

Abstract
Inherited defects in signaling pathways downstream of the insulin receptor have long been suggested to contribute to human Type 2 diabetes mellitus. Here we describe a mutation in the gene encoding the protein kinase AKT2/PKBβ in a family that shows autosomal dominant inheritance of severe insulin resistance and diabetes mellitus. Expression of the mutant kinase in cultured cells disrupted insulin signaling to metabolic end-points and inhibited the function of co-expressed, wild type AKT. These findings demonstrate the central importance of AKT signaling to insulin sensitivity in humans.
Most forms of diabetes are likely to be polygenic in origin, although a number of monogenic forms are being recognised (1, 2). Although rare, these monogenic examples offer insights into the function of the affected gene in humans as well as offering important clues to understanding more common forms.
We have been screening genomic DNA from 104 unrelated subjects with severe insulin resistance for mutations in genes that are implicated in insulin signalling. We identified a missense mutation in the serine/threonine kinase gene AKT2 in one Caucasian proband. AKT2 (also known as PKBβ) is highly expressed in insulin sensitive tissues and is activated in response to growth factors and related stimuli (3, 4) a process that requires its phosphorylation by the phosphoinositide-3 phosphate-dependent kinase activities designated PDK1 and PDK2 (3). The proband, (iii)/1 (Fig. 1D), is a non-obese 34 year old female who developed diabetes mellitus at 30 years of age. The proband, her non-obese mother, (ii)/2, maternal grandmother, (i)/2, and a maternal uncle, (ii)/3, were all heterozygous for a G to A substitution predicted to result in an R to H substitution at amino acid 274 (Fig. 1 A, B). All were markedly hyperinsulinemic (Table S1) and the mother and maternal grandmother developed diabetes mellitus in their late 30′s. Three other first-degree relatives available for study were all clinically normal with normal fasting glucose and insulin and were homozygous for the wild-type AKT2 sequence (Fig. 1D and Table S1). This mutation was not found in genomic DNA of 1500 Caucasian control subjects from the UK.
Fig. 1.

Detection of a non-conservative heterozygous mutation in AKT2 that co-segregates with severe insulin resistance. (A) Direct sequencing of genomic DNA from the proband, subject (iii)/1 (right) and a control subject (left). Asterisk indicates the heterozygous G to A substitution that produces H274. (B) Location of R274 in relation to known functional domains and phosphorylation sites required for activation of AKT2. (C) R274 (in red and marked *) is highly conserved across different AKT isoforms and diverse species. (D) Family pedigree demonstrating co-segregation of clinical phenotype (also see Table S1) with the R274H mutation. All family members heterozygous for the mutation (+/-) are hyperinsulinemic and 3 of 4 have diabetes mellitus. All wild type subjects (+/+) are normoinsulinemic and non-diabetic. Red denotes fasting hyperinsulinemia. Green denotes Diabetes Mellitus.
R274 forms part of an RD sequence motif within the catalytic loop of the AKT2 kinase domain that is invariant in AKT isoforms in all species, and is also highly conserved within the protein kinase family (Fig. 1C) (5). The RD motif includes the invariant D residue (D275 of AKT2) that performs an essential catalytic function in all protein kinases.
R274 is positioned in the core of the catalytic domain, forming critical hydrogen bonds with the phosphate moiety of phosphoT309 in the activation segment permitting correct positioning the substrate peptide relative to the catalytic base and adenosine triphosphate (ATP) (6). A model of the mutant protein AKT2H274 (Fig. 2A) indicates that this mutation would disrupt the conformation of both the activation segment and the catalytic loop, misaligning the substrate peptide relative to catalytic residues and ATP and hence ablating catalytic activity. Consistent with these predictions, unlike the wild type AKT2, AKT2H274 was unable to phosphorylate a peptide substrate based on the AKT target sequence of glycogen synthase kinase 3 (GSK3) in an in vitro kinase assay (Fig. 2B). To examine the effect of the R274H mutation on the signalling ability of AKT2 we studied the regulation of the FOXA2 transcription factor, a substrate of AKT2 (7). FOXA2 activity is inhibited by phosphorylation, which leads to its exclusion from the nucleus. Treatment of HepG2 human liver cells with insulin induced the translocation of endogenous FOXA2 from the nucleus to the cytosol (Fig 3A). Overexpression of wild type AKT2 mimicked this effect (Fig 3A). In contrast, in cells transfected with mutant AKT2H274 FOXA2 remained entirely nuclear (Fig 3A). Consistent with this, although co-transfected wild type AKT2 ablated FOXA2-mediated transcription from the phosphoenolpyruvate carboxykinase (PCK1) promoter in HepG2 cells, AKT2H274 had no effect (Fig 3B). AKT2H274 also prevented wild type AKT2 from inhibiting FOXA2 driven transcription from the PCK1 promoter in a dose-dependent manner, when both AKT2 proteins were co-expressed (Fig 3C). In the same assay AKT2H274 also inhibited the effect of wild type AKT1, which also contributes substantially to AKT activity in the liver (Fig. 3D) (8).
Fig. 2.
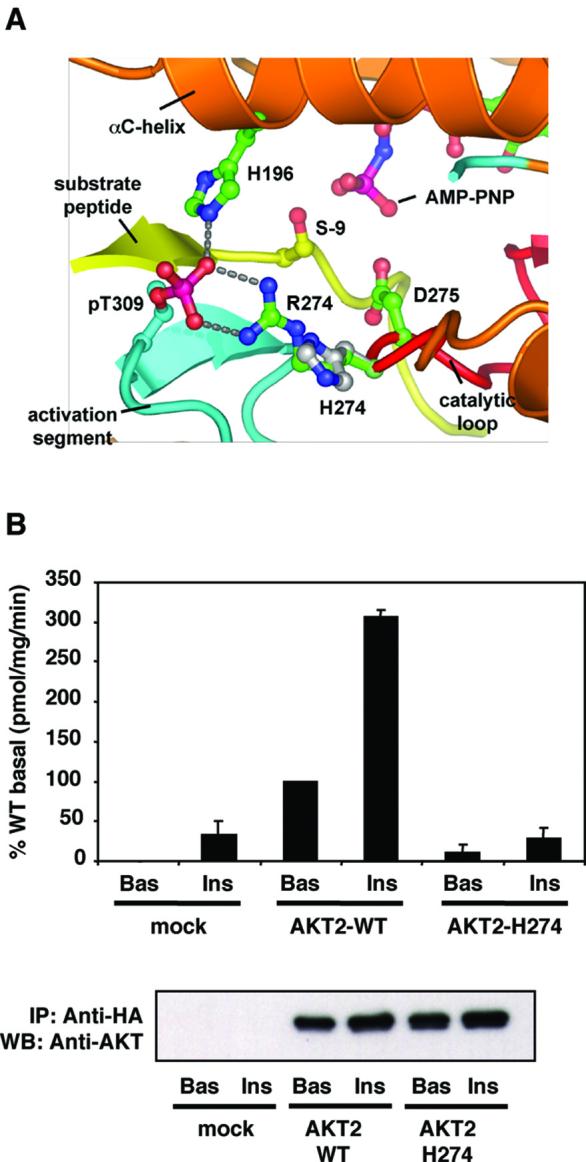
The substitution of R274 by histidine disrupts the kinase domain and abolishes AKT2 kinase activity. (A) Proposed effects of R274H on a structural model of the AKT2 kinase domain. In the wild type protein, R274 contacts phosphoT309 (pT309), organising the activation segment to place the substrate peptide correctly for catalysis. Substitution of H for R274 is predicted to disrupt the conformation of both the activation segment and catalytic loops. (B) HA-AKT2 and HA-AKT2H274 were immunoprecipitated from lysates of appropriately transfected CHO-T cells and enzyme activity was determined by an in vitro kinase assay (upper panel). Mock-transfected cells received empty vector only. Cells were treated without (Bas) or with (Ins) 100nM insulin for 10 min prior to lysis. Data are means±SEM of 4 experiments, Crosstide was used as the substrate. Duplicate immunoprecipitates were also immunoblotted with anti-AKT2 to demonstrate equal expression of the wild type and mutant proteins (lower panel).
Fig. 3.

Functional properties of wild type AKT2 and AKT2H274 in cultured human liver and rodent fat cells (A) HepG2 cells were treated in the absence (panel i) or presence (panel ii) of 50nM insulin, fixed and probed with anti-FOXA2 antibodies to determine intracellular localization (7). The same assay was performed with cells transfected with wild type AKT2 (panel iii) or AKT2H274 (panel iv). Scale bars (40μm each) are shown in white. (B) Luciferase activity was determined in extracts of HepG2 cells transfected with pPCK1 reporter construct with or without FOXA2 in the absence or presence of wild type AKT2 or AKT2H274 (ns denotes no significant difference). Alternatively HepG2 cells were transfected with pPCK1 reporter construct with or without FOXA2 and either wild type AKT2 (125ng) (C) or wild type AKT1 (100ng) (D). In each case AKT2H274 was co-transfected in increasing quantities as indicated. * indicates significant difference from activity in cells transfected with FOXA2 alone (p<0.05). † denotes no significant difference in this comparison. In all cases data are means±SD of four experiments and all data was normalized to co-expressed β-galactosidase activity. (E) 3T3-L1 preadipocytes were stably transfected with empty vector (mock), wild type AKT2 or AKT2H274. 2-day post confluent cells (Day 0) or cells induced to differentiate for 12 days were fixed and stained with oil red O to assess lipid accumulation. Images of day 12 differentiated cells were also obtained by light microscopy (lower panels) and scale bars (200μm each) are shown in white. (F) Lysates were prepared from cells transfected with empty vector (E) wild type AKT2 (W) of AKT2H274 (M) at 2 days post-confluence (Day 0) or at various intervals up to day 12 post-induction of differentiation. Day 0 samples were immunoblotted for AKT2 expression and all lysates were immunoblotted for aP2 expression.
AKT overexpression in preadipocytes augments differentiation to adipocytes (9). We therefore examined the effect of AKT2H274 on adipocyte differentiation. 3T3-L1 mouse preadipocytes overexpressing wild type AKT2 showed increased accumulation of lipid during adipogenesis (Fig. 3E), whereas cells overexpressing the mutant AKT2H274 showed markedly decreased lipid accumulation. The fatty acid transport protein aP2 is a well-defined marker of adipogenesis (10). Expression of aP2 was augmented by expression of wild type AKT2 but reduced by expression of AKT2H274 (Fig. 3F). Thus AKT2H274 may also exert dominant-negative effects over endogenous AKT proteins during adipocyte differentiation.
To allow closer examination of the in vivo consequences of the AKT2H274 mutation the proband underwent a two-step hyperinsulinemic/euglycemic clamp. This revealed extreme insulin resistance (Fig. S1), with the glucose infusion rate remaining very low even in the second step despite an insulin concentration of 7346pmol/l. Examination of the effects of insulin on hepatic glucose production and peripheral glucose use revealed severe insulin resistance in this subject both in the liver and peripheral tissues (supporting online text).
Analysis of the proband’s body composition revealed a -35% difference in total body fat compared to that predicted for her height and weight, consistent with both the ability of AKT2H274 to impair adipogenesis and the recently reported observation that AKT2 knock-out mice develop lipoatrophy as they age (11).
This family provides an example of a monogenic inherited defect in post-receptor insulin signaling leading to human insulin resistance and diabetes mellitus. Genetic variants in IRS1 and PIK3R1 have been previously reported in subjects with insulin resistance and/or type 2 diabetes mellitus but in no case have they clearly segregated with insulin resistance in a pedigree or seriously disrupted signal transduction (12, 13). Dominant negative mutations in peroxisome proliferator-activated receptor gamma (PPARγ) are associated with autosomal dominant severe insulin resistance and diabetes mellitus. However, the mechanisms whereby they result in insulin resistance are unclear and are unlikely to be due to simple impairment of insulin signal transduction (14).
AKT2 (-/-) mice show resistance to insulin’s effects on glucose metabolism in muscle and liver and a subset of these mice go on to develop frank diabetes (11, 15). In view of the abnormal fat distribution in our proband, it is of interest that atrophy of adipose tissue with age has been described in one strain of these mice (11, 15). Notably, AKT2 (-/+) mice show little metabolic phenotype and, even in (-/-) animals, the degree of insulin resistance is only moderate. This contrasts with the extreme hyperinsulinemia and insulin resistance seen in humans heterozygous for the AKT2H274 mutation. This may result from this mutant inhibiting other co-expressed AKT isoforms in a dominant-negative manner or from interference with other functions of upstream kinases such as PDK1 Apart from lipodystrophy, there are no other overt structural or functional alterations in AKT2-expressing tissues from patients with this mutation. This suggests that, at least in those tissues where AKT2 is highly expressed, insulin signal transduction represents the major role for AKT. Frank diabetes mellitus developed in three of the four human carriers of the AKT2 mutation. In the fourth, a middle aged male, marked hyperinsulinemia occurred simultaneously with normal glucose tolerance. Moreover, in our proband, severe hyperinsulinemia preceded diabetes by many years. Although we cannot exclude an effect of the AKT2 mutation on beta-cell function it is clear that the major effect of this mutation was on insulin action.
Germline loss-of-function mutations in genes encoding intracellular signaling kinases are being increasingly recognized as causes of human inherited disease. Thus, JAK3 mutations cause Severe Combined Immunodeficiency (16), RPS6KA3 mutations cause Coffin Lowry syndrome (17) and WNK4 mutations cause an inherited form of hypertension (18). The kindred described here demonstrate that AKT2 can be added to this list, the R274H mutation in this enzyme causing a rare form of human diabetes due to a post-receptor defect in insulin signaling. Importantly, whilst AKT2 mutations are unlikely to explain most common forms diabetes, this mutant uniquely demonstrates the critical role of AKT signaling in maintaining insulin sensitivity in humans.
Supplementary Material
Acknowledgments
Supported by the Wellcome Trust (SOR, JJR, MS, JCW, DBD, AMU), the UK MRC (SG, PRM), the Canadian NSERC (SLG), the Deutsche Forschungsgemeinschaft (SS), the Raymond and Beverly Sackler Foundation (SG), CR-UK (DB) and Diabetes UK (RW). We are also grateful to D. Alessi for AKT2 cDNA and B. Hemmings for helpful discussions.
References and notes
- 1.Gloyn AL, McCarthy MI. Best Pract. Res. Clin. Endocrinol. Metab. 2001;15:293. doi: 10.1053/beem.2001.0147. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy MI, Hattersley AT. Expert Rev. Mol. Diagn. 2001;1:403. doi: 10.1586/14737159.1.4.403. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Alessi DR. Biochem. J. 2000;346(Pt 3):561. [PMC free article] [PubMed] [Google Scholar]
- 4.Brazil DP, Hemmings BA. Trends Biochem. Sci. 2001;26:657. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 5.Johnson LN, Noble ME, Owen DJ. Cell. 1996;85:149. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, et al. Nat. Struct. Biol. 2002;9:940. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 7.Wolfrum C, Besser D, Luca E, Stoffel M. Proc. Natl. Acad. Sci. U S A. 2003;100:11624. doi: 10.1073/pnas.1931483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker KS, et al. Biochem. J. 1998;331(Pt 1):299. doi: 10.1042/bj3310299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. J. Biol. Chem. 1996;271:31372. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 10.Rangwala SM, Lazar MA. Annu. Rev. Nutr. 2000;20:535. doi: 10.1146/annurev.nutr.20.1.535. [DOI] [PubMed] [Google Scholar]
- 11.Garofalo RS, et al. J. Clin. Invest. 2003;112:197. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedersen O. Exp. Clin. Endocrinol. Diabetes. 1999;107:113. doi: 10.1055/s-0029-1212085. [DOI] [PubMed] [Google Scholar]
- 13.Baynes KC, Whitehead J, Krook A, O’Rahilly S. Q.J.M. 1997;90:557. doi: 10.1093/qjmed/90.9.557. [DOI] [PubMed] [Google Scholar]
- 14.Barroso I, et al. Nature. 1999;402:880. [Google Scholar]
- 15.Cho H, et al. Science. 2001;292:1728. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 16.Vihinen M, et al. Clin. Immunol. 2000;96:108. doi: 10.1006/clim.2000.4880. [DOI] [PubMed] [Google Scholar]
- 17.Trivier E, et al. Nature. 1996;384:567. doi: 10.1038/384567a0. [DOI] [PubMed] [Google Scholar]
- 18.Wilson FH, et al. Proc. Natl. Acad. Sci. U S A. 2003;100:680. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.