

Abstract
Recent observations in cell culture provide evidence that negatively charged glycosaminoglycans (GAGs) at the surface of biological cells bind cationic cell-penetrating compounds (CPCs) and cluster during CPC binding, thereby contributing to their endocytotic uptake. The GAG binding and clustering occur in the low-micromolar concentration range and suggest a tight interaction between GAGs and CPCs, although the relation between binding affinity and specificity of this interaction remains to be investigated. We therefore measured the GAG binding and clustering of various mono- and multivalent CPCs such as DNA transfection vectors (polyethylenimine; 1,2-dioleoyl-3-trimethylammonium-propane), amino acid homopolymers (oligoarginine; oligolysine), and cell-penetrating peptides (Penetratin; HIV-1 Tat) by means of isothermal titration calorimetry and dynamic light scattering. We find that these structurally diverse CPCs share the property of GAG binding and clustering. The binding is very tight (microscopic dissociation constants between 0.34 and 1.34 μM) and thus biologically relevant. The hydrodynamic radius of the resulting aggregates ranges from 78 nm to 586 nm, suggesting that they consist of numerous GAG chains cross-linked by CPCs. Likewise, the membrane-permeant monovalent cation acridine orange leads to GAG binding and clustering, in contrast to its membrane-impermeant structural analogs propidium iodide and ethidium bromide. Because the binding and clustering of GAGs were found to be a common denominator of all CPCs tested, these properties might be helpful to identify further CPCs.
INTRODUCTION
The membrane of biological cells basically controls the influx and efflux of molecules. The manipulation of this barrier in biology and medicine with the intention either to promote the permeability, as for drug and gene therapy, or to inhibit the cellular uptake, as during viral infections, is therefore highly relevant. In this respect, cell-penetrating peptides (CPPs) have proven to be very efficient molecules because they cross intact cell membranes within seconds to minutes by a mechanism that is poorly understood (reviewed by Richard et al. (1)).
These molecules are of general interest because they can also facilitate the import of covalently and noncovalently bound drugs, genes, and macromolecules that would not otherwise cross the membrane. Their uptake mechanism proper, however, is controversial because of technical artifacts, limitations of model membranes employed, and noncomparable experimental approaches (1). CPPs have quite different chemical structures, but they share a high cationic charge density (Fig. 1), which rules out their passive permeation through the lipid membrane.
FIGURE 1.

Structures of the compounds investigated. CPCs are classified mainly on the basis of their historical discovery and/or intended use or application. The classes comprise (1, 2) amino acid homopolymers, (3) DNA transfection agents, (4) cationic lipids, and (7, 8) CPPs. Individual structures are (1) nona-l-arginine, (2) hexadeca-l-lysine (PLL16), (3) linear polyethylenimine (LPEI), (4) 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), (5) cell-permeant acridine orange (AOR), (6) cell-impermeant propidium iodide, (7) HIV-1 Tat(47–57), and (8) penetratin. Not shown are monomeric l-arginine and cell-impermeant ethidium bromide (similar to 6). Charges at physiological pH were assigned on the basis of literature values of the pKa. These were 12.5, 10.3, ∼7, and 10.2 for 1, 2, 3, and 5, respectively (19,54).
As an important primary step of CPP uptake, it has recently been observed in living cells that the CPP HIV-1 Tat-PTD leads to the clustering of cell-surface-bound molecules coinciding with the rapid uptake of this CPP into the cell plasma (2). Similarly, a clustering of cell-surface-bound molecules was observed during DNA transfection with the nonpeptidic cell-penetrating compound (CPC) polyethylenimine (3) and cationic lipofectants (4,5). Related in vivo studies suggest that cross-linking of glycosaminoglycan (GAG)-containing proteins is likely an important mediator of the cellular CPP uptake (6–12). This assumption is further substantiated by the observation that enzymatic or genetic removal of GAGs from the surface of living cells reduces or abolishes the uptake of CPPs in vivo (2,12–14). In analogy to such in vivo observations, GAG binding and clustering have recently been substantiated in vitro for the interaction of Tat with heparan sulfate (15).
Our study aimed at investigating the question of whether further CPCs share the property of GAG binding and, in particular, GAG clustering. Furthermore, knowledge of related thermodynamic binding parameters is decisive to understand the delicate balance among the optimum molar ratio of cargo and CPC, extracellular stability of this uptake complex, and intracellular release of the cargo (16).
We investigated various mono- and polycationic CPCs with respect to their properties of GAG binding and clustering in vitro. Clustering is of particular importance in recent discussions of adsorptive endocytosis (6). The CPCs are all known to pass the membrane of intact cells but differ considerably in their primary structure, valency, amine type, and pKa (Fig. 1). The monovalent cation acridine orange was also included in our investigation because of its structural analogy to propidium iodide and ethidium bromide, molecules that are both excluded from living cells (17). In contrast, acridine orange is taken up by living cells (18). In analogy to CPCs, its elevated pKa (19), the energy dependence of its in vivo uptake (20), the need of active loading procedures for inclusion into membrane vesicles (21), and the limited membrane diffusion (22) all make passive diffusion across the membrane unlikely as an uptake mechanism. Heparin was selected as GAG because heparin-like N-sulfo domains provide ligand recognition in cell-surface-associated heparan sulfate (23).
METHODS
Material
Porcine intestinal mucosa heparin (sodium salt; sulfate content of 11.3%; average mol wt 13,000) was from Celsus Laboratories (Cincinnati, OH). l-α-dioleoyl-phosphatidylcholine (DOPC) and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) were obtained from Avanti Polar Lipids (Alabaster, AL). These lipids were used to produce small unilamellar vesicles by sonication. Linear polyethylenimine (LPEI) (mol wt 2500) was purchased from Polysciencences (Eppelheim, Germany), and all other chemicals of HPLC grade were from Sigma-Aldrich (Buchs, Switzerland).
Peptide synthesis
Solid-phase peptide synthesis of HIV Tat-PTD, penetratin, nona-l-arginine, and hexadeca-l-lysine was performed on an Abimed EPS221 peptide synthesizer (Langenfeld, Germany) using Fmoc-protected amino acids and preloaded NovaSyn TGA resins. After synthesis, the peptides were purified by preparative high-pressure liquid chromatography. The mass of the peptides was confirmed by electrospray ionization mass spectrometry, and peptide purity (>98%) was measured by analytical high-pressure liquid chromatography. The effective peptide concentration was measured as amino acid content after acid hydrolysis.
Isothermal titration calorimetry
The heat flow resulting from the binding of CPCs to heparin was measured with high-sensitivity isothermal titration calorimetry using a MicroCal Omega and VP-ITC calorimeter (Northampton, MA) with a reaction cell volume of 1.4 ml. The ITC data were evaluated according to the multisite binding model (24):
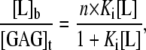 |
(1) |
where [L]b and [L] are the concentrations of bound and free ligand, respectively, [GAG]t is the total concentration of the GAG, Ki is the microscopic binding constant to each individual binding site in the GAG, and n are the number of ligands bound per GAG molecule. Ki, n, and the ligand binding enthalpy,  may be directly determined by a three-parameter least-squares fit to the calorimetric data because the heat released in injection i, δQi, is proportional to the concentration of bound ligand per injection by
may be directly determined by a three-parameter least-squares fit to the calorimetric data because the heat released in injection i, δQi, is proportional to the concentration of bound ligand per injection by
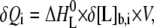 |
(2) |
where δ[L]b,i is the change in bound ligand concentration upon injection i, and V is the actual reaction volume. For a macromolecule with n independent binding sites, the binding constant of the individual binding site varies with the degree of saturation for statistical reasons (24), with the first ligand binding with the macroscopic binding constant K1 = n·Ki, and the last with Kn = Ki/n. For better comparison with literature, dissociation constants Kd are reported that are just the inverse of the binding constants.
Static right-angle light scattering
Static light scattering at a right angle was measured with a Jasco FP 777 fluorimeter (Tokyo, Japan) at a wavelength of 350 nm under constant stirring and at a temperature of 28°C. Quartz cuvettes with inner lengths of 1 cm were filled with 1.4 ml of heparin solution, and 10-μl aliquots of the ligand solution were added at 5-min intervals.
Dynamic light scattering
Dynamic light scattering (DLS) experiments were performed on an ALV/CGS-5022F instrument (ALV, Langen, Germany) equipped with a HeNe laser (λ = 632.8 nm) and an ALV-5000/EPP multi-tau digital correlator. Reactant solutions were filtered (0.22 μm; Millipore, Billerica, MA) before mixing. Measurements were performed in 1 cm (outer diameter) cylindrical cuvettes at 20°C using eight different scattering angles θ (30–150°). The normalized intensity autocorrelation functions were analyzed using a second-order cumulant analysis (25) yielding the collective diffusion coefficient D. The latter was used to calculate the hydrodynamic radius (Rh) using the Stokes-Einstein relationship Rh = kT/6πηD, where k is the Boltzmann's constant, T the absolute temperature, and η the viscosity of water. The reported Rh was obtained by extrapolating Rh of eight different angles to zero scattering angle. The width of the Rh distribution was calculated as full width at half-maximum by means of the diffusion coefficient distribution (25).
To estimate the maximum number of heparin chains within a CPC-GAG cluster, the hydrodynamic volume of the cluster was assumed to be of spherical shape (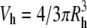 ) and was divided by the volume of a single heparin chain saturated with the defined number of ligands as measured by ITC. The volume of the individual molecules was calculated from known protein database entries as the Connolly solvent-excluded volume (26) using the software Chem3D Pro (Cambridge, MA).
) and was divided by the volume of a single heparin chain saturated with the defined number of ligands as measured by ITC. The volume of the individual molecules was calculated from known protein database entries as the Connolly solvent-excluded volume (26) using the software Chem3D Pro (Cambridge, MA).
RESULTS AND DISCUSSION
Binding affinity
Despite the considerable difference in their overall structures (Fig. 1), all CPCs were found to bind the GAG with a high affinity (Fig. 2) characterized by a distinct number of binding sites and a dissociation constant of the individual binding site in the submicromolar range (Table 1). The observed affinity for GAGs is thus biologically relevant because the CPP concentration generally required for efficient cell penetration (1–10 μM) and physiological GAG concentration are of similar magnitude. For fibroblasts, for example, 2 μg of sulfated GAGs/mg dry tissue are reported (27), corresponding to a macroscopic GAG concentration of ∼7 μM using a typical molecular weight of GAGs found in fibroblasts (28) and an average tissue water content. The effective concentration of individual binding sites is even higher because each GAG can bind several CPPs (Table 1). Compared with CPP binding to GAGs of low sulfate content or short chain length (15,29), the current data indicate an up to fourfold higher binding affinity, supporting earlier observations that both chain length and sulfate content of GAGs influence the binding affinity in the interaction with CPPs (30).
FIGURE 2.

All CPCs investigated bind the sulfated GAG heparin. Isothermal titration calorimetry of various cell-impermeant (A and B) and -permeant compounds (C and D) with heparin. (A–C) Heat flow (same y axis for all three panels). Every 10 min, 5 μl of 15 μM heparin is titrated into the reaction cell (Vcell = 1.4 ml) filled with (A) 100 μM propidium iodide, (B) 100 μM ethidium bromide, or (C) 100 μM acridine orange. (D) Heats of reaction hi (equal to integration of the heat flow peaks) as a function of the number of injections Ni. The heparin concentration in the reaction cell (Vcell = 1.4 ml) is 13.7 μM. Each symbol corresponds to the injection (10 μl every 10 min) of the following compounds: (○) 10.4 mM l-arginine, (•) DOTAP vesicles ([DOTAP]out = 4.8 mM, 30 nm, DOTAP:DOPC = 3:7 (n/n)), (⋄) 650 μM PLL16, (▴) 717 μM LPEI, (★) 1.167 mM nona-l-arginine, (□) 1.302 mM HIV Tat-PTD, and (▪) 1.486 mM penetratin. Symbols represent experimental data. The solid line is a least-squares fit using the binding model described by Eqs. 1 and 2 with the parameters listed in Table 1. Temperature is 28°C. Buffer in all experiments is 30 mM phosphate and 77 mM NaCl at pH 7.40 with a resulting physiological ionic strength of 154 mM.
TABLE 1.
Binding of heparin to diverse CPCs
| ITC
|
DLS
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligand and charge (zCPC) at given pH* | Kd,i (nM)† | n binding sites per heparin‡ (nligands/nheparin) | n × zCPC | Kd,1 (nM)§ | Reaction enthalpy 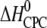 (kcal/mol CPC) (kcal/mol CPC) |
Entropy 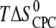 (kcal /mol CPC) (kcal /mol CPC) |
Heat capacity change¶ (cal/mol CPC/K) (cal/mol CPC/K) |
Rh** (nm) | FWHM†† of Rh (nm) |
| HIV Tat-PTD8+ | 443 ± 178 | 6.68 ± 0.57 | 53.4 ± 4.55 | 67.9 ± 30.5 | −12.3 ± 0.33 | −3.47 ± 0.37 | 38.6 | 94.4 ± 5.9 | 36.1 ± 6.6 |
| Penetratin7+ | 338 ± 105 | 6.79 ± 0.45 | 47.5 ± 3.18 | 50.6 ± 19.0 | −11.6 ± 0.12 | −2.63 ± 0.18 | −128 | 129 ± 18.7 | 51.1 ± 8.4 |
| Nona-l-arginine9+ | 459 ± 115 | 5.38 ± 0.62 | 48.4 ± 5.54 | 87.9 ± 33.3 | −12.8 ± 2.65 | −4.02 ± 2.80 | 89.3 | 98.3 ± 6.2 | 36.4 ± 5.2 |
| PLL1616+ | 543 ± 253 | 2.91 ± 0.30 | 46.6 ± 4.86 | 198 ± 108 | −8.50 ± 0.11 | 0.18 ± 0.30 | 11.0 | 77.7 ± 6.8 | 24.8 ± 6.9 |
| LPEI≈20+ (2.5 kDa); pH 7.4 | 741 ± 104 | 3.36 ± 0.51 | 67.3 ± 10.2 | 224 ± 52.1 | −16.5 ± 2.12 | −8.08 ± 2.20 | −235 | 131 ± 14.1 | 28.9 ± 7.2 |
| LPEI≈58+ (2.5 kDa); pH 5.0 | 531 ± 221 | 1.10 ± 0.27 | 64.0 ± 15.9 | 467 ± 94.9 | −37.1 ± 4.98 | −28.4 ± 4.1 | −76.1 | 123 ± 12.2 | 38.1 ± 4.1 |
| DOTAP1+ (as 30-nm small unilamellar vesicles; nDOTAP: nDOPC = 3:7) | 1345 ± 417 | 31.5 ± 3.09 | 31.5 ± 3.09 | 45.7 ± 19.0 | −0.49 ± 0.07 | 7.82 ± 0.53 | −25.7 | 586 ± 149 | 428 ± 136 |
| Acridine orange1+ | 868 ± 293 | 65.7 ± 6.8 | 65.7 ± 6.8 | 13.3 ± 4.6 | −3.82 ± 0.27 | 4.59 ± 0.48 | −32.0 | 311 ± 56.7 | 234 ± 92.5 |
| Ethidium bromide1+ | No binding observed‡‡ | <10 | |||||||
| Propidium iodide2+ | No binding observed | <10 | |||||||
| l-arginine1+ | No binding observed | <10 | |||||||
Conditions and pH (7.4) as described in Fig. 2; LPEI was measured additionally at pH 5.00 (133 mM NaCl, 30 mM acetate). Results are reported as mean ± SD from four individual sample preparations.
Microscopic dissociation constant (Kd,i) of n individual binding sites found per heparin chain.
Stoichiometry as experimentally determined (ITC) for 100% of heparin's multiple binding sites saturated with the specific ligand; heparin had a sulfur content of 11.3%, yielding an average of 45.8 sulfate groups or a total of 68.2 negative charges (including carboxyl groups) per heparin (average mol wt 13,000).
Macroscopic dissociation constant (Kd,1) for the first accessible binding site in the GAG.
Molar heat capacity change at constant pressure (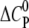 ) as calculated from the slope of
) as calculated from the slope of 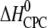 as a function of different temperatures (8, 18, 28, 38, 48, and 58°C).
as a function of different temperatures (8, 18, 28, 38, 48, and 58°C).
Hydrodynamic radius (Rh) as experimentally determined for 50% of heparin's binding sites (13.7 μM, 1.4 ml) saturated with the ligand; except for AOR and DOTAP, which were measured at 10% and 5% saturation of heparin, respectively, because at higher molar ratios, Rh exceeded the laser's wavelength. At current settings, the lower detection limit was ∼10 nm.
Width of the Rh distribution was calculated as full width at half-maximum (FWHM).
Physiological ionic strength apparently prevents binding of ethidium bromide to heparin (32).
Compared with the interaction of protein-based cell receptors with growth factors or cytokines, which usually bind in the low-nanomolar range, present dissociation constants in the high-nanomolar range presumably facilitate the release of the CPC after internalization of the binding site. Accordingly, the intracellular release and redistribution of CPCs to the nucleus are commonly observed after the CPC uptake (2,31), suggesting a tighter interaction of CPCs with intracellular compounds such as DNA (16).
Interestingly, the cationic DNA dyes ethidium bromide and propidium iodide do not bind the GAG at physiological ionic strength and low-micromolar concentration (Fig. 2, Table 1). For instance, ethidium bromide was reported to bind heparin in pure water (Kd ≈ 0.6 M), but not at physiological ionic strength (32). In contrast, acridine orange binds with Kd ≈ 0.87 μM (Table 1) (33), although all three dyes are known to bind DNA with high affinity (34). This remarkable difference and their different optical properties on polyelectrolyte binding (35) suggest that the binding of these cationic dyes to different polyanions follows different mechanisms. It is thus of future interest to clarify whether the pronounced difference in GAG binding is also the basis of their different biological uptake behavior, i.e., no membrane passage for ethidium and propidium (17) but rapid uptake and accumulation of acridine orange in endosomes (18,36).
Binding mechanism
At 28°C, the GAG binding was exothermic for all CPCs (Fig. 1). A negative enthalpy change makes the major contribution to the free energy change for most CPCs (Table 1). For hydrophobic CPCs, such as DOTAP and acridine orange (AOR), however, the reaction entropy provided the main binding force. This is most likely the consequence of the release of hydration water, which is further supported by the observed negative molar heat capacity change (see below).
Interestingly, the cationic charge alone is not sufficient to provide CPC binding to GAGs because a tight binding was found for nona-arginine but not for its monomer arginine compared at identical arginine monomer concentrations (Fig. 2). This suggests that electrostatic interactions at low-micromolar concentrations require a minimum charge density as corroborated by the polyelectrolyte theory (37).
The change in the molar heat capacity 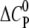 provides a good approximation to distinguish between electrostatic and hydrophobic contribution to the binding. Generally, a negative
provides a good approximation to distinguish between electrostatic and hydrophobic contribution to the binding. Generally, a negative 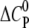 indicates a change in the solvent-accessible surface area (hydrophobic effect) (38), in contrast to a positive heat capacity, which often indicates the dominance of electrostatic interactions (39).
indicates a change in the solvent-accessible surface area (hydrophobic effect) (38), in contrast to a positive heat capacity, which often indicates the dominance of electrostatic interactions (39).
We find that the highly charged CPCs nona-arginine and HIV-1 Tat-PTD bind the GAG with a positive 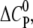 indicating that GAG binding is driven mainly by electrostatic forces. However, the amplitude of
indicating that GAG binding is driven mainly by electrostatic forces. However, the amplitude of 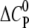 is lower than expected for a pure electrostatic interaction, suggesting additional hydrogen bonds in the binding reaction through interactions of guanidinium groups with sulfates and carboxylates (40–42).
is lower than expected for a pure electrostatic interaction, suggesting additional hydrogen bonds in the binding reaction through interactions of guanidinium groups with sulfates and carboxylates (40–42).
In contrast, the amphipathic CPCs AOR and DOTAP display a negative 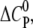 indicating that GAG binding and aggregation are dominated by the decrease in the water-accessible surface area. This change in hydration is further supported by the gain in entropy (Table 1) that most likely results from water and counterion release during binding. These hydrophobic interactions might explain why these two monovalent CPCs bind the GAG in contrast to monovalent arginine. In support, it has been found that soluble acridine orange AOR can self-assemble when interacting with anionic polyelectrolytes or interfaces (33,43,44). It may be noted that the release of solvent water also plays a key role in binding and aggregation of polycations with other polyelectrolytes such as DNA (45–47).
indicating that GAG binding and aggregation are dominated by the decrease in the water-accessible surface area. This change in hydration is further supported by the gain in entropy (Table 1) that most likely results from water and counterion release during binding. These hydrophobic interactions might explain why these two monovalent CPCs bind the GAG in contrast to monovalent arginine. In support, it has been found that soluble acridine orange AOR can self-assemble when interacting with anionic polyelectrolytes or interfaces (33,43,44). It may be noted that the release of solvent water also plays a key role in binding and aggregation of polycations with other polyelectrolytes such as DNA (45–47).
Binding specificity
Despite larger differences in their primary structure, all CPCs were found to bind with high affinity to the GAG. It is therefore suggested that GAG binding requires a specific charge density of the CPC and neglects larger differences in their overall structure. This structural flexibility is further supported by the finding that d-isomers and retro-inverso sequences of CPPs also bind GAGs (48) and are efficiently taken up by living cells (31,49–51).
It is of interest whether a comparable structural flexibility is also allowed for GAGs, or whether the CPP binding requires a specific GAG structure that could be exploited for organ-specific targeting. Such specific GAG epitopes have been postulated but then revised for GAG-mediated growth-factor uptake (52). Likewise, recent studies demonstrated that a given CPP binds to a variety of different GAG types with the binding constant increasing as a function of the sulfate density (15) and chain length of the GAG (30). As a result, a lyase specific for a particular GAG type may be inefficient to completely abolish the CPC binding to GAGs in living cells, which previously led to discussions on whether or not the GAG subtype heparan sulfate is the only mediator of CPP uptake (1). The resulting high structural flexibility in the CPP-GAG interaction combined with the high binding affinity in the submicromolar range might thus explain why CPPs are efficiently internalized by many different cell strains (53), taking into account that GAGs are ubiquitously expressed in vertebrates and invertebrates, in contrast to specific receptor proteins.
Stoichiometry
The stoichiometry of the binding reactions corresponds approximately to charge neutralization (Table 1) with small deviations related to steric constraints. Small molecules such as AOR apparently adapt best to the heterogeneous anion distance in GAGs and thus encounter the greatest number of binding sites; in contrast, large molecules such as 30-nm DOTAP vesicles are less flexible and have the fewest binding sites. For LPEI, the stoichiometry is additionally affected by the pH because of its lower pKa (∼7) (54) compared with other CPCs that are maximally charged at physiological pH (quaternary ions or pKa > 10). As a result, cell-internal pH shifts as encountered during endosome acidification might affect the stoichiometry (Fig. 3) and tightness (Table 1) of the LPEI binding.
GAG clustering
CPPs differ considerably in their primary structure and physicochemical properties, suggesting that a unique conformation of the CPP is unlikely for binding to the biological cell surface receptor. Such interactions of maximized geometrical fit (55) would be stereospecific because of their natural content of l-isomeric amino acids. In contrast, endocytotic receptors react with a wider range of possible ligands (56) where the ligand binding may lead to clustering or capping of integral membrane constituents, such as GAG-containing proteoglycans (6–12) or N-linked GAGs (e.g., sialic acid type) (57,58). Such interactions affect the membrane curvature (59), lateral diffusion (8), and clustering (3,6–11) of their membrane anchor, cytoskeleton bundling (3,7,9,14), and activation of intracellular protein kinases (3,60–63), which are all considered crucial elements in recent views of endocytosis (56). We therefore were interested in whether CPCs not only bind GAGs, reflecting merely adsorption to the cell surface, but also induce the clustering of GAGs with consequences for endocytotic signals.
We find that all CPCs, on GAG binding, produce particles of considerable size as evident from the pronounced increase in light scattering in titration experiments (Fig. 3). The radius of the particles is 70 nm and larger (Fig. 4; Table 1), suggesting that these clusters consist of several GAG chains cross-linked by CPCs rather than of a single GAG chain saturated with the maximum number of ligands (Table 1). The maximum number of heparin molecules per cluster was estimated by dividing the hydrodynamic volume of the particles by the volume of a single heparin chain saturated with the known number of ligands (Table 1). The solvent-excluded volumes of HIV-1 Tat (47–57) and heparin (dp44) employed in this study are 1344 and 7863 Å3, respectively, based on their published structures in the protein database (1TAC and 1hpn (1C4, dp12), respectively). Accordingly, the observed GAG-CPC cluster (Rh = 94.4 nm) comprises up to 2.1 × 105 heparin molecules (each complexed with six or seven Tat molecules) depending on the actual state of hydration.
FIGURE 3.

GAG clustering upon CPC binding. Various cell-impermeant and -permeant compounds are titrated into heparin solution (except control (4b), into pure buffer), and the static right-angle light scattering (λex/λem = 350/350 nm) is recorded. The increase in light-scattering signal demonstrates the formation of larger aggregates of CPCs and GAGs. No comparable scattering signal is produced by the titration of GAGs with structurally related but cell-impermeant compounds 6, 9, and 10. The heparin concentration in the optical cuvette (Vcell = 1.4 ml) is 13.7 μM. Each step corresponds to the injection of 10 μl (every 5 min) of the following compounds: (6) 5.2 mM propidium iodide; (9) 10.4 mM l-arginine; (10) 5.2 mM ethidium bromide; (5) 5.2 mM acridine orange; (8) 1.5 mM penetratin; (4) DOTAP vesicles ([DOTAP]out = 4.8 mM, 30 nm, DOTAP:DOPC = 3:7 (n/n)) into (4a) heparin or (4b) buffer; (2) 650 μM PLL16; (3a–c) 359 μM linear polyethylenimine at (3a) pH 7.4, (3b) pH 5.0, and (3c) pH 3.5; (7) 1.3 mM HIV Tat-PTD; and (1) 1.1 mM nona-l-arginine. Same absolute scale in both panels. Temperature is 28°C. Buffer in all experiments is 30 mM phosphate and 77 mM NaCl at pH 7.40 (ionic strength of 154 mM); except LPEI (3b) at pH 5.0 using 30 mM acetate and 133 mM NaCl (ionic strength of 154 mM), and (3c) at pH 3.5 using 30 mM formate and 141 mM NaCl (ionic strength of 154 mM).
FIGURE 4.

Confirmation of the GAG clustering caused by CPCs. DLS intensity autocorrelation functions were recorded at eight different angles (30–150°). Displayed are recordings at θ = 90° for 50% of heparin's (13.7 μM, 1.4 ml) sulfate charges saturated with indicated cationic compounds. Recordings for AOR (ethidium bromide and propidium iodide) and DOTAP are displayed for 10% and 5% charge saturation of heparin, respectively, so that the particle size did not exceed the sensitive range of the instrument (<1 μm). All CPCs caused a clustering of GAGs as evident from the complex size of >70 nm (Table 1), which is considerably larger than a single GAG molecule saturated with the defined number of CPCs. In contrast, virtually no light scattering was found for the structurally related but non-cell-permeant compounds 10, 9, and 6 even when a 16-times higher receiver gain was used than for the other compounds. Individual tracings are (10) ethidium bromide, (9) l-arginine, (6) propidium iodide, (2) PLL16, (7) HIV Tat-PTD, (1) nona-l-arginine, (3) polyethylenimine, (8) penetratin, (5) acridine orange, and (4) DOTAP vesicles ([DOTAP]out = 4.8 mM, 30 nm, DOTAP:DOPC = 3:7 (n/n)). Temperature was 20°C. Buffer in all experiments is 30 mM phosphate and 77 mM NaCl at pH 7.40 (ionic strength of 154 mM).
The current GAG clustering in vitro relates to the GAG capping recently observed in vivo (2,3). The size of the capping complexes and their visibility in living cells under the light microscope (2) depend on various aspects such as the molar ratio of the reactants (Fig. 3), the type of the CPC (Fig. 4), and also the type and length of the GAG (15). In contrast, no GAG clustering was observed for monomeric arginine, ethidium bromide, and propidium iodide (Fig. 3 and 4), as they do not bind the GAG at physiological ionic strength (Fig. 2). This agrees with their lack of endocytotic uptake (17).
CONCLUSIONS
The results presented here reveal that GAG binding and GAG clustering are common properties of many CPCs. This study shows that CPCs of four chemically unrelated groups react in a very similar fashion. The CPC-GAG interactions are characterized by a high affinity and display a defined number of binding sites. The interactions allow for a structural variability of both CPC and GAG provided a critical charge density of the reactants is met. Although this study reports findings in solution, it follows from cell culture work that CPCs may lead to GAG clustering on the surface of biological cells during the CPC uptake (2,3). Also, GAG clustering and subsequent cellular uptake was observed for specific antibodies against GAGs (9,11). In terms of a screening assay, clustering of GAGs might therefore be helpful to identify new cell-penetrating compounds.
Acknowledgments
We are grateful to Dr. Corinne Vebert (Department of Chemistry) for her precious advice on DLS. We also thank Therese Schulthess for excellent technical assistance.
This work was supported by the Swiss National Science Foundation under grant No. 3100-107793/1.
Editor: Jonathan B. Chaires.
References
- 1.Richard, J. P., K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik, and B. Lebleu. 2003. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 278:585–590. [DOI] [PubMed] [Google Scholar]
- 2.Ziegler, A., P. Nervi, M. Durrenberger, and J. Seelig. 2005. The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry. 44:138–148. [DOI] [PubMed] [Google Scholar]
- 3.Kopatz, I., J. S. Remy, and J. P. Behr. 2004. A model for non-viral gene delivery: through syndecan adhesion molecules and powered by actin. J. Gene Med. 6:769–776. [DOI] [PubMed] [Google Scholar]
- 4.Rejman, J., A. Wagenaar, J. B. Engberts, and D. Hoekstra. 2004. Characterization and transfection properties of lipoplexes stabilized with novel exchangeable polyethylene glycol-lipid conjugates. Biochim. Biophys. Acta. 1660:41–52. [DOI] [PubMed] [Google Scholar]
- 5.Wiethoff, C. M., J. G. Smith, G. S. Koe, and C. R. Middaugh. 2001. The potential role of proteoglycans in cationic lipid-mediated gene delivery. Studies of the interaction of cationic lipid-DNA complexes with model glycosaminoglycans. J. Biol. Chem. 276:32806–32813. [DOI] [PubMed] [Google Scholar]
- 6.Belting, M. 2003. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem. Sci. 28:145–151. [DOI] [PubMed] [Google Scholar]
- 7.Dull, R. O., R. Dinavahi, L. Schwartz, D. E. Humphries, D. Berry, R. Sasisekharan, and J. G. Garcia. 2003. Lung endothelial heparan sulfates mediate cationic peptide-induced barrier dysfunction: a new role for the glycocalyx. Am. J. Physiol. Lung Cell. Mol. Physiol. 285:L986–L995. [DOI] [PubMed] [Google Scholar]
- 8.Fuki, I. V., M. E. Meyer, and K. J. Williams. 2000. Transmembrane and cytoplasmic domains of syndecan mediate a multi-step endocytic pathway involving detergent-insoluble membrane rafts. Biochem. J. 351:607–612. [PMC free article] [PubMed] [Google Scholar]
- 9.Martinho, R. G., S. Castel, J. Urena, M. Fernandez-Borja, R. Makiya, G. Olivecrona, M. Reina, A. Alonso, and S. Vilaro. 1996. Ligand binding to heparan sulfate proteoglycans induces their aggregation and distribution along actin cytoskeleton. Mol. Biol. Cell. 7:1771–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summerford, C., and R. J. Samulski. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tkachenko, E., and M. Simons. 2002. Clustering induces redistribution of syndecan-4 core protein into raft membrane domains. J. Biol. Chem. 277:19946–19951. [DOI] [PubMed] [Google Scholar]
- 12.Tyagi, M., M. Rusnati, M. Presta, and M. Giacca. 2001. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 276:3254–3261. [DOI] [PubMed] [Google Scholar]
- 13.Fischer, R., M. Fotin-Mleczek, H. Hufnagel, and R. Brock. 2005. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. ChemBioChem. 6:2126–2142. [DOI] [PubMed] [Google Scholar]
- 14.Nakase, I., A. Tadokoro, N. Kawabata, T. Takeuchi, H. Katoh, K. Hiramoto, M. Negishi, M. Nomizu, Y. Sugiura, and S. Futaki. 2007. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 46:492–501. [DOI] [PubMed] [Google Scholar]
- 15.Ziegler, A., and J. Seelig. 2004. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: binding mechanism and thermodynamic parameters. Biophys. J. 86:254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziegler, A., and J. Seelig. 2007. High affinity of the cell-penetrating peptide HIV-1 Tat-PTD for DNA. Biochemistry. 46:8138–8145. [DOI] [PubMed] [Google Scholar]
- 17.Bank, H. L. 1987. Assessment of islet cell viability using fluorescent dyes. Diabetologia. 30:812–816. [DOI] [PubMed] [Google Scholar]
- 18.Sugar, J., and E. Gati. 1958. Change of acridine orange uptake by cells of Ehrlich ascites tumors under the effect of different chemotherapeutic agents. Krebsarzt. 13:119–120. [PubMed] [Google Scholar]
- 19.Andrade, S. M., and S. M. Costa. 2002. The aqueous environment in AOT and Triton X-100 (w/o) microemulsions probed by fluorescence. Photochem. Photobiol. Sci. 1:500–506. [DOI] [PubMed] [Google Scholar]
- 20.Dell'Antone, P., and G. F. Azzone. 1974. Inhibition of energy-linked uptake of acridine dyes by permeant anions. FEBS Lett. 39:67–72. [DOI] [PubMed] [Google Scholar]
- 21.Haran, G., R. Cohen, L. K. Bar, and Y. Barenholz. 1993. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta. 1151:201–215. [DOI] [PubMed] [Google Scholar]
- 22.Wirth, M. J., and J. D. Burbage. 1991. Adsorbate reorientation at a water/(octadecylsilyl)silica interface. Anal. Chem. 63:1311–1317. [Google Scholar]
- 23.Mulloy, B., and R. J. Linhardt. 2001. Order out of complexity–protein structures that interact with heparin. Curr. Opin. Struct. Biol. 11:623–628. [DOI] [PubMed] [Google Scholar]
- 24.van Holde, K. E., W. C. Johnson, and P. S. Ho. 1998. Chemical equilibria involving macromolecules. In Principles of Physical Biochemistry. K. E. van Holde, W. C. Johnson, and P. S. Ho, editors. Prentice Hall, Upper Saddle River, NJ. 604–611.
- 25.Koppel, D. E. 1972. Analysis of macromolecular polydispersity in intensity correlation spectroscopy: The method of cumulants. J. Chem. Phys. 57:4814–4820. [Google Scholar]
- 26.Connolly, M. L. 1993. The molecular surface package. J. Mol. Graph. 11:139–141. [DOI] [PubMed] [Google Scholar]
- 27.Schafer, I. A., J. C. Sullivan, J. Svejcar, J. Kofoed, and W. V. Robertson. 1968. Study of the Hurler syndrome using cell culture: definition of the biochemical phenotype and the effects of ascorbic acid on the mutant cell. J. Clin. Invest. 47:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turnbull, J. E., and J. T. Gallagher. 1991. Sequence analysis of heparan sulphate indicates defined location of N-sulphated glucosamine and iduronate 2-sulphate residues proximal to the protein-linkage region. Biochem. J. 277:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goncalves, E., E. Kitas, and J. Seelig. 2005. Binding of oligoarginine to membrane lipids and heparan sulfate: structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry. 44:2692–2702. [DOI] [PubMed] [Google Scholar]
- 30.Rusnati, M., G. Tulipano, D. Spillmann, E. Tanghetti, P. Oreste, G. Zoppetti, M. Giacca, and M. Presta. 1999. Multiple interactions of HIV-I Tat protein with size-defined heparin oligosaccharides. J. Biol. Chem. 274:28198–28205. [DOI] [PubMed] [Google Scholar]
- 31.Brugidou, J., C. Legrand, J. Mery, and A. Rabie. 1995. The retro-inverso form of a homeobox-derived short peptide is rapidly internalised by cultured neurones: a new basis for an efficient intracellular delivery system. Biochem. Biophys. Res. Commun. 214:685–693. [DOI] [PubMed] [Google Scholar]
- 32.Chaudhuri, S., G. O. Phillips, D. M. Power, and J. V. Davies. 1975. Interaction of ethidium bromide with heparin. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 28:345–352. [DOI] [PubMed] [Google Scholar]
- 33.Menter, J. M., R. E. Hurst, N. Nakamura, and S. S. West. 1979. Thermodynamics of mucopolysaccharide-dye binding. III. Thermodynamic and cooperativity parameters of acridine orange-heparin system. Biopolymers. 18:493–505. [DOI] [PubMed] [Google Scholar]
- 34.Beisker, W., and W. G. Eisert. 1989. Denaturation and condensation of intracellular nucleic acids monitored by fluorescence depolarization of intercalating dyes in individual cells. J. Histochem. Cytochem. 37:1699–1704. [DOI] [PubMed] [Google Scholar]
- 35.Giancotti, V., F. Quadrifoglio, and V. Crescenzi. 1973. Polyelectrolyte behaviour of phosvitin. Spectroscopic, microcalorimetric and acridine-orange-binding data. Eur. J. Biochem. 35:78–86. [DOI] [PubMed] [Google Scholar]
- 36.Matteoni, R., and T. E. Kreis. 1987. Translocation and clustering of endosomes and lysosomes depends on microtubules. J. Cell Biol. 105:1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouzina, I., and V. A. Bloomfield. 1996. Macroion attraction due to electrostatic correlation between screening counterions. 1. Mobile surface-adsorbed ions and diffuse ion cloud. J. Phys. Chem. 100:9977–9989. [Google Scholar]
- 38.Tanford, C. 1979. Interfacial free energy and the hydrophobic effect. Proc. Natl. Acad. Sci. USA. 76:4175–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sturtevant, J. M. 1977. Heat capacity and entropy changes in processes involving proteins. Proc. Natl. Acad. Sci. USA. 74:2236–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hileman, R. E., R. N. Jennings, and R. J. Linhardt. 1998. Thermodynamic analysis of the heparin interaction with a basic cyclic peptide using isothermal titration calorimetry. Biochemistry. 37:15231–15237. [DOI] [PubMed] [Google Scholar]
- 41.Thompson, L. D., M. W. Pantoliano, and B. A. Springer. 1994. Energetic characterization of the basic fibroblast growth factor-heparin interaction: identification of the heparin binding domain. Biochemistry. 33:3831–3840. [DOI] [PubMed] [Google Scholar]
- 42.Sakai, N., and S. Matile. 2003. Anion-mediated transfer of polyarginine across liquid and bilayer membranes. J. Am. Chem. Soc. 125:14348–14356. [DOI] [PubMed] [Google Scholar]
- 43.Myhr, B. C., and J. G. Foss. 1971. Acridine orange-poly(alpha-l-glutamic acid) complexes. I. Stoichiometry and stacking coefficients. Biopolymers. 10:425–440. [DOI] [PubMed] [Google Scholar]
- 44.Yao, H., S. Kobayashi, and K. Kimura. 2007. Self-assembly of acridine orange dye at a mica/solution interface: formation of nanostripe supramolecular architectures. J. Colloid Interface Sci. 307:272–279. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, W., J. P. Bond, C. F. Anderson, T. M. Lohman, and M. T. Record, Jr. 1996. Large electrostatic differences in the binding thermodynamics of a cationic peptide to oligomeric and polymeric DNA. Proc. Natl. Acad. Sci. USA. 93:2511–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jonsson, M., and P. Linse. 2003. Monte Carlo simulations of the hydrophobic effect in aqueous electrolyte solutions. J. Phys. Chem. B. 115:3406–3418. [DOI] [PubMed] [Google Scholar]
- 47.Patel, M. M., and T. J. Anchordoquy. 2005. Contribution of hydrophobicity to thermodynamics of ligand-DNA binding and DNA collapse. Biophys. J. 88:2089–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goncalves, E., E. Kitas, and J. Seelig. 2006. Structural and thermodynamic aspects of the interaction between heparan sulfate and analogues of melittin. Biochemistry. 45:3086–3094. [DOI] [PubMed] [Google Scholar]
- 49.Gammon, S. T., V. M. Villalobos, J. L. Prior, V. Sharma, and D. Piwnica-Worms. 2003. Quantitative analysis of permeation peptide complexes labeled with Technetium-99m: chiral and sequence-specific effects on net cell uptake. Bioconjug. Chem. 14:368–376. [DOI] [PubMed] [Google Scholar]
- 50.Shen, W. C., and H. J. Ryser. 1979. Poly (l-lysine) and poly (d-lysine) conjugates of methotrexate: different inhibitory effect on drug resistant cells. Mol. Pharmacol. 16:614–622. [PubMed] [Google Scholar]
- 51.Wender, P. A., D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman, and J. B. Rothbard. 2000. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. USA. 97:13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kreuger, J., P. Jemth, E. Sanders-Lindberg, L. Eliahu, D. Ron, C. Basilico, M. Salmivirta, and U. Lindahl. 2005. Fibroblast growth factors share binding sites in heparan sulphate. Biochem. J. 389:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mann, D. A., and A. D. Frankel. 1991. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 10:1733–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smits, R. G., G. J. M. Koper, and M. Mandel. 1993. The influence of nearest- and next-nearest-neighbor interactions on the potentiometric titration of linear poly(ethylenimine). J. Phys. Chem. 97:5745–5751. [Google Scholar]
- 55.Cramer, F. 1995. Biochemical correctness: Emil Fischer's lock and key hypothesis, a hundred years after—an essay. Pharm. Acta. Helv. 69:193–203. [Google Scholar]
- 56.Herz, J., and D. K. Strickland. 2001. LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108:779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Freeze, H. H., and A. Varki. 1986. Endo-glycosidase F and peptide N-glycosidase F release the great majority of total cellular N-linked oligosaccharides: use in demonstrating that sulfated N-linked oligosaccharides are frequently found in cultured cells. Biochem. Biophys. Res. Commun. 140:967–973. [DOI] [PubMed] [Google Scholar]
- 58.Roux, L., S. Holojda, G. Sundblad, H. H. Freeze, and A. Varki. 1988. Sulfated N-linked oligosaccharides in mammalian cells. I. Complex-type chains with sialic acids and O-sulfate esters. J. Biol. Chem. 263:8879–8889. [PubMed] [Google Scholar]
- 59.McMahon, H. T., and J. L. Gallop. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 438:590–596. [DOI] [PubMed] [Google Scholar]
- 60.Biener, Y., and Y. Zick. 1990. Basic polycations activate the insulin receptor kinase and a tightly associated serine kinase. Eur. J. Biochem. 194:243–250. [DOI] [PubMed] [Google Scholar]
- 61.Khan, N. A., I. Masson, V. Quemener, G. Clari, V. Moret, and J. P. Moulinoux. 1990. Polyamines and polyamino acids regulation of cytosolic tyrosine protein (Tyr-P) kinase from human erythrocytes. Biochem. Int. 20:863–868. [PubMed] [Google Scholar]
- 62.Mohammadi, M., A. Honegger, A. Sorokin, A. Ullrich, J. Schlessinger, and D. R. Hurwitz. 1993. Aggregation-induced activation of the epidermal growth factor receptor protein tyrosine kinase. Biochemistry. 32:8742–8748. [DOI] [PubMed] [Google Scholar]
- 63.Xu, Q. Y., S. L. Li, T. R. LeBon, and Y. Fujita-Yamaguchi. 1991. Aggregation of IGF-I receptors or insulin receptors and activation of their kinase activity are simultaneously caused by the presence of polycations or K-ras basic peptides. Biochemistry. 30:11811–11819. [DOI] [PubMed] [Google Scholar]