

Abstract
Previously, we have shown that endothelial cell chemotaxis to the proangiogenic chemokine MIP-2 (macrophage inflammatory protein-2), is much greater in mouse aortic endothelial cells (EC) than pulmonary arterial endothelial cells (PA EC). This was true despite the observation that both cell types display comparable levels of the ligand receptor, CXCR2 (8). Since the systemic arterial circulation is proangiogenic in the adult lung and the pulmonary circulation is relatively resistant to neovascularization, we questioned whether the observed functional heterogeneity is related to inherent differences in cell signaling cascades of the two EC subtypes. Specifically, we measured activation of Rac1 and RhoA, both thought to be involved in EC cell migration. Rac1 showed inconsistent and minimal changes in both cell types after MIP-2 treatment (p>0.05). However, activated RhoA was increased upon exposure to MIP-2 only in aortic EC (61% increase; p<0.05). Decreased RhoA activation after treatment of aortic EC with specific siRNA for RhoA resulted in a functional decrease in EC chemotaxis to MIP-2 (17% increase; p<0.05). Additionally, increased RhoA activation in PA EC with adenoviral infection of RhoA caused an increase in PA EC chemotaxis to MIP-2 (46% increase; p<0.05). Inhibition of RhoA activity with the Rho kinase inhibitor, Y27632 blocked aortic EC chemotaxis and stress fiber formation. Thus, RhoA activation is increased after MIP-2 treatment in mouse aortic endothelial cells but not in pulmonary artery endothelial cells. We conclude that RhoA is part of a signaling pathway essential for aortic cell migration after CXCR2 ligation. This result provides one explanation for the difference in chemotaxis observed in these two endothelial subtypes that express similar levels of CXCR2.
Keywords: angiogenesis, endothelium, chemokines
INTRODUCTION
ELR+(Glu-Leu-Arg) CXC chemokines are potent promoters of angiogenesis and have been shown to induce chemotaxis in endothelial cells (4, 8, 10, 13). In mice, macrophage inflammatory protein-2 (MIP-2; CXCL2) is one of the predominant pro-angiogenic ELR+ CXC chemokine ligands, which binds to the G-protein coupled receptor, CXCR2 (13). Angiogenesis in the lung is largely a systemic vascular event and the pulmonary circulation does not participate (7). This heterogeneity in endothelial potential has not been explained. We previously have shown that endothelial cell chemotaxis, an essential component of angiogenesis, was markedly enhanced in mouse systemic arterial endothelial cells compared to pulmonary artery endothelial cells in response to MIP-2 (8). Interestingly, however, we measured equivalent levels of CXCR2 expression between aortic and pulmonary endothelial cell subtypes. Thus, enhanced chemotaxis of systemic arterial endothelial cells in the presence of MIP-2 could not be attributed to increased receptor expression and likely depends on inherent differences in intracellular events after receptor ligation.
Numerous studies have focused on defining intracellular signaling cascades leading to cytoskeletal reorganization and subsequent chemotaxis in a variety of cell types (2). However, few studies have focused specifically on intracellular signaling events after CXCR2 ligation that leads to a pro-angiogenic, endothelial cell phenotype. Schraufstatter and colleagues demonstrated that RhoA and Rac1, members of the Rho family of monomeric GTPases, control chemotaxis in human systemic microvascular endothelial cells after ligand binding to CXCR2 (10). Hoang and colleagues identified RhoA activity as a critical regulator of endothelial cell assembly into new blood vessels (3). Given the differences in angiogenic potential between systemic and pulmonary arterial endothelial cells and MIP-2 induced chemotaxis, we questioned whether there exists basic differences in activation of signaling proteins. Specifically, we hypothesized that MIP-2 induced chemotaxis of arterial endothelial cells requires activation of RhoA and/or Rac1, and that this activation is much greater in systemic arterial cells than in pulmonary artery endothelial cells. Our results confirm significant heterogeneity in RhoA activation between the two endothelial cell subtypes.
METHODS
Isolation of mouse aortic & pulmonary artery endothelial cells
The aorta and pulmonary artery from C57Bl/6 mice (n=8) were dissected and placed with the intima side down on Matrigel-coated 35 mm tissue culture dishes. After 4-6 days, endothelial cells that had migrated were replated to gelatinized T25 culture flasks. Cells were cultured in DMEM supplemented with 20% FCS, 15 μg/ml ECGS, 100 μg/ml penicillin/streptomycin, 0.25 μg/ml amphotericin B, and 0.1 mM MEM with non-essential amino acids. Endothelial cell phenotypes were confirmed by immunostaining techniques for PECAM, vWF, and uptake of Dil-ac-LDL. Only cells with positive staining were used for further experiments. All experiments were carried out using endothelial cells between passages 2-10.
Endothelial Cell Chemotaxis Assay
Polycarbonate filters (5 μm pore size, Corning Costar, Cambridge, MA) were coated with 0.2% gelatin. Endothelial cells were detached with 2 mM EDTA/PBS, washed and resuspended in DMEM with 0.1% fatty acid free BSA. Endothelial cells (105) were added to the upper chamber of transwells (Corning Inc, Corning, NY) and incubated (37°C). After 1h, 10 ng/ml MIP-2 was placed in the lower chamber and incubated for 2 hours. This concentration of MIP-2 was selected based on our previous work, which showed an intermediate chemotactic response in aortic endothelial cells (8). Control conditions for all experiments were cells treated with vehicle (PBS) for a similar duration as agonist treatment. Non-migrated cells were removed and migrated cells were fixed then stained (Diff-Quik stain set; Dade Behring, Newark, DE). Membranes were mounted on slides and migrated cells were counted under a microscope (10x objective) in 5 fields across each membrane and averaged.
Western Blot Analysis
Monolayer cells were stimulated and lysed in Laemmli buffer. Cell lysates were resolved by SDS-PAGE, transferred to nitrocellulose membrane, blocked with 5% BSA in TBS-tween and exposed to specific primary antibodies for each experiment. Total RhoA and Rac1 protein and RhoA and Rac1 activity were measured using kits from Upstate Biotechnology (Lake Placid, NY). RhoA and Rac1 activity were measured using rhotekin-RBD and PAK-1 PBD, respectively. These proteins possess binding domains that specifically pull down activated RhoA (Rho-GTP) and Rac1 (Rac1-GTP). After cells were treated and cell lysates were collected, Rho-GTP or Rac1-GTP were precipitated using rhotekin-RBD or PAK-1 PBD immobilized on agarose. Affinity precipitated RhoA, and Rac1 proteins as well as total RhoA and Rac1 were resolved by SDS-PAGE and detected by Western blotting. Each experiment was repeated in multiple different cell isolates. Immunoblots were quantified by densitometry using Un-Scan-It Gel software (Silk Scientific, Orem, UT) and compared to vehicle controls within the same membrane. In preliminary experiments the time course of RhoA activation was assessed at 15, 30, and 45 min. Since the maximum activation of RhoA was complete by 30 min, all subsequent studies were performed for 30 min. Rac1-GTP activation was measured at 5, 15, and 30 min after MIP-2.
Silencing of Endogenous RhoA
Predesigned RhoA-specific small interfering RNA (RhoA/siRNA) of standard purity was purchased from Ambion (Austin, TX) in purified, desalted, deprotected, and annealed double-strand form. The 2 following 21-bp duplexes of siRNA were used: 1) sense 5′-GCAGGAFCCGGUAAAACCUtt-3′ and anti-sense 5′-AGGUUUUACGGCUCCUGCtt-3′ ,2) sense 5′-GGAAGAUUAUGACCGCCUGtt-3′ anti-sense 5′-CAGGCGGUCAUAAUUCCtg-3′. GAPDH siRNA (GAPDH/siRNA; Ambion) was used as a control treatment. Mouse aortic EC were grown to 70% confluence, and the transfection of siRNA (final concentration, 100 nmol/L) was performed using siPORT Amine transfection reagent according to the manufacturer’s protocol (Ambion). Forty-eight hours after transfection, the cells were used for experimentation.
Adenoviral infection
Recombinant adenovirus containing the constitutively active RhoA L63 was purchased from Cell Biolabs (San Diego, CA). Recombinant adenovirus containing beta-galactosidase (adß-gal; Cell Biolabs) was used as a control. PA EC at 70% confluence were infected with adenoviruses at a multiplicity of infection = 100 pfu/cell in DMEM with 2% FBS for 15h. Following washing (DMEM with 20% FBS) cells were incubated for an additional 24 h and used for experimentation.
Inhibitor studies
To further confirm the RhoA pathway, an inhibitor of Rho kinase, a downstream effector of RhoA was used in histologic and functional assays. Monolayer aortic EC were serum-starved for 1hr and then incubated for 30 min with the Rho kinase inhibitor-Y27632 (5 μM, EMD Biosciences, San Diego, CA) followed by 30 min treatment with 10 ng/ml MIP-2. This inhibitor concentration was selected based on the work of others (5). Aortic EC were treated for fluorescence microscopy or counted for chemotaxis.
Fluorescence staining
Aortic EC grown on glass coverslips were fixed (3.7% formaldehyde solution in PBS for 10 min) after agonist treatment (30 min with 10 ng/ml MIP-2), washed with PBS, permeabilized (0.2% triton X-100 for 15 min at room temperature), and blocked (2% BSA in PBS-Tween for 30 min). Actin filaments were stained with Texas Red-conjugated phalloidin (Invitrogen, Carlsbad, California) for 1h at room temperature. Images were acquired with an Olympus IX51 inverted fluorescence microscope (Olympus, Melville, NY) using a 60x oil objective and a digital camera.
Statistical analysis
Differences in outcomes were evaluated by one-way ANOVA. Relevant within group comparisons were made using Fisher’s test for Least Significant Differences. A p value ≤ 0.05 was accepted as significant.
RESULTS
Results were obtained from 8 separate isolations of mouse endothelial cells. No significant differences between aortic and pulmonary endothelial cells were observed in MIP-2 induced activation of Rac1. MIP-2 caused a small, variable (0-30%) response in Rac1 activation in both cell types over the course of 5-30 min (n=4 experiments/cell type). Figure 1 shows a representative immunoblot of activated Rac-1 (Rac1-GTP) and total Rac1 protein under control conditions and after 30 min of MIP-2 treatment. A comparison of the densitometric quantification of immunoblots is presented in Figure 1B. Minimal activation and no difference between cell types were observed (p>0.05). Because of this lack of difference between cell types, all subsequent experiments focused on RhoA. Figure 2A shows representative immunoblots of activated RhoA (Rho-GTP) and total RhoA protein in aortic EC and PA EC under control conditions and 30 min after MIP-2 treatment. A comparison of the densitometric quantification of immunoblots is presented in Figure 2B. MIP-2 caused a significant increase in Rho-GTP only in aortic EC (161% of control; p=0.008). Rho-GTP in PA EC was unaffected by MIP-2 treatment. The increase in Rho-GTP in aortic EC was significantly greater than in PA EC (p=0.002; n=5 experiments/cell type). Total basal RhoA protein normalized to GAPDH did not differ between aortic EC and PA EC (0.69 vs 0.63 arbitrary units, respectively; n=3).
Figure 1.
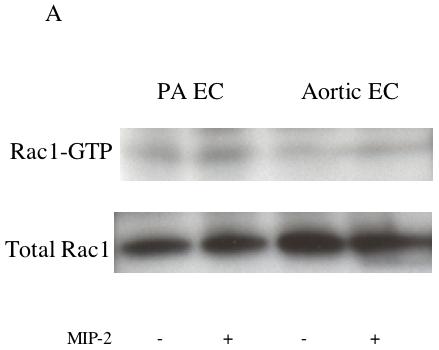
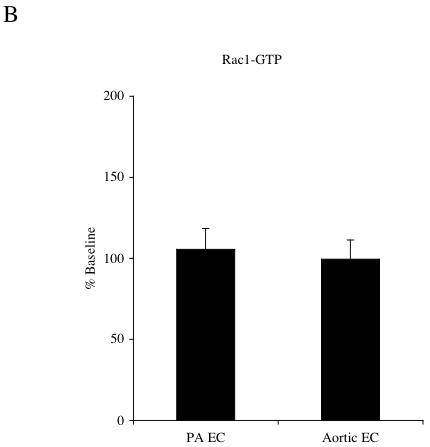
A: Representative immunoblots of activated Rac1-GTP and total Rac1 protein in pulmonary artery endothelial cells (PA EC) and aortic endothelial cells (EC) without (-) and with (+) MIP-2 (10 ng/ml) after a 30 min treatment. B: Average increase in Rac1-GTP after MIP-2 relative to baseline control. MIP-2 had no effect on Rac1-GTP in either cell type (p > 0.05; n=4 experiments / cell type).
Figure 2.
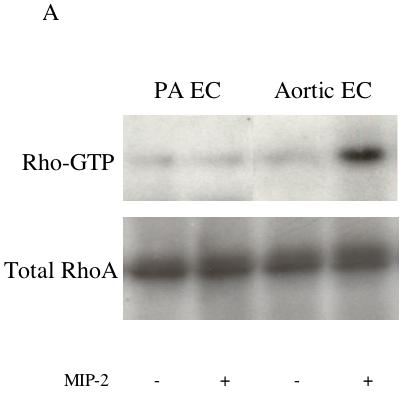
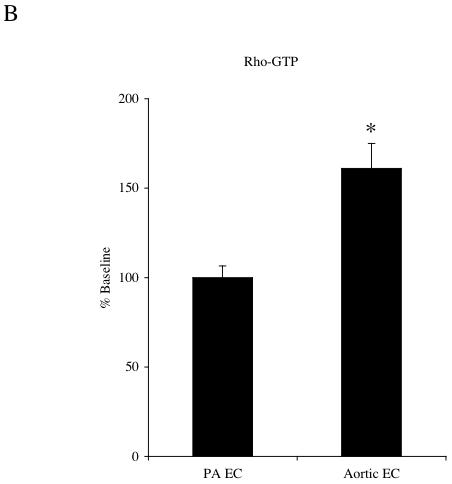
A: Representative immunoblots of activated Rho-GTP and total RhoA protein in pulmonary artery endothelial cells (PA EC) and aortic endothelial cells (EC) without (-) and with (+) MIP-2 (10 ng/ml) after a 30 min treatment. B: Average increase in RhoA-GTP after MIP-2 relative to baseline control. MIP-2 caused a significant increase in Rho-GTP only in aortic EC (* indicates p = 0.008). Rho-GTP in PA EC was unaffected by MIP-2 treatment (n=5 experiments / cell type).
To confirm that MIP-2 caused physical changes in cell morphology, actin stress fiber formation was evaluated by staining aortic EC with Texas Red-conjugated phalloidin. Figure 3A shows normal endothelial cell architecture with cortical actin staining while Figure 3B shows the dramatic appearance of numerous actin stress fibers after cell treatment with MIP-2. When aortic endothelial cells were pretreated with a Rho kinase inhibitor prior to MIP-2, actin stress fibers were not obvious and only peripheral cortical actin staining was observed (Figure 3C). Correlate functional assessment of MIP-2 induced responses are shown in Figure 4. Aortic EC chemotaxis is shown in response to MIP-2 treatment with (n=4) /without (n=3) the Rho kinase inhibitor Y27632. A significant increase in the number of migrated cells was apparent after MIP-2 treatment (p< 0.05). However, there was no change in the number of migrated cells after MIP-2 if the cells were pretreated with the Rho kinase inhibitor (p > 0.05 from baseline control).
Figure 3.
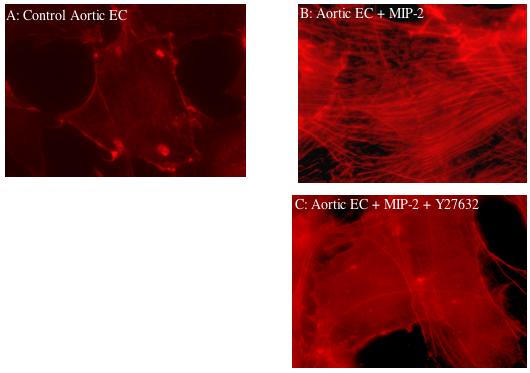
Fluorescence staining (Texas Red-conjugated phalloidin) of actin filaments in aortic endothelial cells; 3A: quiescent control aortic endothelial cell (EC); 3B: aortic endothelial cell after MIP-2 treatment (10 ng/ml; 30 min); note appearance of stress fibers; 3C: aortic endothelial cell pretreated with Y27632 (Rho kinase inhibitor; 30 min) prior to MIP-2 treatment (10 ng/ml; 30 min). Note disappearance of actin stress fibers when cells were pretreated with inhibitor. 600x magnification
Figure 4.
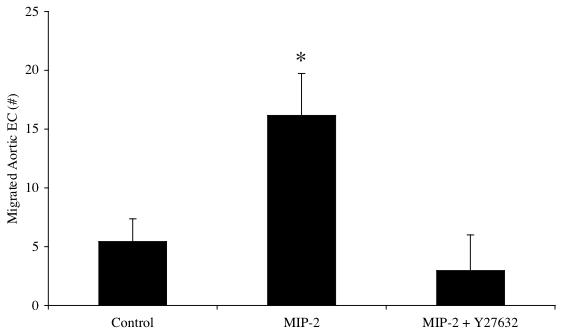
Functional assessment of MIP-2 treatment on aortic endothelial cells showing chemotaxis (# of migrated aortic endothelial cells) and with inhibitor of Rho kinase (Y27632). Aortic endothelial cells were incubated for 30 min with the Rho kinase inhibitor-Y27632 (5 μM) or vehicle control (PBS) followed by a 2 hr treatment with MIP-2 (10 ng/ml). A significant increase in the number of migrated cells was apparent after MIP-2 treatment (n=3-4 experiments /group; * indicates p < 0.05). However, there was no change in the number of migrated cells after MIP-2 if the cells were pretreated with the Rho kinase inhibitor (p > 0.05 from vehicle control without MIP-2).
To further confirm the importance of RhoA activation in CXCR2 signaling, RhoA/siRNA was used to assess the functional significance of this pathway for aortic EC chemotaxis. Figure 5A shows a representative immunoblot demonstrating the successful knock down of Rho-GTP and total RhoA in aortic EC. Additionally, the treatment control shows effective silencing of GAPDH without effect on Rho-GTP. Treatments did not affect total protein as assessed by actin levels. Figure 5B shows the functional chemotactic response of aortic EC to MIP-2 after these silencing strategies. Baseline chemotaxis without MIP-2 did not differ among the groups (n=3/ treatment group; p > 0.05). Average responses (% control) for each treatment are presented. Experiments using GAPDH/siRNA demonstrated chemotaxis (70% increase) equivalent to aortic EC treated only with MIP-2 (65% increase). This increase was significantly reduced in aortic EC treated with RhoA/siRNA (17% increase; p < 0.05 from GAPDH/siRNA group).
Figure 5.
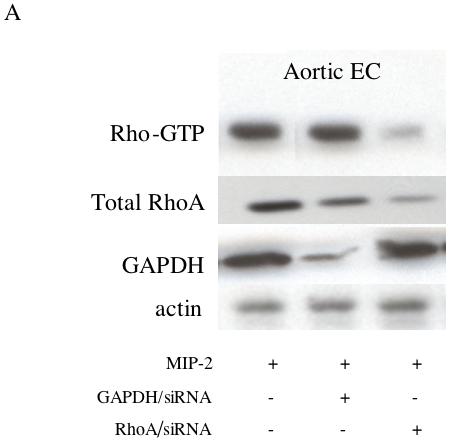
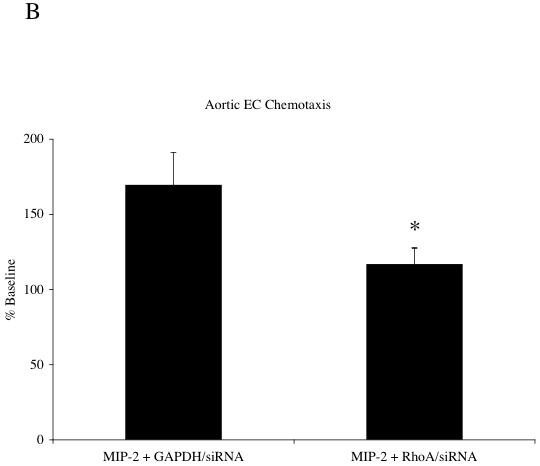
A: A representative immunoblot demonstrating successful knock down of Rho-GTP and RhoA protein in aortic EC with RhoA/siRNA. The treatment control (GAPDH/siRNA) shows effective silencing of GAPDH without effect on Rho-GTP. Total protein as assessed by actin was unaffected by treatments. B: Control chemotactic response (GAPDH/siRNA) of aortic EC after MIP-2 treatment (10 ng/ml; 2 hr) compared to aortic EC with decreased activated RhoA(RhoA/siRNA). Average responses (% baseline control without MIP-2) for each treatment are presented. Experiments using GAPDH/siRNA demonstrated a 70% increase in chemotaxis. This increase was significantly reduced in aortic EC treated with RhoA/siRNA (17% increase; n=4 experiments; * indicates p < 0.05).
To determine whether PA EC could be altered to induce responsiveness to MIP-2, PA EC were modified with adenovirus encoding constituitively active RhoA-L63. Figure 6A demonstrates increased Rho-GTP and total RhoA in PA EC relative to adenoviral vector control after MIP-2 treatment. Baseline level of RhoA-GTP in RhoA-L63 infected cells without MIP-2 treatment was increased by 22% (n=2). GAPDH remained constant across treatment groups. Functional assessment of Rho-GTP in transfected PA EC is presented in Figure 6B. Baseline chemotaxis without MIP-2 was not different among groups (p > 0.05). PA EC exposed to MIP-2 only (25 % increase; data not shown) or after adenoviral ß-gal transfection show minimal chemotaxis (12% increase). However, cells transfected with adRhoA showed increased chemotaxis (46 % increase, n=6 experiments; p < 0.05 from adenoviral ß-gal group).
Figure 6.
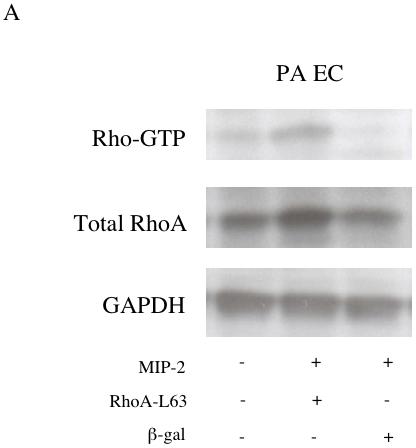
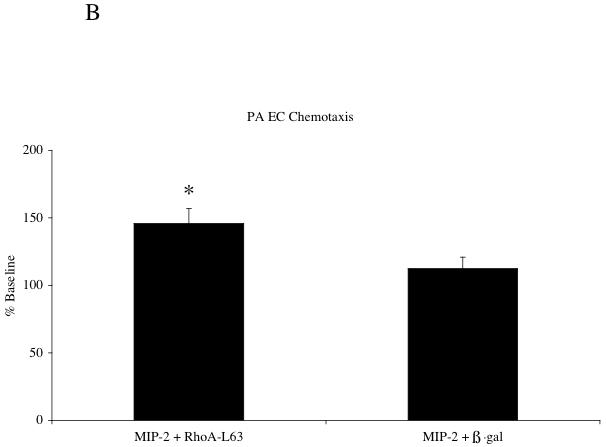
A: A representative immunoblot demonstrating successful adenoviral transfer of RhoA-L63 and activation of Rho-GTP and increased total RhoA in pulmonary artery endothelial cells (PA EC) after MIP-2 treatment (10 ng/ml; 30 min). B shows average chemotaxis responses after MIP-2 treatment (10 ng/ml; 2 hr treatment) expressed as the % baseline control without MIP-2 in PA EC (n=6 experiments; * indicates p < 0.05).
DISCUSSION
We have shown previously that over a wide range of agonist concentrations, mouse aortic EC chemotaxis was markedly greater than that observed in pulmonary artery EC exposed to MIP-2 (8). The goals of the present study were to determine intracellular signaling mechanisms that might account for this functional difference. Specifically, we hypothesized that activation of the small GTPases RhoA and/or Rac1, thought to be involved in CXCR2 signaling, would be significantly greater in aortic EC compared to pulmonary artery EC. Our work confirmed enhanced RhoA activation after MIP-2 treatment in aortic EC compared to pulmonary EC.
EC heterogeneity is increasingly recognized and has been based on differences in anatomic location and physiologic function (1, 9, 12). An important pathophysiologic observation in humans as well as all other animals studied, shows EC heterogeneity in angiogenic potential within the lung. Systemic endothelium within and surrounding the lung responds to a variety of conditions within the lung that signal a need for neovascularization. Interestingly, the pulmonary circulation rarely participates in the rampant neovascularization that occurs in cystic fibrosis, asthma, chronic inflammation, cancer, or pulmonary ischemia (7). In a mouse model of lung angiogenesis, after complete unilateral pulmonary vascular ischemia, the C-X-C chemokines appear to play an essential role in the establishment of new systemic blood vessels to the lung (11, 16). As expected, no evidence of pulmonary endothelial proliferation was observed in this model (6). Thus, in the present study, we specifically sought to determine basic endothelial cell differences with regard to C-X-C chemokine signaling. Although there are several known mouse ELR+, C-X-C chemokines (KC, LIX, MIP-2), we focused on MIP-2, which was the most highly expressed in the lung in this in vivo model (11). It is thought that MIP-2 binds primarily to CXCR2 to result in EC proliferation and chemotaxis (10, 13). Since we previously showed in vitro that CXCR2 receptor numbers did not differ between aortic and pulmonary artery EC (8), we speculated that activation of intracellular signaling cascades differed between endothelial cell subtypes. Based on the work of others, we focused on small GTPases that are activated upon CXCR2 ligation, namely RhoA and Rac1. Our results showed similar minimal activation of Rac1 in both endothelial cell subtypes over a time period in which we expected to see a response. Consequently we focused on differences in RhoA activation. By measuring activated protein levels, we showed activation of RhoA after chemokine treatment only in aortic endothelial cells (Figure 2). Pulmonary EC did not show RhoA activation after MIP-2 until cells were manipulated to express a constituitively active RhoA. MIP-2 induced chemotaxis after adenoviral infection of active RhoA was significantly increased relative to adenoviral controls (Figure 6). Interestingly, baseline chemotaxis without MIP-2 treatment was not altered in pulmonary EC after infection with constituitively active RhoA-L63, thus suggesting that signaling factors upstream of RhoA are critical for chemotaxis. The results of this series of experiments, however, provide strong support for differential activation in systemic arterial EC compared to pulmonary EC of a key signaling pathway important for angiogenesis.
We further confirmed the importance of RhoA in aortic EC by silencing RhoA (Figure 5) and by inhibiting a downstream effector protein of RhoA, Rho kinase (Figure 4). Overall, the results consistently demonstrated an important role for RhoA in MIP-2 induced cell chemotaxis, an essential component of the angiogenic phenotype. Furthermore, these results are consistent with several other studies that have confirmed the importance of the RhoA signal transduction pathway in angiogenesis by inhibition studies and the assessment of tube formation (3, 15). Schraufstatter and colleagues demonstrated in human dermal microvascular cells that migration of cells in response to the C-X-C chemokine, IL-8, could be completely inhibited with anti-CXCR2 antibody (10). Although in their study there was early activation (<5 min) of RhoA, the predominant and longer lasting (5-60 min) effect of IL-8 treatment was through Rac1 activation. Since migration was evaluated over the longer time course, the authors concluded that CXCR2 ligation signaled predominantly through Rac1. In contrast, our results demonstrated a minimal and variable change in Rac1 activation, which did not differ between endothelial cell subtypes. The differences in responses in the two studies may highlight basic heterogeneity of responses with conduit vessel endothelial cells showing different patterns of response than microvascular cells as well as species-dependent differences. Additionally, these investigators used a 10-fold greater agonist concentration than in the present study, as well as quantifying RhoA and Rac1 activity indirectly by fluorometric evaluation of stress fibers with/without inhibitors. However, both studies confirmed the overall importance of the small GTPases in endothelial cell migration in response to a C-X-C chemokine and inherent endothelial cell heterogeneity.
In summary, we have shown that RhoA activity is increased after MIP-2 treatment of mouse aortic endothelial cells but not in pulmonary artery endothelial cells. We conclude that RhoA is part of a signaling pathway essential for aortic cell migration after CXCR2 ligation. This result provides one explanation for the difference in chemotaxis observed in these two endothelial subtypes that express similar levels of CXCR2. However, we can only speculate as to why pulmonary endothelial cells lack the activation of this essential pathway. Several possibilities exist, including differences in CXCR2 receptor binding capacity and internalization, and differences in accessory proteins that control active GTP- bound and inactive GDP-bound states of RhoA (14).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Aird WC. Mechanisms of endothelial cell heterogeneity in health and disease. Circ Res. 2006;98:159–162. doi: 10.1161/01.RES.0000204553.32549.a7. [DOI] [PubMed] [Google Scholar]
- 2.Curnock AP, Logan MK, Ward SG. Chemokine signalling: pivoting around multiple phosphoinositide 3-kinases. Immunology. 2002;105:125–136. doi: 10.1046/j.1365-2567.2002.01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoang MV, Whelan MC, Senger DR. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc Natl Acad Sci U S A. 2004;101:1874–1879. doi: 10.1073/pnas.0308525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–1801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 5.Loovers HM, Postma M, Keizer-Gunnink I, Huang YE, Devreotes PN, van Haastert PJ. Distinct Roles of PI(3,4,5)P3 during Chemoattractant Signaling in Dictyostelium: A Quantitative In Vivo Analysis by Inhibition of PI3-Kinase. Mol Biol Cell. 2006;17:1503–1513. doi: 10.1091/mbc.E05-09-0825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitzner W, Lee W, Georgakopoulos D, Wagner E. Angiogenesis in the mouse lung. Am J Pathol. 2000;157:93–101. doi: 10.1016/S0002-9440(10)64521-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitzner W, Wagner EM. Vascular remodeling in the circulations of the lung. J Appl Physiol. 2004;97:1999–2004. doi: 10.1152/japplphysiol.00473.2004. [DOI] [PubMed] [Google Scholar]
- 8.Moldobaeva A, Wagner EM. Difference in Proangiogenic Potential of Systemic and Pulmonary Endothelium: Role of CXCR2. Am J Physiol Lung Cell Mol Physiol. 2005 doi: 10.1152/ajplung.00370.2004. [DOI] [PubMed] [Google Scholar]
- 9.Peters DG, Ning W, Chu TJ, Li CJ, Choi AM. Comparative SAGE Analysis of the Response to Hypoxia in Human Pulmonary and Aortic Endothelial Cells. Physiol Genomics. 2006 doi: 10.1152/physiolgenomics.00152.2005. [DOI] [PubMed] [Google Scholar]
- 10.Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1094–1103. doi: 10.1152/ajplung.2001.280.6.L1094. [DOI] [PubMed] [Google Scholar]
- 11.Srisuma S, Biswal SS, Mitzner WA, Gallagher SJ, Mai KH, Wagner EM. Identification of genes promoting angiogenesis in mouse lung by transcriptional profiling. Am J Respir Cell Mol Biol. 2003;29:172–179. doi: 10.1165/rcmb.2002-0276OC. [DOI] [PubMed] [Google Scholar]
- 12.Stevens T. Molecular and cellular determinants of lung endothelial cell heterogeneity. Chest. 2005;128:558S–564S. doi: 10.1378/chest.128.6_suppl.558S. [DOI] [PubMed] [Google Scholar]
- 13.Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–27357. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- 14.Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- 15.Uchida S, Watanabe G, Shimada Y, Maeda M, Kawabe A, Mori A, Arii S, Uehata M, Kishimoto T, Oikawa T, Imamura M. The suppression of small GTPase rho signal transduction pathway inhibits angiogenesis in vitro and in vivo. Biochem Biophys Res Commun. 2000;269:633–640. doi: 10.1006/bbrc.2000.2315. [DOI] [PubMed] [Google Scholar]
- 16.Wagner EM, Petrache I, Schofield B, Mitzner W. Pulmonary ischemia induces lung remodeling and angiogenesis. J Appl Physiol. 2006;100:587–593. doi: 10.1152/japplphysiol.00029.2005. [DOI] [PubMed] [Google Scholar]