

Abstract
Thymidylate synthase (TS) is an important target of several chemotherapeutic agents, including 5-FU and raltitrexed (Tomudex). During TS inhibition, TTP levels decrease with a subsequent increase in dUTP. Uracil incorporated into the genome is removed by base excision repair (BER). Thus, BER initiated by uracil DNA glycosylase (UDG) activity has been hypothesized to influence the toxicity induced by TS inhibitors. In this study we created a human cell line expressing the Ugi protein inhibitor of UNG family of UDGs, which reduces cellular UDG activity by at least 45-fold. Genomic uracil incorporation was directly measured by mass spectrometry following treatment with TS inhibitors. Genomic uracil levels were increased over 4-fold following TS inhibition in the Ugi-expressing cells, but did not detectably increase in UNG proficient cells. Despite the difference in genomic uracil levels, there was no difference in toxicity between the UNG proficient and UNG-inhibited cells to folate or nucleotide-based inhibitors of TS. Cell cycle analysis showed that UNG proficient and UNG-inhibited cells arrested in early S-phase and resumed replication progression during recovery from RTX treatment almost identically. The induction of γ-H2AX was measured following TS inhibition as a measure of whether uracil excision promoted DNA double strand break formation during S-phase arrest. Although γ-H2AX was detectable following TS inhibition, there was no difference between UNG proficient and UNG-inhibited cells. We therefore conclude that uracil excision initiated by UNG does not adequately explain the toxicity caused by TS inhibition in this model.
Keywords: base excision repair, uracil DNA glycosylase, thymidylate deprivation, antifolates
INTRODUCTION
Thymidylate synthase (TS) is a therapeutic target for the cancer chemotherapeutic drugs 5-fluorouracil (5-FU), capecitabine (prodrug of 5-FU), and raltitrexed (Tomudex, RTX) [1]. TS converts dUMP to TMP using N5, N10-methylenetetrahydrofolate as a coenzyme and provides the only de novo source of TMP for DNA synthesis and repair. While 5-FU can also be incorporated into RNA and DNA, the anti-folate RTX appears to be specific for TS. During TS inhibition, the level of TMP decreases and dUTP increases, which presumably increases uracil levels in DNA [2]. Base excision repair (BER) initiated by uracil DNA glycosylases actively removes uracil from the genome [3]. However, during thymidylate deprivation uracil would presumably be reincorporated during repair synthesis, thus leading to futile cycling of BER.
Four known genetic loci in humans encode for uracil DNA glycosylases [3]. Biochemical characterization of the proteins suggests specialized roles that combat two sources of uracil introduction into the genome, namely deamination of cytosine and incorporation of dUMP during replication. The UNG genetic locus encodes mitochondrial (UNG1) and nuclear (UNG2) forms of uracil DNA glycosylase [3]. The nuclear form of UNG appears to account for the bulk of cellular UDG activity; more specifically, the primary role of UNG2 seems to be counteracting uracil misincorporation during replication [4,5]. Despite the attractiveness of the futile cycling hypothesis, there is little direct evidence in mammalian cells demonstrating that futile cycling of BER contributes to the toxicity of TS inhibitors. Sensitivity to RTX was not influenced by UNG overexpression [6]. Ung+/+ andUng−/− murine embryonic fibroblasts (MEFs) showed no difference in the cytotoxic effects of 5-FU or fluorodeoxyuridine (FdUrd, the deoxynucleoside derivative of 5-FU), despite an increased accumulation of AP sites [7]. The SMUG1 genetic locus encodes a DNA glycosylase that has been proposed to serve as a backup for UNG, although SMUG1 excises a broader range of damaged pyrimidines [3]. It was shown that the SMUG1 DNA glycosylase can remove 5-FU from DNA and that this activity protects MEFs from 5-FU toxicity [8]. Interpreting the causes of 5-FU toxicity is complicated by the fact that 5-FU incorporated into DNA can be recognized by mismatch repair [9], and two additional DNA glycosylases of BER, namely TDG and MBD4 [10,11]. Thus, the precise role of BER during thymidylate deprivation remains unclear.
Our investigations seek to define the role of BER during chemotherapy-induced thymidylate deprivation. Previous results in DNA polymerase β deficient MEFs suggested that BER pathway activation by uracil excision was not contributing to the strand breaks and cell death observed during thymidylate deprivation induced by TS inhibitors [12,13]. These and other studies were performed in MEFs [7,8], which raises questions about the broader applicability of these observations. In this study, we directly examined the influence of inhibiting intracellular UNG activity in human cells. RTX, FdUrd, and 5-FU were used to induce thymidylate deprivation. To our knowledge, this is the first study that directly measured endogenous genomic uracil following treatment with TS inhibitors.
MATERIALS AND METHODS
Drugs and Cell culture
Raltitrexed (RTX) was generously supplied by AstraZeneca, U.K. 5-Fluoro-2'-deoxyuridine, 5-fluorouracil and Sulforhodamine B (SRB) were purchased from Sigma (St. Louis, MO). Human embryonic kidney (HEK) 293 cells were obtained from ATCC and maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% regular or dialyzed fetal bovine serum (Hyclone, Logan, UT) and 1% penicillin/ streptomycin (Sigma) at 37°C in a humidified 5% CO2 incubator. We have verified that the HEK293 cells used in this study are uninfected with mycoplasma.
Generation of stable GFP and GFP-hUgi -expressing cell lines
The pLGCX and pLGC-hUgi plasmids were a kind gift from Shari Kaiser in the laboratory of Michael Emerman (University of Washington). The pLGC-hUgi plasmid contains a codon-optimized Ugi for expression in human cells [14]. The pLGCX and pLGC-hUgi plasmids were transfected into HEK293 cells by a Gene Pulser Xcell electroporation system according to the manufacturer's instructions (Bio-Rad, Hercules CA). The cells were resuspended with 200 μl of DMEM medium without serum and antibiotic in 2-mm long cuvettes and electroporated with the following settings: 110 V, 25ms pulse length and 1 pulse. One week after electroporation, GFP-positive cells were enriched by FACS on an EPICs XL-MCL flow cytometer (Beckman Coulter, Fullerton, CA) in the Instrumentation Resource Facility at the South Carolina Cancer Center. Multiple rounds of sorting were performed until greater than 99% of the cell population was GFP positive.
Cytotoxicity
Cytotoxicity was performed by SRB assay as described previously [12]. In brief, 1000 cells were plated on 96-well plates 24 h prior to treatment. The cells were treated with various concentrations of RTX, FdUrd, or 5-FU for 24 h, and then grown in drug- free medium for 3 days. Cells were then fixed, washed, and stained. Absorbance was measured using a plate reader at 560 nm (Bio-Tek UV808 Microplate Reader, Winooski, VT). Colony-forming assays were performed as previously described [12].
Uracil DNA Glycosylase activity
UDG activity in the GFP and GFP-hUgi cells was measured using an oligodeoxynucleotide-based assay [13]. The analysis was identical to that previously described, except that the oligo containing a single uracil (5'-GACTACTACATGUTTGCCGACCATT-3', (Midland Certified Reagents, Midlands, TX) also contained a 5'-HEX label and was directly visualized and quantitated using a Bio-Rad Molecular Imager® FX and Quantity One® software.
Analysis of Genomic Uracil
Uracil levels in DNA were analyzed as described [15] with modifications [13]. Briefly, 24 h after seeding, the cells were exposed to 100 nM RTX or 50 nM FdUrd for 24 hour, then washed of drug-containing media. At the times listed, the cells were washed by PBS, harvested, and genomic DNA was extracted using the DNAeasy kit according to manufacturer's instructions (Qiagen, Valencia, CA). To release genomic uracil from DNA, 3 μg of genomic DNA was incubated with 0.2 Units of UDG (NEB, Beverley, MA). Following digestion, 300 pg of a labeled uracil internal standard (13C4H4O215N2; Cambridge Isotope Laboratories, Andover, MA) was added to each tube, the samples were vacuum dried and derivatized with 3,5-Bis (trifluoromethyl) benzyl bromide. The samples were analyzed by GC-MS on a Thermo-Finnigan TSQ triple quadrupole mass spectrometer equipped with a Trace GC 2000 gas chromatograph and AS 2000 autosampler (all Thermo-Fisher, Waltham MA). Separation was performed on a 30 meter Rtx-5 column (Restek, Bellefonte PA) using a column head pressure of 10 psi and an oven temperature program as follows: start at 100°C, ramp 15°C /min to 210°C, 5°C/min to 260°C, 15°C/min to 300°C. The uracil derivative elutes at ∼15 min. Mass spectrometry was done in negative ionization mode with methane (4000 millitorr) as the reagent gas. The mass spectrometer was run in select reaction monitoring mode with the first quadrupole alternating between m/z 337 (natural uracil) and m/z 343 (heavy labeled uracil). The collision cell was set for 35eV collisions with 3 millitorr of argon and the third quadrupole was set on m/z 213 for both natural and heavy labeled uracil. A standard curve was established by measuring standards containing 5, 20, and 50 pg of unlabeled uracil. Standards consisting of 54 and 108 pg of uracil-containing oligo described in the UDG activity assay above served as an additional control to insure that the UDG incubation conditions quantitatively removed uracil from DNA.
Western blot analysis
Western blots were performed as described previously [12,13]. Anti-phospho-Histone-H2AX (Ser 139) was purchased from Upstate (Temecula, CA). Anti-DUT-n antibody to detect the nuclear form of dUTPase was purchased from Abcam (Cambridge, MA). Equal protein loading was confirmed by β-actin (Abcam, Cambridge, MA).
Cell Cycle Analysis
Cell cycle experiments were performed as previously described with minor modifications [13]. Exponentially growing cells were exposed to 100 nM RTX for 24 h and allowed to recover in drug-free medium at time points indicated in the text. Adherent and floating cells were collected in medium, fixed by chilled 100% ethanol, and stored at 4 °C until processed for analysis. Fixed cells were stained with 0.3 mL PI / RNase A solution which containing propidium iodide (50 μg/mL), RNase A (0.1 mg/mL) and 1%BSA in PBS. DNA content was determined using an EPICs XL-MCL flow cytometer (Beckman Coulter, Fullerton, CA) in the Instrumentation Resource Facility at the South Carolina Cancer Center.
RESULTS
We created sublines of 293 human embryonic kidney cells expressing either GFP only as a control or a GFP-hUgi fusion with pLGCX and pLGC-hUgi vectors, respectively [14]. Ugi is a well- known inhibitor specific to the UNG family of proteins but not other UDGs such as SMUG1, TDG, or MBD4 [16,17]. The Ugi coding sequence in this case was codon-optimized for human expression [14] and GFP-positive cells were selected by FACS (materials and methods). Cell extracts were prepared and examined for UDG activity using an oligodeoxynucleotide-based assay. UDG activity is readily detectable in the extracts from UNG proficient cells for uracil in single-stranded DNA (Figure 1A, left) or double stranded DNA (Figure 1B, left). In contrast, UNG-inhibited cells contained undetectable levels of UDG activity (Figure 1, right), an estimated difference of at least 45-fold lower compared to UNG proficient cells. Although both cell lines contain hSMUG1 detectable by western blot (data not shown), the activity assays demonstrate that UDG from the UNG locus provides the predominant UDG activity, in agreement with previous studies [4].
Figure 1. hUgi inhibits UNG activity in 293 cells.
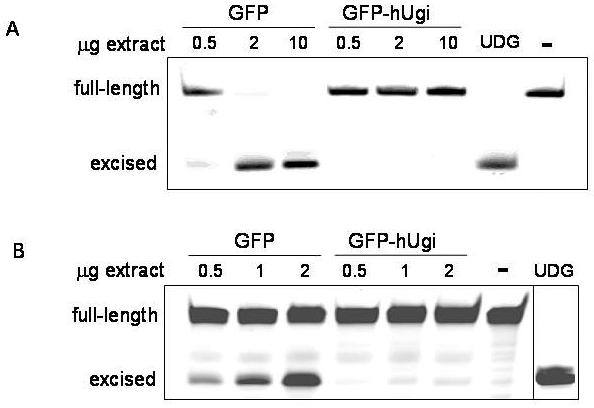
UNG-proficient cells (GFP) have a higher UDG activity than cells expressing hUgi (GFP-HUgi) for uracil in single-strand DNA (A) or double strand DNA (B). Increasing amounts of cell lysate for each cell line were incubated with 0.1 pmol of labeled oligonucleotide containing a single uracil. Cleavage results in generation of a truncated product. The negative control (−) reaction contained oligo in the absence of lysate. The lane labeled “UDG” was oligo incubated with purified UDG.
In order to directly test whether hUgi is inhibiting UNG activity in the nucleus, we measured genomic uracil levels before and after treating the cells with IC90 doses of RTX (Figure 2a) or FdUrd (Figure 2b). In untreated UNG proficient or UNG-inhibited cells, genomic uracil levels are the same (∼2 picograms per μg of genomic DNA), near the limits of accurately quantitating the peaks on the spectrograms. When the cells were treated with RTX or FdUrd, the levels of uracil remained the same in UNG proficient cells, which indicated that UNG activity is robust enough to prevent an accumulation of genomic uracil during thymidylate deprivation. In contrast, the UNG-inhibited cells accumulated over 4 times the level of genomic uracil following 24 h RTX treatment (Figure 2a) or 24 h FdUrd treatment (Figure 2b). To determine whether recovery from TS inhibition altered genomic uracil incorporation, cells were placed in drug-free medium for 3 and 24 hours following 24 h RTX or FdUrd treatment. The uracil levels in UNG-inhibited cells remained elevated at 3 and 24 hour recovery time points following removal of RTX (Figure 2a). At a 24 h recovery time point following FdUrd treatment, the levels of genomic uracil had dropped by approximately half (Figure 2b). Taken together, the results suggest that hUgi is indeed inhibiting UNG in the cells during treatment with TS inhibitors and causes an accumulation of genomic uracil in the hUgi-expressing cells.
Figure 2. Genomic uracil levels following treatment with RTX (A) or FdUrd (B).
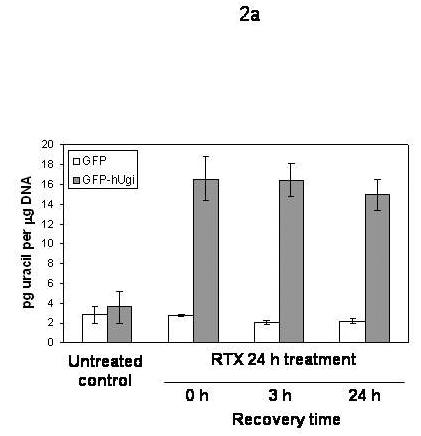
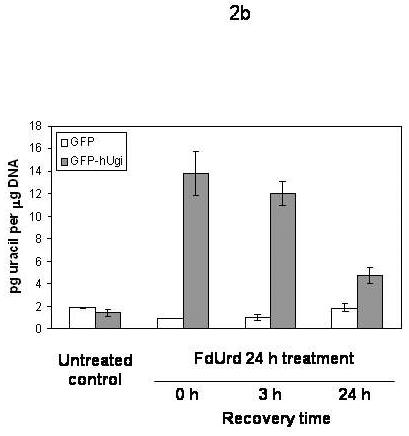
A. UNG proficient (GFP) and UNG-inhibited (GFP-hUgi) cells were treated with 100 nM RTX for 24 h, and then incubated in drug-free media for 0, 3, or 24h. The results shown are the average of four independent experiments (error bars are the standard deviation). B. UNG proficient (GFP) and UNG-inhibited (GFP-hUgi) cells were treated with 50 nM FdUrd for 24 h, and then incubated in drug-free media for 0, 3, or 24h. The results shown are the average of two independent experiments and two GC-MS injections per sample (error bars are the standard deviation).
The sensitivity of the UNG proficient and UNG-inhibited cells to RTX, FdUrd, and 5-FU was determined (Figure 3). The results in Figure 3 show that there was no significant difference in sensitivity to RTX (A), FdUrd (B), or 5-FU (C) between the UNG proficient and UNG-inhibited cells. Dialyzed serum was also used because it is commonly known that thymidine salvaged from serum can significantly influence sensitivity to folate-based TS inhibitors. There was no difference in sensitivity between the UNG-proficient and UNG-inhibited cells when grown in regular or dialyzed serum. This suggests that the availability of salvageable thymidine does not selectively influence the sensitivity of UDG proficient or UDG inhibited cells. Colony forming assays were also performed with FdUrd to insure that the viability assay was not missing a substantially delayed onset of death. At a dose of 10 μM FdUrd, survival was 19% (±3.5, n=3) for the UNG proficient cells and 36% (±11.2, n=3), a less than 2-fold difference that was insignificant. It is known that elevated dUTPase expression can influence cell death caused by TS inhibitors [18-21]. The results in Figure 4 show that nuclear dUTPase protein levels are similar in the UNG proficient and UNG-inhibited cells. Thus, changes in dUTPase levels do not account for the differences seen in genomic uracil incorporation (Figure 2).
Figure 3. The sensitivity to TS inhibitors is similar in the UNG proficient and UNG-inhibited cells.
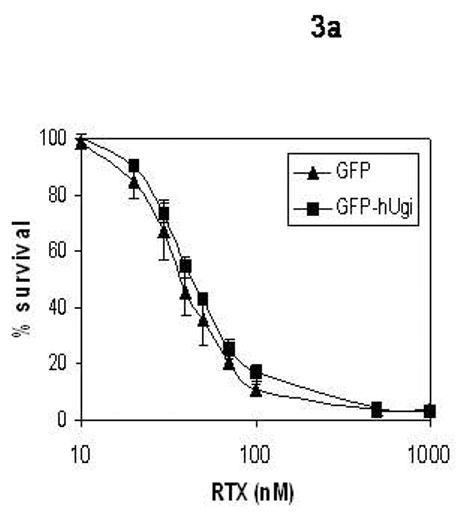
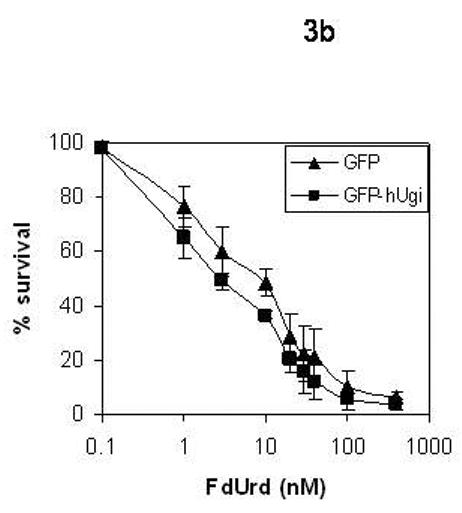
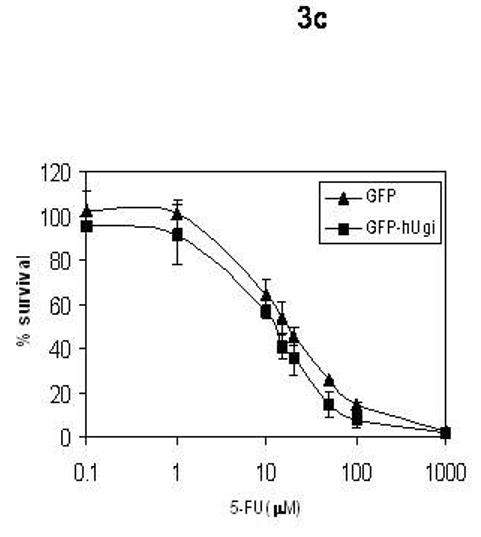
UNG-proficient (GFP, ▲) and UNG-inhibited (GFP-hUgi, ■) cell lines were exposed to RTX (A), FdUrd (B), and 5-FU (C) for 24 hours, followed by incubation in drug-free media for 72 h. Cytotoxicity was determined as described (materials and methods). The data are an average of at least three independent experiments, error bars represent standard deviations.
Figure 4. The protein levels of dUTPase are the same in the UNG proficient and UNG-inhibited cells.
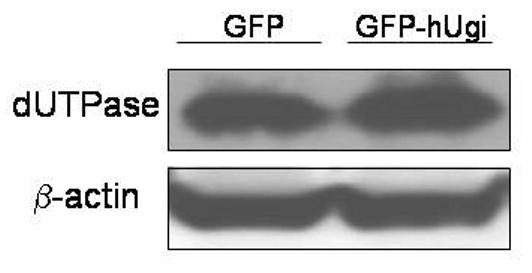
Cell lysates from UNG proficient (GFP) and UNG-inhibited (GFP-hUgi) cells were analyzed by SDS-PAGE, and the immunoblots probed with an antibody specific to nuclear dUTPase. β-actin served as the loading control.
It is well known that TS inhibition induces cell cycle arrest in early S-phase concomitant with TTP depletion. We examined cell cycle progression by FACS in the UNG proficient and UNG-inhibited cells to determine whether accumulation of genomic uracil or its excision by UNG influenced the initial S-phase arrest. When treated with RTX for 24 hours, UNG proficient and UNG-inhibited cells displayed an identical early S-phase arrest (Figure 5, middle panels). The early S-phase arrest seen was essentially identical to the arrest previously observed in pol β proficient and deficient MEFs [12]. To examine the cellular recovery from S-phase arrest, cells were placed in drug-free medium for 24 h following 24 h RTX treatment. The UNG proficient and UNG-inhibited cells resumed S-phase progression in an almost identical manner (Figure 5, bottom panels). In comparing S-phase progression seen at 24 h recovery with genomic uracil levels in the UNG-inhibited cells (Figure 2a), the results suggest that no significant additional uracil incorporation is occurring during the resumption of replication.
Figure 5. Cell cycle arrest induced by RTX is similar in the UNG proficient and UNG-inhibited cells.
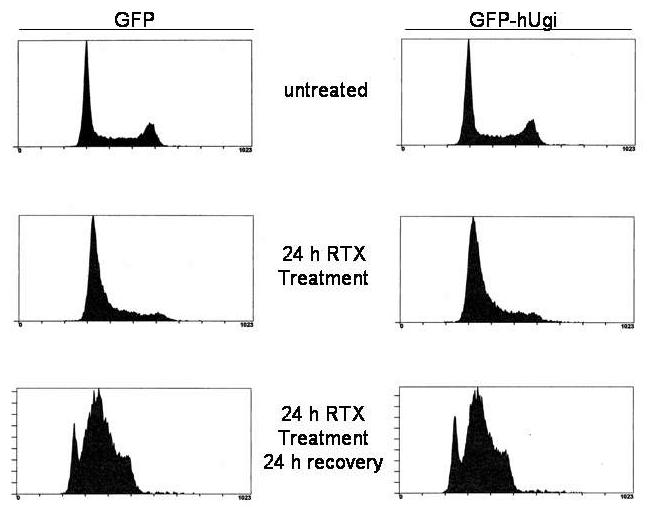
UNG proficient (GFP, left panels) and UNG-inhibited (GFP-hUgi, right panels) cells were treated with 100 nM RTX for 24 h (middle panel), and then incubated in drug-free media for 24 h (bottom panel). Cells were harvested and analyzed as described (Materials and Methods). Cell cycle analysis was performed four times; results shown are representative from one experiment.
The formation of DNA double strand breaks has been reported following TS inhibition, although double strand breaks have not uniformly correlated with death in response to folate-based TS inhibitors [20,22]. We previously found that RTX induced sister chromatid exchanges at subtoxic doses, which suggested that homologous recombination was invoked during thymidylate deprivation [13]. To determine whether uracil excision by UNG might contribute to the formation of double strand breaks during thymidylate deprivation, the induction of γ-H2AX was measured (Figure 6). Phosphorylation of the H2AX histone protein is a presumptive marker of double strand break induction and is also seen as a consequence of stalled replication forks [23]. As shown in Figure 6, a 24 h treatment with RTX (A) or FdUrd (B) induced γ-H2AX, yet little difference was seen between UNG proficient and UNG-inhibited cells. This suggests that although H2AX phosphorylation occurs following TS inhibition, the formation of γ-H2AX was not altered by the activity of UNG (or uracil incorporation into DNA in the hUgi-expressing cells). Taken together, the results do not suggest that BER intermediates generated by UNG are being produced, or if produced, do not substantially contribute to double strand breaks that occur following TS inhibition.
Figure 6. The induction of γ-H2AX does not differ between UNG-proficient or UNG-inhibited cells.
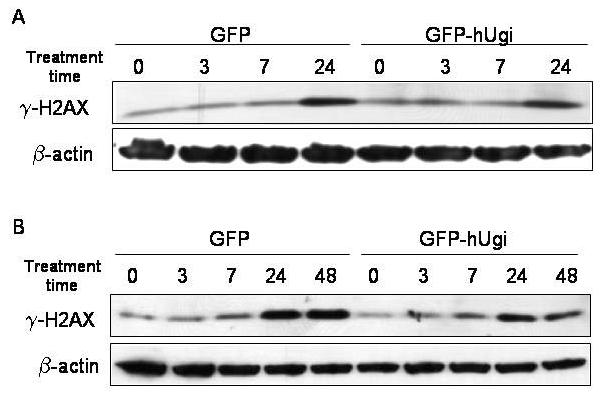
UNG-proficient (GFP) and UNG-inhibited (GFP-hUgi) cells were treated with RTX 1 μM (A) and FdUrd 0.5 μM (B) for the indicated periods of time. Cell lysates were analyzed by SDS-PAGE, and the immunoblots probed with an antibody specific to γ-H2AX. β-actin served as the loading control.
Discussion
The futile cycling hypothesis of BER during thymidylate deprivation proposes that uracil incorporated into DNA is excised by uracil DNA glycosylases, but repair synthesis causes uracil reincorporation because of elevated dUTP levels. The accumulation of BER single strand break intermediates could thus be converted to double strand breaks during replication and lead to cell death. Although we observed that genomic uracil accumulated following the inhibition of TS by RTX or FdUrd and UDG inhibition by hUgi, this did not appear to influence killing by antifolate or nucleotide-based TS inhibitors in this cell line. In comparing the data in Figures 2 and 5, genomic uracil incorporation likely occurred very early upon entry into S-phase, near simultaneously with the S-phase arrest seen. It is important to note that because the arrest in UNG proficient and UNG inhibited cells appears to be identical (Figure 5), this suggests that BER intermediates generated by UNG do not contribute to causing the initial S-phase arrest. During recovery from TS inhibition when replication has resumed, there was no further increase in genomic uracil (Figure 2, 24 h time point), thus suggesting that dUTP incorporation is not contributing to replication restart. The reduction in genomic uracil seen following recovery from TS inhibition (more pronounced in the FdUrd treated cells) is likely due to the extent of replication restart with TTP, thus diluting genomic uracil. The induction of γ-H2AX was also examined to determine whether double strand breaks were induced by one of two scenarios. The first is that the presence of BER intermediates created by UNG activity might be converted to double strand breaks during replication. The second is that the presence of genomic uracil itself might halt replication forks. The fact that the induction of γ-H2AX occurred to the same extent in UNG proficient or UNG-inhibited cells suggests that thymidylate deprivation likely induces replication fork arrest independent of UNG activity or uracil incorporation. In either case it is not clear precisely how BER intermediates, which occur on a single strand, might be converted into double strand breaks during TS inhibition. Uracil incorporation during replication would only occur in the newly synthesized strands, i.e., uracil excision and AP endonuclease activity would cause single strand breaks. Double strand break formation resulting from BER activity under conditions of TS inhibition would require a more complex interaction with the replication fork (e.g., a replication fork collapse), or would invoke an additional repair response such as homologous recombination, which proceeds through DSB intermediates.
Several studies have examined the influence of BER in S. cerevisiae in response to antifolates or 5-FU [24-26]. The influence of UDG activity on sensitivity to thymidylate deprivation in S. cerevisiae appeared to be transient [24,26]. S. cerevisiae lack SMUG1, MBD4, and TDG homologues [27], all of which are reported to remove uracil and 5-FU at least in vitro [8,10,11]. The absence of APN1, the major AP endonuclease in S. cerevisiae, leads to heightened sensitivity to 5-FU or antifolates [24,25]. Interestingly, S. cerevisiae lack a paralog of pol β, which removes the 5'-dRP group caused by AP endonuclease activity. Instead, S. cerevisiae seem to rely on RAD27 to remove the 5'-dRP as part of the displaced strand, analogous to FEN1-dependent long-patch BER in mammalian cells. It is intriguing to note that a rad27 null strain was markedly resistant to 5-FU [25], which is analogous to our finding that pol β deficient MEFs were resistant to TS inhibitors [12].
Our results agree with other studies in mammalian cells that do not support the model that excision of uracil by UNG contributes to toxicity caused by FdUrd or RTX [6-8]. The contribution of genomic 5-FU to the toxicity of 5-FU or FdUrd relative to the inhibition of TS (as FdUMP) remains incompletely understood. Note that our studies do not rule out a role for BER initiated by other DNA glycosylases such as SMUG1, TDG, and MBD4 in response to genomic 5-FU incorporation [8]. It has been reported that the nuclear isoform of UNG (UNG2) is down-regulated following FdUrd treatment in certain human cell lines, which corresponded to resistance [28]. One possible explanation of the phenotypic differences seen between cells lacking UNG2 versus hUgi-expressing cells in which UNG2 is inhibited might lie in the reported interactions between UNG2 and replication-associated proteins [5,29]. To our knowledge, our study is the first to provide direct evidence that genomic uracil incorporation occurs during TS inhibition by FdUrd or RTX. There is no issue of genomic 5-FU incorporation in RTX-treated cells. Although an effect of uracil incorporation into mitochondrial DNA has not formally been excluded, it is highly unlikely to contribute to the genomic uracil detected (Figure 2), given the much smaller size of the mitochondrial genome and typical number of mitochondria in most cell types.
It is well known that unrepaired BER intermediates caused by an imbalance of too much DNA glycosylase activity relative to the downstream activities of AP endonuclease and 5'-dRP lyase are problematic [30]. However, amelioration of futile BER cycling during TS inhibition by limiting UDG activity has its own limitation, namely that the presence of genomic uracil itself is dramatically problematic. The inviability of dUTPase deficient strains of E. coli and S. cerevisiae even in the absence of UDG activity suggest that excessive genomic uracil cannot be tolerated [31,32]. Studies that manipulated dUTPase levels during chemotherapy-induced thymidylate deprivation indicate that increased dUTPase activity can only delay death [18-21]. The time dependence implied that cytotoxicity did not ultimately depend on the DNA damage resulting from uracil incorporation [20,21]. Furthermore, in some cell types death was seen in TTP-depleted cells despite little or no dUTP accumulation [33], while changes in dATP levels have also been implicated [34]. As shown in Figure 4, dUTPase levels appear to be the same in the UNG proficient and deficient cells. The activity of dUTPase alone is not sufficient to prevent uracil incorporation because UNG-inhibited cells accumulate genomic uracil (Figure 2). In all of the above cited studies, the amount of uracil incorporated into DNA was not directly measured. It is not known among mammalian cell types what the threshold tolerance is for genomic uracil, or whether cancer cells in particular have adapted to tolerate a higher level of genomic uracil. We are investigating these possibilities.
The DNA damage endpoint most commonly measured in studies of TS inhibition is double strand breaks, predominantly measured by pulsed-field gel electrophoresis or the comet assay. The induction of γ-H2AX following RTX and FdUrd treatment is evident (Figure 4), yet the lack of difference between UNG proficient and UNG inhibited cells suggests that the more likely source of γ-H2AX induction is stalled or collapsed replication forks. It is known that altered S-phase progression occurs in cancer cells, and that defects in S-phase kinase signaling affects sensitivity to 5-FU [35-37]. Evaluation of such phenomena is particularly challenging in human cancer cell lines with known (and likely as yet unknown) defects in checkpoint signaling. One prominent example is that of the commonly used HCT 116 colon cancer cells, which in addition to being mismatch repair defective also contain a defective MRN signaling complex for double strand breaks [38] and a mutation in TS [39]. Homologous recombination is thought to be important for the resolution of collapsed replication forks. We are exploring the influence of homologous recombination pathways on S-phase progression and the sensitivity to TS inhibitors.
Acknowledgements
The authors gratefully thank Drs. Shari Kaiser and Michael Emerman (University of Washington) for the pLGCX and pLGC-hUgi plasmids. Dr. Sondra Berger, Department of Pharmaceutical and Biomedical Sciences, South Carolina College of Pharmacy is thanked for helpful discussions. Paul Meeh is gratefully acknowledged for assistance with the FACS analysis. AstraZeneca is acknowledged for a gift of RTX. This research was supported by a grant from the NIH (1 R01 CA100450) to MDW.
Abbreviations
- TS
thymidylate synthase
- BER
base excision repair
- 5-FU
5-fluorouracil
- FdUrd
5-fluoro-2'-deoxyuridine
- RTX
raltitrexed (Tomudex)
- UDG
uracil DNA glycosylase
- UNG
uracil DNA glycosylase from the UNG locus
- UNG2
nuclear isoform of uracil DNA glycosylase from the UNG locus
- hUgi
human codon-optimized inhibitor of UNG family of DNA glycosylases
- SMUG1
uracil DNA glycosylase from the SMUG1 locus
- dUTPase
deoxyuridine-triphosphate pyrophosphatase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 2.Aherne GW, Brown S. The role of uracil misincroporation in thymineless death. In: Jackman AL, editor. Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press Inc; Totowa, NJ: 1999. pp. 409–421. [Google Scholar]
- 3.Krokan HE, Drablos F, Slupphaug G. Uracil in DNA--occurrence, consequences and repair. Oncogene. 2002;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 4.Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell. 2000;5:1059–1065. doi: 10.1016/s1097-2765(00)80271-3. [DOI] [PubMed] [Google Scholar]
- 5.Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. EMBO J. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welsh SJ, Hobbs S, Aherne GW. Expression of uracil DNA glycosylase (UDG) does not affect cellular sensitivity to thymidylate synthase (TS) inhibition. Eur J Cancer. 2003;39:378–387. doi: 10.1016/s0959-8049(02)00610-x. [DOI] [PubMed] [Google Scholar]
- 7.Andersen S, Heine T, Sneve R, König I, Krokan HE, Epe B, Nilsen H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 2005;26:547–555. doi: 10.1093/carcin/bgh347. [DOI] [PubMed] [Google Scholar]
- 8.An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- 9.Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- 10.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, Bellacosa A. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000;275:32422–32429. doi: 10.1074/jbc.M004535200. [DOI] [PubMed] [Google Scholar]
- 11.Hardeland U, Bentele M, Jiricny J, Schar P. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J Biol Chem. 2000;275:33449–33456. doi: 10.1074/jbc.M005095200. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Berger SH, Wyatt MD. Involvement of base excision repair in response to therapy targeted at thymidylate synthase. Molecular Cancer Therapeutics. 2004;3:747–753. [PubMed] [Google Scholar]
- 13.Li L, Connor EE, Berger SH, Wyatt MD. Determination of apoptosis, uracil incorporation, DNA strand breaks, and sister chromatid exchanges under conditions of thymidylate deprivation in a model of BER deficiency. Biochemical Pharmacology. 2005;70:1458–1468. doi: 10.1016/j.bcp.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 14.Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80:875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mashiyama ST, Courtemanche C, Elson-Schwab I, Crott J, Lee BL, Ong CN, Fenech M, Ames BN. Uracil in DNA, determined by an improved assay, is increased when deoxynucleosides are added to folate-deficient cultured human lymphocytes. Analytical Biochemistry. 2004;330:58–69. doi: 10.1016/j.ab.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 16.Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW, Tainer JA. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–708. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Mosbaugh DW. Uracil-DNA glycosylase inhibitor gene of bacteriophage PBS2 encodes a binding protein specific for uracil-DNA glycosylase. J Biol Chem. 1989;264:1163–1171. [PubMed] [Google Scholar]
- 18.Canman CE, Radany EH, Parsels LA, Davis MA, Lawrence TS, Maybaum J. Induction of resistance to fluorodeoxyuridine cytotoxicity and DNA damage in human tumor cells by expression of Escherichia coli deoxyuridinetriphosphatase. Cancer Res. 1994;54:2296–2298. [PubMed] [Google Scholar]
- 19.Koehler SE, Ladner RD. Small interfering RNA-mediated suppression of dUTPase sensitizes cancer cell lines to thymidylate synthase inhibition. Mol Pharmacol. 2004;66:620–626. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 20.Parsels LA, Parsels JD, Wagner LM, Loney TL, Radany EH, Maybaum J. Mechanism and pharmacological specificity of dUTPase-mediated protection from DNA damage and cytotoxicity in human tumor cells. Cancer Chemother Pharmacol. 1998;42:357–362. doi: 10.1007/s002800050829. [DOI] [PubMed] [Google Scholar]
- 21.Webley SD, Welsh SJ, Jackman AL, Aherne GW. The ability to accumulate deoxyuridine triphosphate and cellular response to thymidylate synthase (TS) inhibition. Br J Cancer. 2001;85:446–452. doi: 10.1054/bjoc.2001.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsui SI, Arredondo MA, Wrzosek C, Rustum YM. DNA damage and p53 induction do not cause ZD1694-induced cell cycle arrest in human colon carcinoma cells. Cancer Res. 1996;56:4715–4723. [PubMed] [Google Scholar]
- 23.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 24.Dornfeld K, Johnson M. AP endonuclease deficiency results in extreme sensitivity to thymidine deprivation. Nucleic Acids Res. 2005;33:6644–6653. doi: 10.1093/nar/gki975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seiple L, Jaruga P, Dizdaroglu M, Stivers JT. Linking uracil base excision repair and 5-fluorouracil toxicity in yeast. Nucleic Acids Res. 2006;34:140–151. doi: 10.1093/nar/gkj430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tinkelenberg BA, Hansbury MJ, Ladner RD. dUTPase and uracil-DNA glycosylase are central modulators of antifolate toxicity in Saccharomyces cerevisiae. Cancer Res. 2002;62:4909–4915. [PubMed] [Google Scholar]
- 27.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, D.C.: 2006. [Google Scholar]
- 28.Fischer JA, Muller-Weeks S, Caradonna SJ. Fluorodeoxyuridine Modulates Cellular Expression of the DNA Base Excision Repair Enzyme Uracil-DNA Glycosylase. Cancer Res. 2006;66:8829–8837. doi: 10.1158/0008-5472.CAN-06-0540. [DOI] [PubMed] [Google Scholar]
- 29.Parlanti E, Locatelli G, Maga G, Dogliotti E. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007;35:1569–1577. doi: 10.1093/nar/gkl1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.el-Hajj HH, Wang L, Weiss B. Multiple mutant of Escherichia coli synthesizing virtually thymineless DNA during limited growth. J Bacteriol. 1992;174:4450–4456. doi: 10.1128/jb.174.13.4450-4456.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gadsden MH, McIntosh EM, Game JC, Wilson PJ, Haynes RH. dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. Embo J. 1993;12:4425–4431. doi: 10.1002/j.1460-2075.1993.tb06127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Webley SD, Hardcastle A, Ladner RD, Jackman AL, Aherne GW. Deoxyuridine triphosphatase (dUTPase) expression and sensitivity to the thymidylate synthase (TS) inhibitor ZD9331. Br J Cancer. 2000;83:792–799. doi: 10.1054/bjoc.2000.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Houghton JA, Tillman DM, Harwood FG. Ratio of 2′-deoxyadenosine-5′-triphosphate/thymidine-5′-triphosphate influences the commitment of human colon carcinoma cells to thymineless death. Clin Cancer Res. 1995;1:723–730. [PubMed] [Google Scholar]
- 35.Parsels LA, Parsels JD, Tai DC, Coughlin DJ, Maybaum J. 5-fluoro-2′-deoxyuridine-induced cdc25A accumulation correlates with premature mitotic entry and clonogenic death in human colon cancer cells. Cancer Res. 2004;64:6588–6594. doi: 10.1158/0008-5472.CAN-03-3040. [DOI] [PubMed] [Google Scholar]
- 36.Robinson HM, Jones R, Walker M, Zachos G, Brown R, Cassidy J, Gillespie DA. Chk1-dependent slowing of S-phase progression protects DT40 B-lymphoma cells against killing by the nucleoside analogue 5-fluorouracil. Oncogene. 2006;25:5359–5369. doi: 10.1038/sj.onc.1209532. [DOI] [PubMed] [Google Scholar]
- 37.Xiao Z, Xue J, Sowin TJ, Zhang H. Differential roles of checkpoint kinase 1, checkpoint kinase 2, and mitogen-activated protein kinase-activated protein kinase 2 in mediating DNA damage-induced cell cycle arrest: implications for cancer therapy. Mol Cancer Ther. 2006;5:1935–1943. doi: 10.1158/1535-7163.MCT-06-0077. [DOI] [PubMed] [Google Scholar]
- 38.Giannini G, Ristori E, Cerignoli F, Rinaldi C, Zani M, Viel A, Ottini L, Crescenzi M, Martinotti S, Bignami M, Frati L, Screpanti I, Gulino A. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002;3:248–254. doi: 10.1093/embo-reports/kvf044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berger SH, Barbour KW, Berger FG. A naturally occurring variation in thymidylate synthase structure is associated with a reduced response to 5-fluoro-2′-deoxyuridine in a human colon tumor cell line. Mol Pharmacol. 1988;34:480–484. [PubMed] [Google Scholar]