

Abstract
Statistical power calculations constitute an essential first step in the planning of scientific studies. If sufficient summary statistics are available, power calculations are in principle straightforward and computationally light. In designs, which comprise distinct groups (e.g., MZ & DZ twins), sufficient statistics can be calculated within each group, and analyzed in a multi-group model. However, when the number of possible groups is prohibitively large (say, in the hundreds), power calculations on the basis of the summary statistics become impractical. In that case, researchers may resort to Monte Carlo based power studies, which involve the simulation of hundreds or thousands of replicate samples for each specified set of population parameters. Here we present exact data simulation as a third method of power calculation. Exact data simulation involves a transformation of raw data so that the data fit the hypothesized model exactly. As in power calculation with summary statistics, exact data simulation is computationally light, while the number of groups in the analysis has little bearing on the practicality of the method. The method is applied to three genetic designs for illustrative purposes.
Keywords: Simulation, Power
Introduction
The importance of statistical power in (behavior) genetic analyses is evident in the number of articles devoted to power calculations. Power has been studied in virtually all research designs, ranging from the classical twin design (Martin et al. 1978; Neale et al. 1994), to extended family designs (e.g., Heath et al. 1985; Heath and Eaves 1985; Posthuma and Boomsma 2000), to sibpair and family linkage and association designs, either in- or excluding gene by environment interaction (Abecasis et al. 2000a, b; Boomsma and Dolan 1998; Dolan et al. 1999; Fulker and Cherny 1996; Purcell 2002; Purcell and Sham 2002; Sham et al. 2000; Sham and Hewitt 1999; Sham et al. 2002; Van den Oord 1999). For a wide range of genetic designs, the Genetic Power Calculator1 (Purcell et al. 2003) can be used to calculate power. However, for customized designs and specific research questions, researchers may have to resort to their own procedure to carry out power calculations.
Power calculation based on the likelihood with the general Pearson–Nyman statistical decision theory takes two forms. First, the non-centrality parameter λ of the non-null χ2-distribution can be calculated in the analysis of exact sufficient statistics (e.g., Dolan et al. 1999). If the distribution of the data is multivariate normal, the expected variance covariance matrix Σ and the means vector μ are sufficient statistics, as they define the likelihood of the data up to an arbitrary constant (Azzelini 1996). Second, when Σ and μ are not sufficient statistics, the non-centrality parameter λ of the non-null χ2-distribution can be estimated on the basis of the analysis of simulated data using Monte Carlo simulation methods (e.g., Fulker and Cherny 1996; Abecasis et al. 2002a, b; Purcell 2002; van den Oord 1999). The latter is computationally intensive, but does not require the presence of sufficient summary statistics, whereas the former is computationally light, but does require sufficient summary statistics.
The aim of the present note is to discuss a third method of power calculation, which we refer to as exact data simulation. This method is suitable when data are multivariate normal, and sufficient summary statistics are in principle available, but the number of possible groups is prohibitively large (say, in the hundreds). The large number of distinct groups renders power calculations on the basis of the summary statistics impractical. Usually, researchers resort to Monte Carlo based power studies under such circumstances. However, exact data simulation, in combination with the definition variable facilities in packages like Mplus (fourth edition, Muthén and Muthén 1998–1997) or the freely available Mx program2 (Neale et al. 2003), is also applicable, and is more efficient than raw data simulation. Exact data simulation was used by Dolan et al. (2005) to evaluate the effects of missing data on the power in structural equation modeling, and by Van der Sluis et al. (Under revision) to evaluate the power to detect gene by environment interaction in sib-pair association studies. Although the technique of exact simulation is in itself not new (Bollen and Stine provided the basics in 1993, and the technique of exact data simulation has recently been integrated as a distinct function in the freely available R-program3), we wish to bring it to the attention of geneticists since this method of calculating power has general value in the field of genetic modeling. Below, we shortly recapitulate the basics of power analysis, and then outline the procedure of exact simulation, which may be implemented readily (we use the freely available R program). The method is illustrated in three genetic designs. Although we confine ourselves in this paper to illustrations in the context of genetic designs, we stress that this form of simulation can be used for power calculations in a wide range of other designs such as random effects models, growth curve and simplex models, and structural models.
Power calculation
The concept of power is closely related to the two types of statistical errors: the Type I error (i.e., the probability of rejecting a true hypothesis, α), and the Type II error (i.e., the probability of accepting a false hypothesis, β). Power is defined as 1 − β, i.e., the probability of rejecting a false hypothesis, or the probability of not making a Type II error. The basic aim of a power study is to determine the sample size N, which is required to achieve adequate power, given chosen α and a particular effect size.
For example, suppose that we want to fit a classical univariate ACE twin-model (see Fig. 1), and we expect that additive genetic effects (a) account for 40% of the phenotypic variance, besides effects of shared (c) and unique (e) environment. We denote this model 1, or hypothesis 1, H1. Under model 1, four parameters are estimated: the path coefficient for the additive genetic effects A (a), the path coefficient for the shared environment C (c), the path coefficient for the unique environment E (e, which includes measurement error), and the means of the twins (μ), which are usually set to be equal within twin pairs and across MZ and DZ twins. The parameter vector θ for this model with df1 degrees of freedom, is θ1 = {a, c, e, μ}, from which the covariance matrices Σ1mz and Σ1dz and mean vector μ1 can be derived. Now consider a second model, which we denote model 0, or H0, in which the additive genetic effects are assumed to account for only 5% of the phenotypic variance. This alternative model will be characterized by parameter vector θ0 = {c, e, μ}. Note that in model 0, parameter a is not estimated (and thus not part of θ) but fixed at a value that corresponds exactly to 5% of the phenotypic variation being explained by additive genetic effects. This model has df0 degrees of freedom, covariance matrices Σ0mz & Σ0dz, and mean vector μ0. This alternative model H0 is nested in the null-model H1, because the parameters in θ0 represent a subset of the parameters in θ1 (e.g., Bollen 1989; Satorra and Saris 1985). The study of power is subsequently concerned with the probability of rejecting the false model H0 in favor of the true model H1, given α, the true value of parameter a (the effect size), and sample size N.
Fig. 1.

Classical univariate ACE-twin model
To calculate power, we adopt the method of Satorra and Saris (1985, see also Saris and Satorra 1993), which is based on the normal theory log-likelihood ratio test statistic T. In a single group, T is calculated as follows:
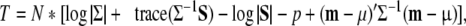 |
1 |
where N is the sample size, p denotes the number of variables in the analysis, and Σ and μ, and S and m represent the theoretical and observed variance covariance matrix and the means vector, respectively. Given that the assumptions of normal theory maximum likelihood are met (e.g., multivariate normality, and a large sample of independently and identically distributed cases), and under the assumption that Σ and μ represent the true model (Σ1 and μ1), the test statistic T follows a χ2 distribution with df1 degrees of freedom, i.e., T ∼ χ2(df1) (Azzeline 1996; Bollen 1989). If Σ and μ do not represent the true model but the alternative model (Σ0 and μ0), and given regularity conditions are satisfied (practically amounting to multivariate normality, limited misfit and large sample size N), the test statistic T follows a non-central χ2 distribution with df0 degrees of freedom and non-centrality parameter λ, i.e., T ∼ χ2(df0, λ), where λ > 0.
Given the significance level of the test α, and the difference in degrees of freedom between the true model and the alternative model, df1 − df0, the criterion level cα can be obtained from a χ2 table. If the test statistic T exceeds this criterion level, i.e., T > cα, then the alternative model is rejected in favor of the true model (i.e., the fit of the alternative model to the data is significantly worse than the fit of the true model). The aim of power studies is to determine the probability of observing T > cα, i.e., P(χ2(df0, λ) > cα), given Σ0, μ0, Σ1, μ1, N, and α, i.e., the probability of rejecting the alternative model in favor of the true model.
The non-centrality parameter λ can be obtained by fitting the alternative model to the true Σ1 and μ1, whereby λ equals the difference in the χ2 fit statistic of the model H1 and the χ2 fit statistic of the alternative model H0. That is (again in a single group),
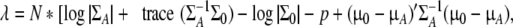 |
2 |
and the non-null distribution of this test statistic is χ2(df1 − df0,λ). A variety of programs can subsequently be used to integrate the non-null distribution to obtain the power (e.g., R, Mx; see also Hewitt and Heath 1988). Note that some packages, such as the Mx program, also compute the total sample size that would be required (given the reported proportion of subjects in each group) to reject the hypothesis at various power levels.
As stated in the introduction, power calculations usually take on one of two forms. First, one may be in the position that all information present in the raw data can be summarized in the covariance matrix Σ and the means vector μ, in which case Σ and μ are sufficient statistics, because they define the likelihood of the data up to an arbitrary constant (Azzelini 1996). In that case, one can derive the expected population statistics Σ and μ for every group in the study design under H1 and H0, and base the power calculations on these summary statistics.
The second method of power calculation is applied when sufficient summary statistics are not available. For example, the population statistics Σ and μ do not summarize all information present in the raw data when the continuous data are a mixture (i.e., a convex combination of different distributions; McLachlan and Peel 2002), or when data are missing at random (MAR; Shafer and Graham 2002). In family studies, gene by environment interaction may render the summary statistics insufficient. For instance, Purcell (2002) showed how environmental moderation on the means and variances can be modeled (see Fig. 2). For both MZ and DZ twins, the variance of twin i is calculated as:
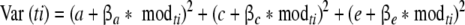 |
3 |
while the for MZ twins, covariance between twin i and twin j is calculated as:
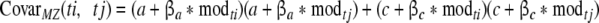 |
4 |
and for DZ twins as:
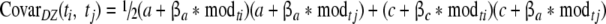 |
5 |
The expected values of means for all twins are calculated as:
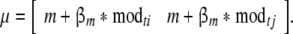 |
6 |
Fig. 2.

Univariate ACE-twin model including moderation on the variances and the means
If the environmental moderator is categorical or ordinal (e.g., gender or affection status), the sufficient statistics (Σ and μ) are available, assuming that the data are normally distributed conditional on the levels of the moderator. In that instance, the twins in a pair may be concordant with respect to the moderator (i.e., both twins score 0, or both twins score 1), or the twins may be discordant with respect to the moderator (i.e., scoring 0 and 1, respectively). For both MZ and DZ twins, Σ and μ can be formulated for all possible combinations, such that all information present in the raw data is summarized with 6 different variance covariance matrixes Σ (3 for the MZ twins, and 3 for the DZ twins) and 3 different means vectors μ (assuming no relation between zygosity and mean), as such distinguishing 6 different groups. By comparing the fit of the model including moderator effects on both the means and the variances, to a model in which the moderator only affects the means (say), one can obtain an estimate of non-centrality parameter λ, from which the power to detect the effect of the moderator can be derived.
However, if the moderator is continuously distributed, sufficient statistics cannot be calculated. In the absence of sufficient statistics, power calculation may be conducted by means of Monte Carlo simulation. This design implies determination of the values of the parameters of interest (e.g., based on previous studies or corresponding to realistic effect sizes), and subsequent (quasi-) random data generation according to the true model H1, with realistic sample size N. By fitting the false model H0 to the data simulated according to the true model H1, an indication of the power is obtained. However, in contrast to the situation in which sufficient summary statistics are available, parameter values are not recovered exactly when the H1 model is fitted, as the random data are the outcome of a stochastic sample process. Therefore, the difference in the χ2 statistic of the model H0 and the model H1 cannot be taken as an exact estimate of the non-centrality parameter λ. To solve this, a large number of datasets are usually generated, and λ is estimated as the mean of the difference in χ2 obtained in these data sets minus the number of degrees of freedom (df1 − df0). Since power studies often concern multiple parameters with multiple values, such Monte Carlo simulation studies can be prohibitively intensive. As an alternative, simulated sample sizes may be chosen very large to induce asymptotic behavior of the χ2 statistic. However, how large a sample size should be chosen depends on the study design in question, and very large sample sizes also render the analyses computationally intensive.
Exact data simulation
Power calculations based on sufficient summary statistics are computationally relatively efficient to carry out. However, the actual feasibility of this type of power calculation depends on the number of distinct groups. If the number of groups is large (i.e., >100), it may be more convenient to carry out Monte Carlo based power calculations. We now introduce the concept of exact data simulation, which shares the virtues of the power studies based on summary statistics, but is more practicable given a large number of distinct groups.
The idea of exact data simulation is that data, which are randomly generated to begin with, can subsequently be transformed to fit the null-model H0 exactly. That is, first a data file is generated using a normal distribution quasi-random number generator. These data are then transformed, using a transformation proposed by Bollen and Stine (1993), so that the variance covariance matrix and means are exactly as specified under the model H0.
Assume a total sample size of N, and k distinct groups with known probability pk. Let Y denote the Nk × q data matrix for group k, where Nk is N*pk (possibly rounded to the nearest integer) and q is the number of variables. Let m denote the q × 1 vector of observed means and S = YtY/(N − 1)−mmt be the observed covariance matrix, Σ the expected covariance matrix implied by model H1, and μ the expected means vector implied by the model H1. Let S1/2 and Σ1/2 then denote the square root factorization of the positive definite matrices S and Σ such as given by a Cholesky factorization. It can then be shown that the covariance matrix and mean vector of data matrix Z, which is obtained through the following transformation of Y
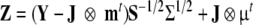 |
7 |
equals Σ and μ, exactly (Bollen and Stine 1993). In Eq. 7, J is a unit vector of length q, and ⊗ denotes the Kronecker product. This transformation allows one to create raw data for numerous groups that fit the null-model exactly. Consequently, when the null-model used for the generation of the data is fitted to these transformed data, all parameter values used for the simulation are recovered exactly. Let Log L0 and Log LA denote the maximum values of log-likelihood functions. The difference 2Log LA − 2Log L0 equals the non-centrality parameter λ.
Compared to multi-group power calculation with summary statistics, which becomes unpractical when the number of groups is large, the practicality of exact data simulation is unaffected by the number of groups in the analysis. However, one issue does require attention. When the number of groups is large (e.g., 256, see Illustration 1 below), the probability pk that a subject belongs to a group k may be relatively small. In order to calculate the Cholesky decomposition of the observed variance covariance matrix S, the number of observations in a group Nk should equal at least q + 1 (i.e., to ensure that S is positive definite and the Cholesky decomposition is possible). There are two ways to handle this problem. First, one can choose N to be sufficiently large that all groups, even those with small probabilities, by choosing a very large overall sample size N for the simulation. Power analyses based on this very large sample size produce non-centrality parameters which can subsequently be used to calculate power for other, more realistic sample sizes. Second, one can choose a smaller overall sample size, and accept that not all possible groups will be represented in the power calculations. This choice is usually justified since very small groups (e.g., including 2 subjects out of a possible 10,000) do not contribute much to the power. However, power calculations are more precise when all groups are represented in the simulation, i.e., overall N is large. Furthermore, it is in principle possible that the presence of all groups is required for model identification.
Note that packages like Mx and R will estimate the number of data vectors required for a power of e.g. 80%, given the proportion of subjects in each group. So while Nk ≥ q + 1 is required for exact simulation, Mx will return an overall sample size N, in which many groups may represented by fewer than q + 1 observations, which is in line with what one would expect to observe in research practice. Using the exact data simulation script subsequently to simulate data with the sample size advertised by Mx would not result in a power of 80% since the groups with Nk < q observations are not represented in the simulated data and thus do not contribute to the power.
Having discussed the concept of power, and the exact data simulation procedure, we will now illustrate the virtues of exact data simulation with three behavior genetics examples. We chose Mx to analyze the simulated data because of the program’s inbuilt option to calculate the sample size required for different power levels given the non-centrality parameter. However, the non-centrality parameter can also be obtained through other software (e.g., LISREL, Mplus). The calculation of sample sizes required for different power levels can then be done using other programs like R. The R-scripts used to simulate the data, and the Mx scripts used to analyze the data are available in the Mx scripts library.4 A small R-script for power computations based on non-centrality parameters can be downloaded from the library as well.
Illustration 1: multivariate ACE-model with data MCAR
Let us consider a four-variate ACE-model with data obtained from MZ and DZ twins (no additional family members). We assume a model with one common genetic factor, one common shared environmental factor, and specifics for A, C and E for all four traits. The model is illustrated in Fig. 3. Parameter values are chosen such that additive genetic influences, shared environmental influences, and non-shared environmental influences explain 50%, 30% and 20% of the total variance, respectively. Of the additive genetic variance, 60% is attributable to the common genetic factor (i.e., 30% of the total variance), and 40% to the specifics of A (i.e., 20% of the total variance). Of the shared environmental variance, 50% is attributable to the common shared environmental factor (i.e., 15% of the total variance), and 50% to the specifics of C (i.e., 15% of the total variance). Table 1 contains the correlation matrices for the MZ and DZ twins in the case that data are not missing. Means for all traits are equal to zero. We consider the power to reject the alternative hypothesis that the genetic specifics for all four traits explain not 20% but only 5% of the total variance,5 i.e., one common genetic factor is (almost) sufficient to explain all genetic variance and covariance in the four traits. If there are no missing data (situation S1), then one could simply use summary statistics to obtain power information, as this is a 2-group analysis. However, suppose we want to study the influence of data missing completely at random (MCAR) on the power to reject the hypothesis that all genetic specifics are zero. Here we consider two scenarios. First, we study the case that the probability of data being MCAR is 20% for all variables (situation S2). Given that we have q = 2 × 4 observations per family, this kind of missingness could yield 28 − 1 = 255 possible data patterns, i.e., 255 different groups (we discard the group in which all data are missing). In that case, power calculations using summary statistics are impractical, whereas exact data simulation is feasible. Note that some data patterns are rather unlikely, e.g., the probability of observing a valid observation for the first trait of the first twin only, while all other observations in the family are missing, is .8*(.27) = 1.024−05. Remember that the Cholesky decomposition cannot be calculated if the number of observations in a group Nk does not equal at least q + 1, so the simulated N needs to be very large if one wants all data patterns to be present in the simulated data set (about (q + 1)/1.024−05 ≈ 900,000). Yet, as very rare observations will hardly contribute to the power (5 of the 900,000 cases in the present example), one can just as well adopt a smaller sample size, and accept the fact that some groups (i.e., patterns of observations) will not be represented, and calculate power given the most likely patterns of observations.
Fig. 3.

Four-variate ACE-model with common factors for additive genetic and shared environmental effects, and specifics for A, C and E
Table 1.
Illustration 1: Four-variate cross-trait-cross-twin MZ correlations (below diagonal) and DZ correlation (above diagonal) for data without missingness
| Twin 1 | Twin 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Trait1 | Trait2 | Trait3 | Trait4 | Trait1 | Trait2 | Trait3 | Trait4 | ||
| Twin 1 | Trait1 | 1.00 | .45 | .45 | .45 | .55 | .30 | .30 | .30 |
| Trait2 | .45 | 1.00 | .45 | .45 | .30 | .55 | .30 | .30 | |
| Trait3 | .45 | .45 | 1.00 | .45 | .30 | .30 | .55 | .30 | |
| Trait4 | .45 | .45 | .45 | 1.00 | .30 | .30 | .30 | .55 | |
| Twin 2 | Trait1 | .80 | .45 | .45 | .45 | 1.00 | .45 | .45 | .45 |
| Trait2 | .45 | .80 | .45 | .45 | .45 | 1.00 | .45 | .45 | |
| Trait3 | .45 | .45 | .80 | .45 | .45 | .45 | 1.00 | .45 | |
| Trait4 | .45 | .45 | .45 | .80 | .45 | .45 | .45 | 1.00 | |
Note: Sample size N is not reported; as the simulations are exact, this correlation matrix should result independent of the sample size chosen for the simulations when data are not missing
Second, we study the case where the probability of data being MCAR is 40% for the first two variables, and zero for the second two (situation S3). This could be a realistic scenario in practice, for example, when a questionnaire study that measures two traits is extended during the data collection to include two additional traits, or when data from different studies are combined (one study in which all four traits were measured, while another study only included measurements of two traits for, e.g., economical reasons). Given that we have q = 2 × 4 observations per family of which only four variables show missingness, this would yield 24 = 16 possible data patterns, i.e., 16 different groups. With only 16 groups, power calculations using summary statistics would be feasible. However, one efficiently setup exact data simulation script can handle both this simple pattern of missingness, and more complex.
For the simulations we chose an overall sample size of 50,000 families (1/3 MZ, 2/3 DZ), which means that for situations S1 and S3, all groups (data patterns) are represented (50,000 and 49,999 cases simulated, respectively), while for situation S2, only the 163 most likely groups of the possible 255 are represented (49,999 cases simulated).
The three simulated data sets were subsequently analyzed in Mx. In the Mx-script, we specify different groups for the MZ and DZ twins. Because we use full information maximum likelihood to accommodate the missingness, we do not need to specify different groups for all possible missing data patterns. The Mx command ‘option power’ (α = .05, df = 4) was used to obtain an estimation of the total sample size that would be required for a power of 80%, given the current proportions of subjects in each group.
We find that for situation S1 (no missingness), 302 families are required for 80% power to reject the alternative hypothesis that the genetic specifics for all four traits explain 5% rather than 20% of the variance each, while for situations S2 (20% missingness for all variables) and S3 (40% missingness for only variable 1 and 2) the number of families required to obtain 80% power is estimated at 494 and 474, respectively. We hasten to note that these results are not informative for the case that data are missing at random (MAR), rather than MCAR (see Schafer and Graham 2002, for a comprehensive review on missingness and statistical procedures for handling missing data).
These power calculations took about 2 min for each situation S. Within the Monte Carlo framework, acquisition of similar power results would take at least T times as long for each situation S (where T is the number of replications one chooses to do). Given that the time required to write the data simulation script is equal for Monte Carlo simulation and exact simulation, it is clear that exact simulation saves a lot of time.
Illustration 2: gene by environment interaction with latent G and measured, categorical E
Gene by Environment (G × E) interaction is an important issue. From the perspective of the power study, a problem with the presence of G × E when the E is continuously distributed is that it renders single summary statistics insufficient; in the presence of G × E, (co)variances and means depend on the level of the environmental moderator, as we have seen in Eqs. 3–6. Purcell (2002) showed how G × E on the means and variances can be modeled if G is latent, and E is measured.
In power calculations in the G × E context, one can adopt a multi-group design, if the environmental moderator is categorical. For example, consider a classical ACE-twin design. If the environmental moderator is dichotomous (e.g., males versus females, young versus old, smoking versus non-smoking), the sample consisting of MZ and DZ twin pairs can be split up into twin pairs who are concordant with respect to the moderator (e.g., both twins do, or do not, smoke), and twin pairs who are discordant with respect to the moderator (only one of the twins in a pair smokes). With a dichotomous moderator and only two subjects per family, power calculations using summary statistics are feasible as there are only three distinguishable groups (not accounting for the distinction between MZ and DZ twins). However, suppose that you have measured an environmental moderator with 4 levels (coded 0, 1, 2, 3) in twin-pairs and their parents. With four persons per family and four possible moderator levels, there are 44 = 256 possible family configurations. In that case, multi-group analysis with summary statistics is impractical, and exact data simulation may be used instead.
For this illustration, parameters a, c, and e were all set to 1, such that the total variance equaled 3 (excluding moderating and main effects). The moderator, which was assumed independent of genotype in this illustration, was coded 0 to 3, such that the group with 0 on the moderator can be considered the baseline condition. The probability for all moderation levels was set to .25, and moderation levels were modeled as independent across family members (i.e., the probability for each family member’s moderation level was independent of the moderation levels of the other family members). Moderation in C, E and the means was fixed to 0, but the regression weight of the moderator was set to .2 for the additive genetic effects, such that the moderator explained 20% of the variance in the total population (i.e., 0%, 13%, 24%, and 34% of the variance, respectively, depending on the level of the moderator). The model is illustrated in Fig. 4. For the simulation, we chose an overall sample size N of 10,000 (1/3 MZ and 2/3 DZ twins).
Fig. 4.

Univariate ACE-model for parents and twin-offspring, including moderation on the variances and the means
Given these simulated data, we want to estimate the power to reject the alternative hypothesis that all moderator effects on the variances are zero (i.e., no G × E, or C × E, or E × E). In practice, one would fix all regression weights concerning the moderating effects on the variances (βa, βc, and βe) to zero at once, resulting in a test with 3 degrees of freedom.
The data were analyzed in Mx: different groups were specified for the MZ and DZ twins, and the moderator featured as a so-called definition variable. The ‘option power’ command (α = .05, df = 3) was again used to obtain an estimation of the total sample size that would be required for a power of 80%, given the current proportions of subjects in each group.
With the probability for all moderation levels fixed to .25, all 256 groups were represented in both the MZ and the DZ twins in the simulated data file (9,984 cases simulated), and for the chosen values, the analysis shows that we would need 165 families for a power of 80%. If we were to change the moderator level probabilities from .25 for every level to .4, .3, .2, and .1 for levels 0, 1, 2 and 3 respectively, then 179 and 225 groups would be represented in MZ and DZ twins, respectively (9,705 cases simulated). In that case, 206 families would be required for a power of 80% even though the moderator effect (βa) is unchanged.
These power calculations took at most 1 minute in total. Again, acquisition of similar power results would take at least T times as long within the Monte Carlo framework (where T is the number of replications). Assembling the data simulation script takes equally long for both types of simulation, so overall, exact simulation saves time.
Illustration 3: association for a tri-allelic locus with different allele frequencies
The aim of association studies is to determine whether genetic variation is associated with the risk for disease or the expression of a continuously distributed trait. Association studies may produce false positives, i.e., significant association in the absence of any true genetic effects. Population stratification is one source of false positives, i.e., the mixture of two populations with different allele frequencies and different phenotypic means. Fulker et al. (1999) showed that this type of spurious association can be avoided in a family-based study design. In this illustration, we focus on the situation in which data are available for pairs of siblings. Although this design allows for the simultaneous modeling of linkage and association, we limit the analysis to the association, but note that linkage information (i.e., IBD sharing estimation) could be included in exact data simulation scripts. For the present illustration, however, we assume that the locus under study is the QTL itself and not a marker in linkage disequilibrium with the QTL.
If a locus is diallelic, 22 = 4 genotypes can be distinguished: AA, AB, BA and BB (of course, in practice, there are only 2 + 1 = 3 distinguishable groups as AB and BA are the same, but when simulating the data exactly, it is convenient simply to treat them as different groups). These 22 genotypes give rise to (22)2 = 16 possible combinations of siblings (not accounting order), i.e., a 16 group analysis. Note that this is the simplest case: with 3 alleles, the number of possible sib-pairs is already (32)2 = 81, and when the locus under study is a polymorphic marker, with, say, 15 possible alleles, the number of distinguishable sib-pairs is (152)2 = 50,625. Clearly, multi-group analyses with sufficient summary statistics quickly become impractical as the number of alleles––or loci––increases.
We illustrate the use of exact data simulation in the context of the sib-pair association design, for a tri-allelic locus with alleles A, B, and C, with frequencies p, q, and r, respectively. The aim of this particular power calculation is to determine the influence of the allele frequencies on the power to detect a QTL. The biometrical model for a tri-allelic locus is summarized in Table 2. As with the more familiar diallelic case, the expected genotypic value 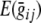 is assumed zero, so that everything is scaled in terms of deviations. In the case of three alleles, 2 genotypic values are distinguished, which were both fixed to .206, so that AA was associated with an increase of .206, BB with an increase of .206, and CC with a decrease of−.206 to .206 = −.412. Dominance was assumed to be absent, so the genotypic effect for the heterozygous genotypes AB, AC and BC was calculated as the mean of the effects of the homozygous groups. In the case of equal allele frequencies (p = q = r = 1/3), this QTL explains 2.5% of the variance (as determined using regular regression with the phenotype as dependent variable and genotype as predictor). Note that the variance explained by the QTL depends on the allele frequencies, so even though the genotypic values remain the same across all simulations, varying the allele frequencies affects the effect size of the QTL effect. For all simulations, background variance was decomposed such that additive genetic effects explained 30%, and unique environmental influences (E) explained 70% of the variance that remained after the QTL-effects was taken into account. Overall sample size N was fixed to 10,000 (note that because of rounding, the actual N modeled will not be equal to the overall sample size N of 10,000; see Table 3).
is assumed zero, so that everything is scaled in terms of deviations. In the case of three alleles, 2 genotypic values are distinguished, which were both fixed to .206, so that AA was associated with an increase of .206, BB with an increase of .206, and CC with a decrease of−.206 to .206 = −.412. Dominance was assumed to be absent, so the genotypic effect for the heterozygous genotypes AB, AC and BC was calculated as the mean of the effects of the homozygous groups. In the case of equal allele frequencies (p = q = r = 1/3), this QTL explains 2.5% of the variance (as determined using regular regression with the phenotype as dependent variable and genotype as predictor). Note that the variance explained by the QTL depends on the allele frequencies, so even though the genotypic values remain the same across all simulations, varying the allele frequencies affects the effect size of the QTL effect. For all simulations, background variance was decomposed such that additive genetic effects explained 30%, and unique environmental influences (E) explained 70% of the variance that remained after the QTL-effects was taken into account. Overall sample size N was fixed to 10,000 (note that because of rounding, the actual N modeled will not be equal to the overall sample size N of 10,000; see Table 3).
Table 2.
Expectations for a tri-allelic locus following the standard biometric model when dominance is assumed absent
| Genotype | AA | AB | BB | AC | BC | CC |
|---|---|---|---|---|---|---|
| Genotype frequency fij | p2 | 2pq | q2 | 2pr | 2qr | r2 |
| Genotypic value gij | x | (x + y)/2 | y | x + z/2 = −y/2 | y + z/2 = −x/2 | z |
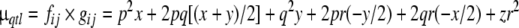 | ||||||
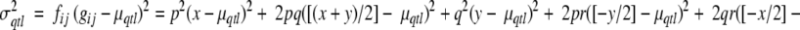 | ||||||
Note: p, q, and r denote the frequencies of alleles A, B and C, respectively; x is the genotypic value associated with genotype AA, y the genotypic value associated with genotype BB. As 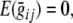 the genotypic value for genotype CC is z = −x − y (i.e., x + y + z = 0). μqtl denotes the expected contribution of the QTL to the population mean, and σ2qtl denotes the expected contribution of the QTL to the population variance (adapted from Falconer and Mackay 1996)
the genotypic value for genotype CC is z = −x − y (i.e., x + y + z = 0). μqtl denotes the expected contribution of the QTL to the population mean, and σ2qtl denotes the expected contribution of the QTL to the population variance (adapted from Falconer and Mackay 1996)
Table 3.
Results for illustration 3: Power calculations for sib-pair association with a tri-allelic locus with fixed genotypic values
| Frequencies alleles A, B, C | Effect size (%) | Actual N | Nr of groups represented | χ2(6) | Observed power | N required for power of 80% |
|---|---|---|---|---|---|---|
| .33/.33/.33 | 2.5 | 9,639 | 81 | 837.824 | 1 | 157 |
| .25/.5/.25 | 1.7 | 9,985 | 81 | 734.357 | 1 | 185 |
| .45/.45/.1 | .6 | 9,993 | 81 | 352.963 | 1 | 386 |
| .1/.45/.45 | 2.8 | 9,992 | 80 | 966.698 | 1 | 141 |
Note: Effect size is defined as % of variance explained by QTL; Actual N refers to actual number of sib-pairs in the analysis; Nr of groups represented refers to the number of groups, of the possible 81, that were represented in the analysis; χ2(6): the χ2-value of the test for association when the genotypic effects for all 6 distinguishable genotypes are fixed to zero; Observed power refers to the power observed for the modeled sample size N
Note that between and within effects were exactly equal (i.e., B = W) as we did not model population stratification; all between and within parameters can thus be fixed to be equal without loss of fit. The overall test for genetic association then involves fixing the genotypic effects for all 6 distinguishable genotypes (AA, AB, AC, BB, BC, and CC) to zero, i.e., 6 degrees of freedom.
The simulated sib-pair data were analyzed in Mx, using the ‘option power’ command (α = .05,df = 6) to obtain the sample size required for a power of 80%, given the current proportions of subjects in each group. The results presented in Table 3 show that the power to detect a QTL with certain genotypic values depends on the allele frequencies. As expected, the power is greatest when the frequency for the allele with the largest genotypic value (allele C) is highest.
With exact data simulation, these power calculations took about 2 min for each choice of allele frequencies. Again, it would take at least T times as long to obtain similar power results within the Monte Carlo framework (where T is the number of replications), while the time required to write the data simulation script takes equally long for both types of simulation.
Conclusion
In this paper we discussed a third method of power calculation, which can be useful when sufficient summary statistics are available in principle, but the number of possible groups is so large to render a multi-group analysis impractical. The illustrations presented in this paper represent only a few of the possible (behavior genetics) designs in which exact data simulation may prove useful. Other models for which exact data simulation can be used include random-effects models, latent growth curve models, simplex models, and (hierarchical) structural models, either or not in the context of genetics, just to name a few. Exact data simulation does not require more programming skills, or programming time, than Monte Carlo simulation, but one may save a lot of time analyzing the simulated data and calculating power, especially when one wishes to construct graphs of power vs. effect size.
In this paper, we used the Mx program to analyze the simulated data because of its inbuilt power calculation function. Another useful option of Mx in this context is the possibility to output individual likelihood statistics for each raw data group. This information can be used to identify the groups that contribute most to the power to detect the effects of interest. Of course, various other statistical software packages (e.g., QTDT, LISREL, MPlus, R) can also be used in combination with exact data simulation to obtain the non-centrality parameters required for power calculations.
We emphasize that the power results obtained through exact data simulation are exactly similar to power results obtained through the analysis of summary statistics, and, just like power calculation using summary statistics, asymptotically similar to results obtained through Monte Carlo simulation (depending on the number of runs used in Monte Carlo). Differences between those two customary method of power calculation and exact data simulation only occur when subgroups have very low probabilities and the simulated overall sample size is not large enough to include all possible groups to sufficient extent; these small groups may then not be represented in the exact power simulation, while they may be (more or less) represented in other methods. However, as stated previously, the ensuing differences with respect to the power results, are very small as such small groups hardly contribute to the power anyway. Even so, to avoid the exclusion of small groups, one should choose a sufficiently large overall sample size in exact simulation, such that all groups are represented. This is perfectly doable, and does not alter the practicability of the method as it still involves analyzing a single (yet larger) dataset. The non-centrality parameter obtained in the analysis of the large simulated data set can subsequently be used to calculate the power for smaller, more realistic sample sizes. Alternatively, one may decide to accept the absence of certain groups, and the implied slight underestimation of power. Happily, the discrepancy between the intended N and the realized N is simple to calculate (as demonstrated in the R script available in the Mx scripts library), so that one can readily obtain an impression of the implications of this decision.
Throughout the paper, we have assumed that the data, conditional on group, are normally distributed, so that sufficient statistics are in principle available. With respect to situations that preclude sufficient statistics, the present method may still have some use. For instance, a continuous moderator in a G × E model, as discussed by Purcell (2002), might be approximated by a 5 point or 7 point Likert scale, which would render exact simulation possible in principle (see illustration 2).
Finally we note that the extension of this method to discrete data would obviously be very useful, and does seem feasible.
Acknowledgements
Preparation of this manuscript was financially supported by NWO/MaGW VIDI-016-065-318, MH-65322, and DA-18673
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Note that typically, one will want to know whether the specifics can be discarded from the model altogether, i.e., whether the values of the specifics deviate significantly from zero. However, fixing variance parameters to zero, i.e., on the boundary of the parameter space, causes the null distribution of the test statistic T to follow a mixture of central χ2 distributions, rather than the usual central χ2(df) distribution (see e.g., Carey 2005; Dominicus et al. 2006). In determining the critical value given the choice of α, one would then have to refer to this mixture distribution, rather than to the central χ2(df). To keep things simple, we therefore chose to fix the variance parameter not to zero, but to a value much smaller than the actual value. If this value is not too close to the boundary, the null distribution of the test statistic T is the standard central χ2(df).
Edited by Stacey Cherny.
References
- Abecasis GR, Cardon LR, Cookson WOC (2000a) A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66:279–292 [DOI] [PMC free article] [PubMed]
- Abecasis GR, Cookson WOC, Cardon LR (2000b) Pedigree tests of transmission disequilibrium. Eur J Hum Genet 8:545–551 [DOI] [PubMed]
- Azzelini A (1996) Statistical inference based on the likelihood. Chapman and Hall, London
- Bollen KA (1989) Structural equations with latent variables. Wiley, New York
- Bollen KA, Stine RA (1993) Bootstrapping goodness-of-fit measures in structural equation models. In: Bollen KA, Long JS (eds) Testing structural equation models. Sage, Newbury Park, CA, pp. 111–135
- Boomsma DI, Dolan CV (1998) A comparison of power to detect a QTL in sib-pair data using multivariate phenotypes, mean phenotypes, and factor scores. Behav Genet 28(5):329–340 [DOI] [PubMed]
- Carey G (2005) Cholesky problems. Behav Genet 35(5):653–665 [DOI] [PubMed]
- Dolan CV, Boomsma DI, Neale MC (1999) A note on the power provided by sibships of sizes 2, 3, and 4 in genetic covariance modeling of a codominant QTL. Behav Genetics 29(3):163–170 [DOI] [PubMed]
- Dolan CV, van der Sluis S, Grasman R (2005) A note on normal theory power calculation in SEM with data missing completely at random. Struct Eq Model 12(2):245–262 [DOI]
- Dominicus A, Skrondal A, Gjessing HK, Pedersen NL, Palmgren J (2006) Likelihood ratio tests in behavior genetics: problems and solutions. Behav Genet 36(2):331–340 [DOI] [PubMed]
- Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th edn. Pearson Education Ltd., Essex, England
- Fulker DW, Cherny SS (1996) An improved multipoint sib-pair analysis of quantitative traits. Behav Genet 26:527–532 [DOI] [PubMed]
- Fulker DW, Cherny SS, Sham PC, Hewitt JK (1999) Combined linkage and association sib-pair analysis for quantitative traits. Am J Hum Genet 64:259–267 [DOI] [PMC free article] [PubMed]
- Heath AC, Eaves LC (1985) Resolving the effects of phenotype and social background on mate selection. Behav Genet 15(1):15–30 [DOI] [PubMed]
- Heath AC, Kendler KS, Eaves LC, Markell D (1985) The resolution of cultural and biological inheritance: informativeness of different relationships. Behav Genet 15(5):439–465 [DOI] [PubMed]
- Hewitt JK, Heath AC (1988) A note on computing the chi-square noncentrality parameter for power analysis. Behav Genet 18:105–108 [DOI] [PubMed]
- Martin NG, Eaves LC, Kersey MJ, Davies P (1978) The power of the classical twin design. Heredity 40:97–116 [DOI] [PubMed]
- McLachlan G, Peel D (2002) Finite mixture models. Wiley, New York
- Muthén, LK, Muthén, BO (1998–2007) Mplus User’s guide, 4th edn. Muthén & Muthén, Los Angeles, CA
- Neale MC, Eaves LJ, Kendler KS (1994) The power of the classical twin study to resolve variation in threshold traits. Behav Genet 24:239–258 [DOI] [PubMed]
- Neale MC, Boker SM, Xie G, Maes HH (2003) Mx: statistical modeling, 6th edn. Department of Psychiatry, Richmond, VA
- Posthuma D, Boomsma DI (2000) A note on the statistical power in extended twin designs. Behav Genet 30(2):147–158 [DOI] [PubMed]
- Purcell S (2002). Variance components models for gene-environment interaction in twin analysis. Twin Res 5:554–571 [DOI] [PubMed]
- Purcell S, Sham P (2002) Variance components models for gene-environment interaction in quantitative trait locus linkage analysis. Twin Research 5(6):572–576 [DOI] [PubMed]
- Purcell S, Cherny SS, Sham PC (2003) Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19(1):149–150 [DOI] [PubMed]
- Saris WE, Satora A (1993) Power evaluations in structural equation models. In: Bollen KA, Long JS (eds) Testing structural equation models. Sage, Newbury Park, CA, pp 181–204
- Satorra A, Saris WE (1985) The power of the likelihood ratio test in covariance structure analysis. Psychometrika 50:83–90 [DOI]
- Schafer JL, Graham JW (2002) Missing data: our view of the state of the art. Psychological Methods 7:147–177 [DOI] [PubMed]
- Sham PC, Cherny SS, Purcell S, Hewitt JK (2000) Power of Linkage versus Association Analysis of Quantitative Traits, by Use of Variance-Components Models, for Sibship Data. Am J Hum Genet 66:1616–1630 [DOI] [PMC free article] [PubMed]
- Sham P, Purcell S, Cherny SS, Abecasis GR (2002) Powerful regression-based quantitative-trait linkage analysis of general pedigrees. Am J Hum Genet 71:238–253 [DOI] [PMC free article] [PubMed]
- Van den Oord EJCG (1999) Method to detect genotype-environment interactions for quantitative trait loci in association studies. Am J Epidemiol 150(11):1179–1187 [DOI] [PubMed]
- Van der Sluis S, Dolan CV, Neale MC, Posthuma D (2007) A general test for gene-environment interaction in family-based association analysis of quantitative traits. Manuscript under revision [DOI] [PMC free article] [PubMed]