen receptor. Proc. Natl. Acad. Sci. USA. 87:4358–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neel, B. G., H. Gu, and L. Pao. 2003. The “Shp”ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28:284–293. [DOI] [PubMed] [Google Scholar]
- 37.Stefanova, I., B. Hemmer, M. Vergelli, R. Martin, W. E. Biddison, and R. N. Germain. 2003. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 4:248–254. [DOI] [PubMed] [Google Scholar]
- 38.Hubbard, S. R. 1999. Src autoinhibition: let us count the ways. Nat. Struct. Biol. 6:711–714. [DOI] [PubMed] [Google Scholar]
- 39.Frearson, J. A., and D. R. Alexander. 1998. The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T-cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T-cells. J. Exp. Med. 187:1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davidson, D., B. Schraven, and A. Veillette. 2007. PAG-associated FynT regulates calcium signaling and promotes anergy in T-lymphocytes. Mol. Cell Biol. 27:1960–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wylie, D. C., J. Das, and A. K. Chakraborty. 2007. Sensitivity of T-cells to antigen and antagonism emerges from differential regulation of the same molecular signaling module. Proc. Natl. Acad. Sci. USA. 104:5533–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohtake, H., N. Ichikawa, M. Okada, and T. Yamashita. 2002. Transmembrane phosphoprotein Csk-binding protein/phosphoprotein associated with glycosphingolipid-enriched microdomains as a negative feedback regulator of mast-cell signaling through the FcεRI. J. Immunol. 168:2087–2090. [DOI] [PubMed] [Google Scholar]
- 43.Torigoe, C., J. Inman, and H. Metzger. 1998. An unusual mechanism for ligand antagonism. Science. 281:568–572. [DOI] [PubMed] [Google Scholar]