

Abstract
ATP-sensitive K+ (KATP) channels are activated by several vasodilating hormones and neurotransmitters through the PKA pathway. Here, we show that phosphorylation at Ser1387 of the SUR2B subunit is critical for the channel activation. Experiments were performed in human embryonic kidney (HEK) 293 cells expressing the cloned Kir6.1/ SUR2B channel. In whole cell patch, the Kir6.1/SUR2B channel activity was stimulated by isoproterenol via activation of β2 receptors. This effect was blocked in the presence of inhibitors for adenylyl cyclase or PKA. Similar channel activation was seen by exposing inside-out patches to the catalytic subunit of PKA. Because none of the previously suggested PKA phosphorylation sites accounted for the channel activation, we performed systematic mutational analysis on Kir6.1 and SUR2B. Two serine residues (Ser1351, Ser1387) located in the NBD2 of SUR2B were critical for the channel activation. In vitro phosphorylation experiments showed that Ser1387 but not Ser1351 was phosphorylated by PKA. The PKA-dependent activation of cell-endogenous KATP channels was observed in acutely dissociated mesenteric smooth myocytes and isolated mesenteric artery rings, where activation of these channels contributed significantly to the isoproterenol-induced vasodilation. Taken together, these results indicate that the Kir6.1/SUR2B channel is a target of β2 receptors and that the channel activation relies on PKA phosphorylation of SUR2B at Ser1387.
Keywords: K+ channel, second messenger, protein kinase A, vascular tones
ATP-SENSITIVE K+ (KATP) CHANNELS play an important role in vascular tone regulation (18, 32, 36). The Kir6.1/SUR2B channel is the major KATP channel isoform in vascular smooth myocytes (VSM) (8, 24, 30, 44), although mRNA of Kir6.2 has been detected (4, 19). The Kir6.1/SUR2B channel has a single-channel conductance of 35–40 pS (24, 35, 44) and is activated by nucleotide diphosphates (38). These properties are similar to those of VSM-endogenous KATP channels (48, 49). Disruption of the abcc9 (SUR2) gene leads to coronary artery vasospasm and elevated resting blood pressure (7). Kir6.1-null mice show a phenotype of Prinzmetal angina with impaired response to vasodilators (28). These KATP channel knockout (SUR2 or Kir6.1) animals also have a high rate of sudden death (7, 28) and fatal susceptibility to endotoxemia (21).
The vascular KATP channel is activated by PKA. Blockade of the PKA signaling pathway eliminates the channel modulation by several vasodilators, such as adenosine, calcitonin gene-related peptide, and epoxyeicosatrienoic acids (23, 31, 45, 47). The PKA signaling pathway can be activated by β-adrenergic receptors (β-ARs). Stimulation of the β-ARs hyperpolarizes VSMs leading to vasodilation (14, 20). Although both β1 and β2 receptors may be involved, the β2 receptor is known to play a major role. Mice lacking the β2 receptor develop hypertension during exercise or adrenaline challenges (6). Abnormalities in β-adrenergic responses are seen in rats with spontaneous hypertension (13). In humans, single nucleotide polymorphisms in the β2-AR gene are associated with increased vasoconstriction and stage-2 hypertension (11, 12). Pharmacological studies have suggested that K+ channels, especially the ATP-sensitive K+ (KATP) channels, play a role in the β-AR-mediated vasodilation (5, 10, 15, 20, 29, 34, 40).
Although the PKA-dependent activation of vascular KATP channels has been the focus of other studies, questions about how PKA stimulation leads to the channel activation remain open. For example, is the channel directly phosphorylated by PKA? Which subunit (Kir6.1, SUR2B, or both) is the target of PKA phosphorylation? To address these questions, we performed studies on the cloned Kir6.1/SUR2B channel. Our data suggest that phosphorylation of SUR2B underscores the channel activation by β-adrenergic receptor signaling pathway.
MATERIALS AND METHODS
Rat Kir6.1 (GenBank #D42145) and mouse SUR2B (GenBank #D86038) were used in the present study. Both cDNAs were cloned in the eukaryotic expression vector pcDNA3.1 and used for mammalian cell expression (44). Site-specific mutations were created with a site-directed mutagenesis kit using the Pfu DNA polymerase (Stratagene, La Jolla, CA). The human β2-adrenergic receptor (ADRB2) (GenBank #NM_000024) was purchased from Origene (Rockville, MD).
Human embryonic kidney cells
Human embryonic kidney cell line (HEK293, CRL-1573, Batch #2187595; American Type Culture Collection, Rockville, MD) were chosen to express the KATP channels.The HEK293 cells were cultured as a monolayer in the DMEM with 10% FBS and penicillin/streptomycin added. Cultured at 37°C with 5% CO2 in the atmosphere, the cells were split twice weekly. The HEK293 cells were transfected using lipofectamine2000 (Invitrogen, Carlsbad, CA) with 1 μg Kir6.1 and 3 μg SUR2B per 3-mm petri dish. To facilitate the identification of positively transfected cells, 0.5 μg green fluorescent protein (GFP) cDNA (pEGFP-N2; Clontech, Palo Alto, CA) was added to the cDNA mixture. Cells were disassociated from the monolayer using 0.25% trypsin ~24 h posttransfection. A few drops of the cell suspension were added on to 5 × 5 mm2 cover slips in a 35-mm petri dish. The cells were then cultured for 24–48 h before experiments.
Mesenteric arterial rings
Mesenteric arterial rings were obtained from Sprague-Dawley rats (250–350 g) in accordance with the guidelines for the care and use of laboratory animals by Georgia State University. The rats were anesthetized by inhaling saturated halothane vapor followed by cervical dislocation. The mesenteric arteries were dissected free and transferred to ice-cold Krebs solution containing (in mM): 118.0 NaCl, 25.0 NaHCO3, 3.6 KCl, 1.2 MgSO4, 1.2 KH2PO4, 11.0 glucose, and 2.5 CaCl2 (44). The arteries were cut into 6–8 endothelium-intact rings of 2 mm in length and stored in Krebs solution. Endothelium-denuded rings were also used in which the endothelium was removed by a rough plastic tube and tested by the loss of response to ACh. During the experiment, a ring was mounted on a force-electricity transducer (Model FT-302, iWorx/CBSciences, Dover, NH) in a tissue bath. With a 0.8 g preload, the ring was allowed to equilibrate in the tissue bath for 30 min when the tension was reduced to ~0.6 g. The tissue bath was filled with Krebs solution and perfused with 5% CO2 at 36°C. Arterial tone was measured as changes in isometric force. Only rings that showed a clear vasoconstriction response to 1.0 μM phenylephrine were used in the study.
Dissociated vascular smooth cells
Acutely dissociated vascular smooth muscle cells were prepared with a two-step enzyme digestion. Mesenteric arteries were obtained as previously described, cut into small segments (1–2 mm), and placed in a 5-ml saline solution containing (in mM): 140 NaCl, 5.4 KCl, 1 MgCl2, 0.1 CaCl2, 10 HEPES, and d-glucose 10 at room temperature for 10 min. The tissues were then placed in 1 ml solution containing 20 units of papain (Worthington Biochemical, Lakewood, NJ) and 1.25 mg DTT. After incubation for 30 min at 35°C, the tissue was washed once and then transferred to 1 ml solution containing 440 U of collagenase (CLS II; Worthington Biochemical) and 1.25 mg trypsin inhibitor (Sigma, St. Louis, MO) for 15 min. After a thorough wash, the tissue was moved to a 1-ml solution containing 20% FBS and triturated with a fire-polished Pasteur pipette to yield single smooth muscle cells. The cells were stored on ice and used within 8 h. A drop of cells was placed in a 35-mm tissue culture dish where the cells were allowed to attach to the surface. The cells that had typical smooth muscle morphology and did not show evident swelling or shrinkage were used for further studies.
Patch-clamp experiments
Patch-clamp experiments were performed at room temperature, as described previously (44). In brief, fire-polished patch pipettes were made with 1.2-mm borosilicate glass capillaries. Whole cell recording was performed using single-cell voltage clamp with recording pipettes of 4–6 MΩ. Current records were low-pass filtered (2 kHz, Bessel 4-pole filter, −3 dB), digitized (10 kHz, 16-bit resolution), and stored on computer disk for later analysis using the Clampfit 6 and 9 software (Axon Instruments, Sunnyvale, CA). The bath solution contained (in mM) 10 KCl, 135 potassium gluconate, 5 EGTA, 5 glucose, and 10 HEPES (pH = 7.4). The pipette solution contained (in mM) 10 KCl, 133 potassium gluconate, 5 EGTA, 5 glucose, 1 K2ATP, 0.5 NaADP, and 10 HEPES (pH = 7. 4), in which the free Mg2+ concentration was adjusted to 1 mM using MgCl2. Because the variation of Cl− concentrations in solutions was rather small, the resulting liquid junction potential was less than 1 mV, according to the Henderson equation, and was thereby not corrected. Inside-out patches were performed with symmetric high K+ in the bath and pipette (in mM): 10 KCl, 135 potassium gluconate, 5 EGTA, 5 glucose, and 10 HEPES (pH = 7.4), with [Mg2+] adjusted to 1 mM using MgCl2. After formation of a giga-seal, the patch was excised, and the intracellular side was exposed to bath solution. The holding potential was 0 mV and a constant single voltage of −60 mV was applied to the patch. All currents recorded from the inside-out patches were digitized in a higher sampling rate (20 kHz).
PKA phosphorylation sites
PKA phosphorylation sites were predicted using two online programs Kinasephos, (http://kinasephos.mbc.nctu.edu.tw/) (17) and NetPhosK (http://www.cbs.dtu.dk/services/NetPhosK) (3). A serine or threonine was considered for further studies as a putative PKA site if there were an alkaline amino acid at the −2 or −3 position.
In vitro phosphorylation
In vitro phosphorylation was performed on a SUR2B peptide fused to maltose-binding protein (MBP). A short cDNA sequence corresponding to residues 1308–1399 of SUR2B was produced and amplified using standard PCR. The sequence was then inserted into the pMal-c2 × vector (New England Biolabs, Ipswich, MA) that contains MBP sequence using HindIII and EcoR I. Mutations were created in the sequence with a site-directed mutagenesis kit (Stratagene). The wild-type and mutants were then transformed into protease-deficient Escherichia coli BL21, in which MBP-fusion peptides were induced with 0.3 mM isopropylthiogalactoside for 2 h. The MBP-fusion peptides were purified using amylose resin according to the protocol (New England Biolabs). The fusion peptides were subsequently incubated with the catalytic subunit of PKA (cPKA, P2645 from Sigma) in the following reaction: 5 μg fusion peptides in 5 μl elution buffer, which consisted of (in mM) 200 NaCl, 30 Tris3 HCl, 1 EDTA, 10 maltose, at pH 7.4, 5 μl of 5× reaction buffer, which consisted of (in mM) 125 Tris·HCl, 0.1 EGTA, at pH 7.5, 5 μl Mg-ATP solution, which consisted of (in mM) 20 MOPS, 25 β-glycerophosphate, 5 EGTA, 1 Na3VO4, 1 DTT, 75 MgCl2, 0.5 ATP, at pH 7.2, 10 units of cPKA in 10 μl H2O and 1 μl of 5 μCi/μl of 32P-γ-labeled ATP (Perkin-Elmer, San Francisco, CA). One hour later, 5 μl of 5× protein loading buffer were added to each sample to terminate the reaction. The samples were then subjected to electrophoresis in 10% SDS-PAGE gel, stained with Coomassie blue, and photographed. The gel was then fixed and dried. Autoradiography was carried out using a Fuji BAS 2500 Imaging Plate. This experiment was repeated twice.
Chemicals
Chemicals used in this study were purchased from Sigma unless otherwise stated. All chemicals were prepared as high-concentration stocks in double-distilled H2O or DMSO and were diluted in the recording solution to experimental concentrations immediately before usage. In cases in which DMSO was used, its concentration was maintained at less than 0.1% in the experimental solutions. This concentration of DMSO does not affect the Kir6.1/ SUR2B channel. Isoproterenol, glibenclamide, pinacidil, and forskolin were applied to cells using a perfusion system. Adrenergic β1 and β2 antagonists were administrated at least 2 min before and during the isoproterenol exposure. PKA inhibitors RP-cAMP was included in the pipette solution (200 μM) and added to the perfusion solution (100 μM). PKA inhibitory peptide (PKI5–24, 10 μM) was applied to the pipette solution. To avoid ATP degradation, all ATP-containing solutions were made immediately before experiments and used no longer than 4 h.
Statistics
Data are presented as means ± SE. Differences in means were tested with the ANOVA or Student’s t-test and were accepted as significant if P ≤ 0.05.
RESULTS
Baseline Kir6.1/SUR2B channel activity
The Kir6.1/ SUR2B channel was transiently expressed in HEK293 cells. Whole cell voltage-clamp was performed on GFP-positive cells. Without any treatment, the currents remained small or were slightly increased over a period of 8–10 min. Strong current activation was seen when the cell was exposed to 10 μM pinacidil. The pinacidil-activated currents were strongly inhibited by 10 μM glibenclamide. The currents did not show clear inward rectification (Fig. 1A). These characteristics are consistent with the Kir6.1/SUR2B currents reported previously (38, 46). HEK293 cells transfected with the expression vector alone were used as a negative control, in which small inward rectifier currents were seen. The currents were insensitive to pinacidil and glibenclamide (Fig. 1B).
Fig. 1.
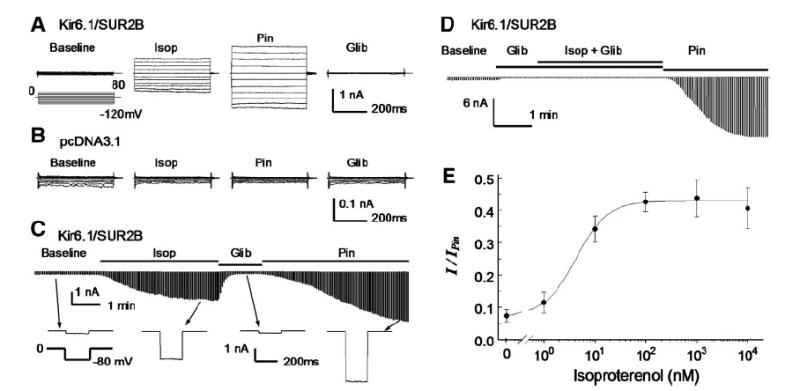
Kizr6.1/SUR2B channels expressed in human embryonic kidney (HEK)293 cells. A: whole cell currents were recorded from a cell transfected with Kir6.1/SUR2B. Symmetric concentrations of K+ (145 mM) were applied to both sides of cell membranes. The cell was held at 0 mV, and pulse voltages from −120 to 80 mV with a 20-mV increment were applied. The current amplitude increased in response to isoproterenol (Isop; 100 nM). The isoproterenol-activated currents were further activated by pinacidil (Pin; 10 μM) and inhibited by glibenclamide (Glib; 10 μM). B: currents recorded from another cell transfected with the expression vector alone were insensitive to isoproterenol, pinacidil, and glibenclamide. C: time course for the Kir6.1/SUR2B channel modulation. Whole cell currents were recorded with a holding potential at 0 mV and command pulses of −80 mV in every 3 s. After whole cell configuration was formed, the cell was perfused with extracellular solution for a 4-to 6-min baseline recording. Note that the baseline record was shortened in the figure. The currents were strongly activated by isoproterenol, and the maximum activation was reached during 3–4 min of the exposure. The currents were inhibited by glibenclamide (10 μM) and further activated by pinacidil (10 μM). The lower panel shows individual currents produced by a single command pulse. D: in the presence of glibenclamide, isoproterenol failed to activate the Kir6.1/SUR2B channel. E: concentration-dependent activation of Kir6.1/SUR2B currents by isoproterenol. The effect of isoproterenol was measured and normalized between the maximum channel inhibition by 10 μM glibenclamide and the maximum channel activation by 10 μM pinacidil. Baseline currents with no isoproterenol were 7.4 ± 1.9% (n = 18) of full channel activation by pinacidil. Evident activation of the Kir6.1/SUR2B currents was seen with 1 nM (11.5 ± 3.2%, n = 4), and the maximum activation was reached with 100 nM (42. 6 ± 3. 0%, n = 8). Further increase in isoproterenol concentration had no further activation, 1 μM (43.8 ± 5.7%, n = 6) and 10 μM (39.4 ± 5.7%, n = 7). The concentration-current relationship was described using the Hill equation y = 0.074 + 0.355/(1 + (EC50/[Isop])h), where y is normalized Kir6.1/SUR2B currents, [Isop] is isoproterenol concentration, EC50 (4.3 nM) is the Isop concentration for 50% channel activation, and h (1.4) is the Hill coefficient.
Activation of the Kir6.1/SUR2B channel by β2 receptor stimulation
The β2-AR was overexpressed in HEK293 cells together with Kir6.1 and SUR2B. The β-AR agonist isoproterenol was applied to the cell following stabilization of the baseline currents for 4–6 min. The isoproterenol exposure activated K+ currents that were sensitive to both pinacidil and glibenclamide. We found that without β2-AR transfection, the Kir6.1/SUR2B currents remained to be activated by isoproterenol. This observation is consistent with the previous finding that β2-AR is endogenously expressed in HEK293 cells (9, 39). Therefore, further experiments were conducted in the HEK293 cells without exogenous β-AR. The currents activated by isoproterenol were further activated by pinacidil and inhibited by glibenclamide (Fig. 1, A and C). The channel activation was totally eliminated in the presence of 10 μM glibenclamide (Fig. 1D) and not seen in cells transfected with the expression vector alone (Fig. 1B).
After currents were normalized between maximum channel inhibition by 10 μM glibenclamide and maximum activation by 10 μM pinacidil, the baseline currents averaged 7.4 ± 1.9% (n = 18) of the maximum channel activity. Isoproterenol (100 nM) increased the currents to 42.6 ± 3.0% (n = 8). The effect showed clear concentration dependence (Fig. 1E). Evident current activation occurred with 1 nM isoproterenol, and the maximum effect was reached with 100 nM. The EC50 was 4.3 nM with a Hill coefficient of 1.4 (Fig. 1E).
To identify the receptor subtype involved, we applied specific β-AR antagonists to the cells 2 min before and during the administration of isoproterenol. The isoproterenol effect was almost completely blocked by β2-AR antagonist ICI-118551 (100 nM), whereas β1-AR antagonist atenolol (1 μM) had no effect (Fig. 2, A-C). These results thus indicate that isoproterenol activates the Kir6.1/SUR2B channel through endogenous β2-AR of the HEK293 cells.
Fig. 2.
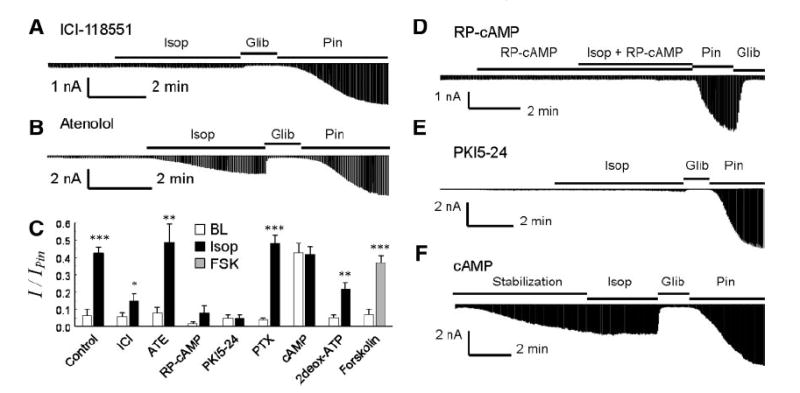
Dissection of signal pathway for Kir6.1/SUR2B channel activation by isoproterenol. A: in the presence of β2 receptor antagonist ICI-118551 (100 nM), isoproterenol had very little effect on the Kir6.1/SUR2B currents. B: with β1 antagonist atenolol (1 μM), isoproterenol remained to activate the Kir6.1/SUR2B currents. Note that β-AR antagonists were perfused to cells 5 min before and during the isoproterenol exposure. C: currents were normalized to pinacidil and glibenclamide effects. Open bar, baseline current; black bar, isoproterenol; gray bar, forskolin. 2Deox-ATP, 2′,5′-dideoxyadenosine-3′-triphosphate; PTX, pertusis toxin. *P < 0.5; **P < 0.01; ***P < 0.001 (n = 5 to 14). BL, baseline; FSK, forskolin. D: RP-cAMP, a potent PKA inhibitor, was applied in both pipette solution (200 μM) and perfusion solution (100 μM). The current’s activation by isoproterenol was almost completely blocked. E: similar blockade of the channel activation was observed with a PKA inhibitory peptide (PKI5–24, 10 μM) in the pipette solution. F: effect of cAMP (100 μM) in pipette solution on the Kir6.1/SUR2B currents. After formation of whole cell configuration, the current amplitude gradually increased and became plateaued at ~40% of the maximum activation by pinacidil in ~5 min. Application of isoproterenol did not produce further activation of the Kir6.1/SUR2B currents.
PKA dependence
The Kir6.1/SUR2B activation by isoproterenol was blocked when 8-(4-chlorophenylthio) adenosine-3′, 5′ -cyclic monophosphorothioate RP-isomer (RP-cAMP), a PKA inhibitor, was applied in both the pipette solution (200 μM) and the perfusion solution (100 μM) (Fig. 2, C and D). Similar blockade was seen in the presence of the specific PKA inhibitory peptide (PKI5–24, 10 μM) added in pipette solution, suggesting that PKA is involved in the activation of the channel by isoproterenol (Fig. 2, C and E).
Since activation of Gs stimulates adenylyl cyclase, we studied the effect of forskolin, a potent adenylyl cyclase activator, on the Kir6.1/SUR2B currents. Exposure to 10 μM forskolin activated the channel to almost the same degree as isoproterenol (Fig. 2C). The forskolin-activated currents also showed identical characteristics to those activated by isoproterenol. When the adenylyl cyclase inhibitor, 2′,5′-dideoxyadenosine 3′-triphosphate (2deox-ATP) was included in the pipette solution, the channel activation by isoproterenol was diminished to 21.3 ± 7.2% with 2 μM (n = 4) and 21.9 ± 4.9% with 10 μM (n = 4), respectively. Because no difference was seen in these two concentrations, they were pooled together to compare with the experimental control. We found that these values were significantly lower than those of the control (P < 0.01, n = 14; Fig. 2C), suggesting that Gs-activated adenylyl cyclase is involved.
The Kir6.1/SUR2B currents were gradually activated when 100 μM cAMP was included in pipette solution (Fig. 2, C and F). The currents averaged 42.8 ± 5.4% (n = 5) of the total currents activated by 10 μM pinacidil. Under such a condition, application of 100 nM isoproterenol produced no further increase in the current amplitude (Fig. 2, C and F).
It is known that persistent β2-AR stimulation switches the intracellular signaling from a Gs cascade to Gi (9). To determine whether Gi also affects the Kir6.1/SUR2B channel activation by isoproterenol, we pretreated the cells overnight with pertussis toxin, a potent Gi inhibitor. Such a treatment had no effect on the Kir6.1/SUR2B channel activation by 10 μM isoproterenol (Fig. 2C), indicating that Gi is not involved in the Kir6.1/SUR2B activation by isoproterenol.
To further demonstrate the PKA dependence, experiments were performed in excised inside-out patches. In the absence of nucleotide, the channels were mostly closed. Significant increase in the current amplitude was seen when the patches were exposed to perfusion solution containing Mg2+ and nucleotide, that is, 1.0 mM Mg2+, 0.5 mM ADP, and 1.0 mM ATP (Fig. 3), consistent with previous reports (38). When the internal membrane of inside-out patches was exposed to the catalytic subunit of PKA in the presence of the same concentrations of Mg2+, ADP, and ATP, the Kir6.1/SUR2B channel was further activated (Fig. 3). Such channel activation was mediated by the augmentation of the open-state possibility (NPo) with no evident effect on the single-channel conductance. These data thus indicate that the channel activation is independent of other cytosolic soluble factors.
Fig. 3.
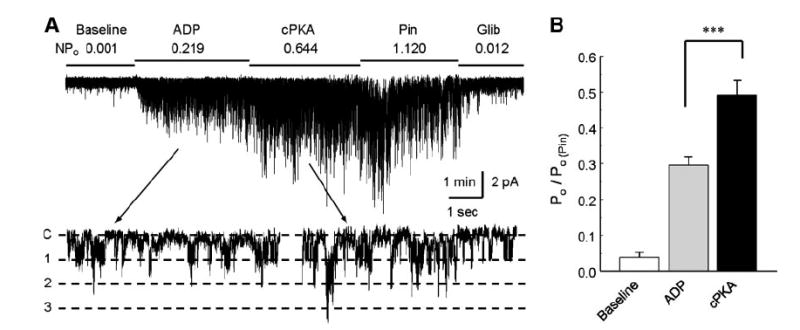
Augmentation of Kir6.1/SUR2B channel activation by the catalytic subunit of PKA. A: Kir6.1/SUR2B currents were recorded in an inside-out patch obtained from an HEK cell with a holding potential of −60 mV and equal concentrations of K+ applied to both sides of the patch membranes. The channel activity was low in the baseline. Exposure of the internal patch membrane to 1.0 mM ATP and 0.5 mM ADP led to activation of the channels that showed a unitary conductance ~35 pS (lower trace). The channels were further activated with an application of cPKA (100 U/ml) to the internal solution in the presence of same concentrations of ATP and ADP. B: summary of the experiment. Channel activity (NPo) was normalized to the level of pinacidil [Po/Po(Pin)]. The channel activity was rather low at baseline, increased markedly with ADP/ATP, and was further augmented with addition of cPKA. ***P < 0.001 (n = 9 patches).
PKA phosphorylation sites
A previous study suggested three PKA phosphorylation sites in the Kir6.1/SUR2B channel, that is, Ser385 in the Kir6.1 subunit, and Thr633 and Ser1465 in the SUR2B (33). To further understand the KATP channel activation by β-ARs, we mutated these three residues either individually or jointly. The Kir6.1_S385A/SUR2B mutant, in which the Ser385 was mutated to alanine, was activated by 100 nM isoproterenol (48.7 ± 3.0%, n = 10) and 10 μM forskolin (39.2 ± 2.3%, n = 6) to almost the same degree as the wild-type (WT) channel (Fig. 4, A and D). The channel was still activated when the Ser385 was mutated to asparagine or glutamate, though to a less degree in the S385E mutation (Fig. 4D). A channel with all three residues mutated (Kir6.1_S385A/ SUR2B_T633A_S1465A or 3AA) responded to isoproterenol (44.6 ± 4.3%, n = 6) and forskolin (42.0 ± 3.0%, n = 10) and showed no significant difference from the WT channel under our experimental condition (Fig. 4, B and D). Besides Ser385, Thr234 is another potential PKA site, as suggested by its counterpart residue (Thr224) in Kir6.2 (26). Neither Kir6.1_T234A nor Kir6.1_T234N mutation affected the channel activation by forskolin (Fig. 4, C and D).
Fig. 4.
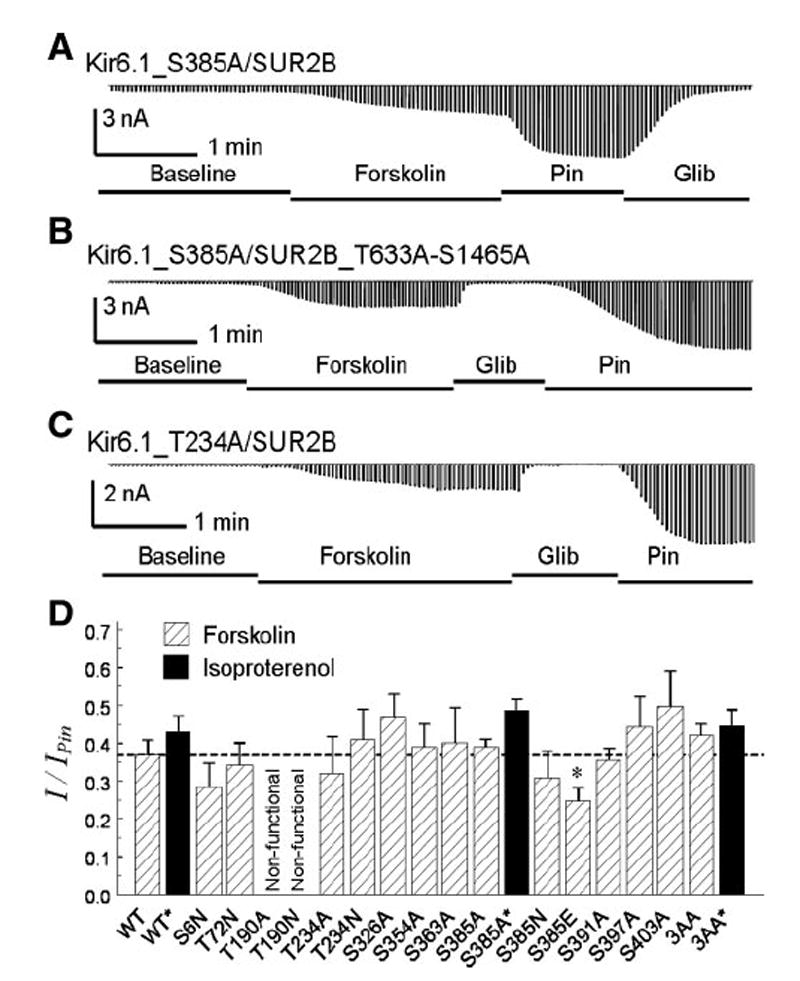
Mutation on potential PKA sites. A: with a mutation of Ser385 to alanine, the Kir6.1_S385A/SUR2 currents were strongly activated by forskolin. B: channel remained to be activated when three potential PKA sites; that is, Ser385 in the Kir6.1, and Ser1465 and Thr633 in the SUR2B subunit, were all mutated to alanine. C: Thr234 mutation on Kir6.1 did not abolish the channel activation by forskolin. D: summary of mutagenesis analysis of potential PKA sites. All mutations were constructed on Kir6.1 expect 3AA in which Ser385 in Kir6.1, and Ser1465/Thr633 in SUR2B were mutated to alanine. All mutant data were obtained from 4–7 cells except two, the 3AA with forskolin and the S385A with isoproterenol, which were obtained from 10 cells. The dashed line indicates the level of WT channel activation by forskolin. *P < 0.05.
Because none of the PKA sites suggested by previous studies seems to be involved in the activation of the Kir6.1/ SUR2B channel by isoproterenol, we performed systematic mutational analysis on both subunits. In the Kir6.1 subunit, nine other residues were predicted based on the PKA consensus sequences (3, 17). Site-specific mutations of eight of them had no effect on the channel activation by forskolin, while mutation of Thr190 to either alanine or asparagine failed to produce pinacidil-sensitive currents (Fig. 4D), consistent with a previous observation that the T190A-mutant channel is nonfunctional (42).
Subsequently, we performed systematic mutational analysis of the SUB2B subunit. Ten PKA consensus sequences were found in the NBD1 and NBD2. Each site was mutated to alanine. All of these mutants expressed functional currents similar to the WT channel, that is, small baseline currents stimulated by pinacidil and inhibited by glibenclamide (Table 1). Two residues (Ser1351 and Ser1387) were found to be critical. Mutation of either residue to alanine caused severe disruption of the channel activation by isoproterenol and forskolin that was independent of the pinacidil effect (Fig. 5, A, B, and E; Table. 1). The effect of cAMP dialysis on Kir6.1/ SUR2B_S1387A was also tested (Fig. 5, D and E). With the mutation, cAMP failed to activate the channel. Ser1351 is located in the NBD2 immediately following the Walker A sequence, and 36 residues away lies the Ser1387. Two serine residues are found in the NBD1 at corresponding locations, that is, Ser710 and Ser748 (Fig. 5F). Mutation of either residue did not affect the channel activation by forskolin (Fig. 5, C and E). Mutation of the remaining potential PKA sites had no effect on the channel sensitivity to forskolin either (Fig. 5E).
Table 1.
Effect of forskolin on Kir6.1/SUR2B and SUR2B mutants
| Baseline | Forskolin | Glib | Pinacidil | n | |
|---|---|---|---|---|---|
| WT | 0.64±0.34 | 1.89±0.60 | 0.39±0.11 | 4.08±0.98 | 7 |
| T633A | 0.69±0.27 | 3.58±1.59 | 0.34±0.11 | 6.63±2.71 | 5 |
| S710A | 0.76±0.12 | 3.65±0.82 | 0.22±0.05 | 9.04±1.96 | 5 |
| S745A | 0.64±0.09 | 1.59±0.30 | 0.33±0.07 | 3.92±0.71 | 9 |
| S748A | 0.57±0.10 | 2.41±0.54 | 0.24±0.09 | 5.76±0.66 | 5 |
| T782A | 0.78±0.26 | 1.79±0.70 | 0.34±0.05 | 4.50±1.67 | 5 |
| S1347A | 0.71±0.30 | 1.96±0.60 | 0.20±0.08 | 4.56±0.81 | 6 |
| S1351A | 0.37±0.15 | 0.48±0.15* | 0.28±0.14 | 3.58±0.67 | 5 |
| S1351N | 0.29±0.12 | 0.25±0.11* | 0.23±0.11 | 3.44±0.69 | 4 |
| S1387A | 0.26±0.07 | 0.48±0.16* | 0.16±0.06 | 4.99±1.60 | 6 |
| S1464A | 0.88±0.46 | 5.88±1.91 | 0.50±0.24 | 11.26±2.49 | 5 |
| S1465A | 0.94±0.15 | 1.79±0.46 | 0.68±0.19 | 2.81±0.87 | 5 |
All of the mutants showed small baseline currents (nA) similar to the wild-type (WT) Kir6.1/SUR2B channel. The currents were activated by pinacidil and inhibited by glibenclamide to a similar degree. The currents of the Ser1351 and Ser1387 mutants appeared to be smaller at the baseline, but they were not significantly different from the WT. After exposure to forskolin, the Ser1351 and Ser1387 mutants had smaller currents than the WT and other mutants (*P < 0. 01). The normalized currents with forskolin exposure are shown in Fig. 5E.
Fig. 5.
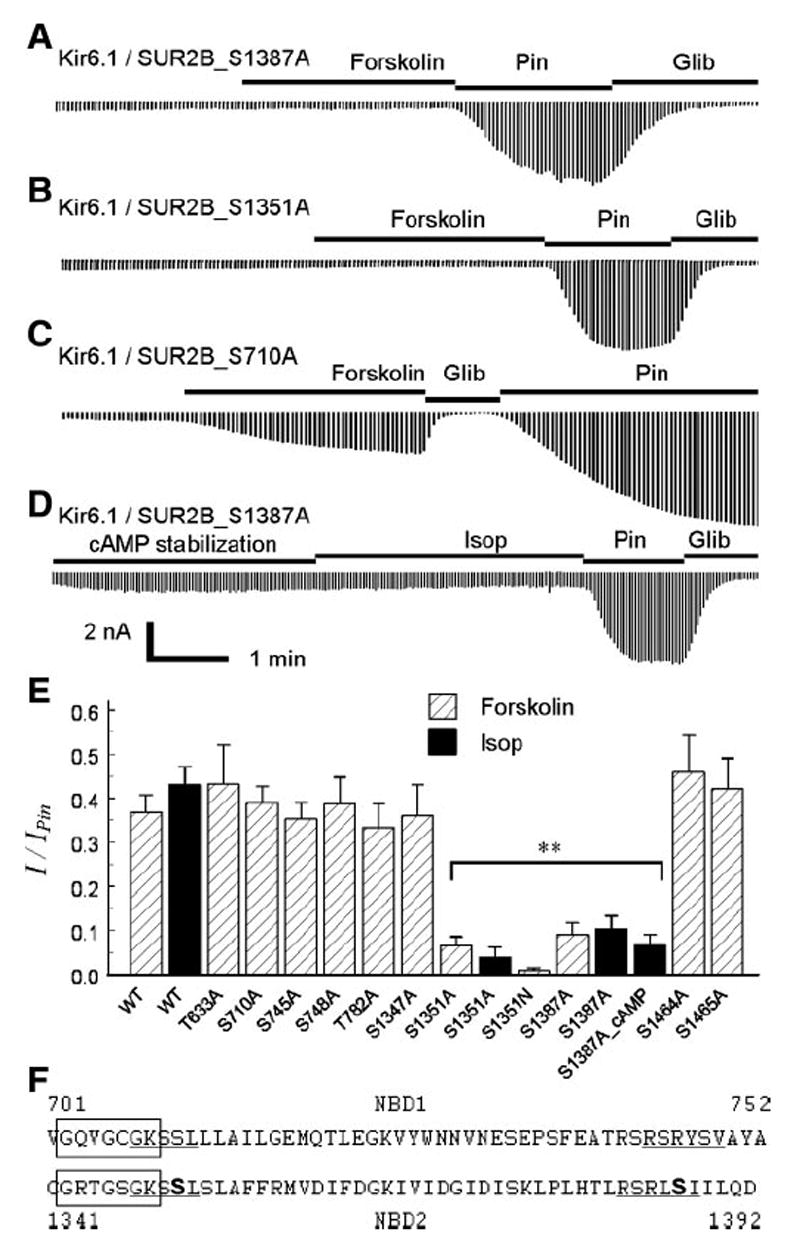
Identification of PKA phosphorylation sites in SUR2B. Mutants were coexpressed with WT Kir6.1 in HEK cells. A and B: site-specific mutation of Ser1387 and Ser1351 in SUR2B abolishes the channel activation by 10 μM forskolin. C: similar mutation at Ser710 had no effect on the forskolin sensitivity. D: intracellular dialysis of cAMP (100 μM in pipette solution) showed only modest stimulation of the S1387A currents, in sharp contrast to the WT channel shown in Fig. 2F. E: compared with WT, mutations of Ser1351 and Ser1387 caused a loss of channel activation by forskolin. The Ser1351 and Ser1387 mutants failed to be activated by isoproterenol either (**P < 0. 01, n = 4 to 6). F: alignment of amino acid sequences around Walker A in NBD1(top) and NBD2 (bottom). Boxed are Walker A sequences. Ser1351 and Ser1387 are bold, and the proposed PKA consensus sequence is underlined. Similar sequences are seen in NBD1, while both Ser710 and Ser748 are not functional PKA sites.
To address the question of whether these two serine residues in SUR2B can be phosphorylated by PKA, we performed in vitro phosphorylation experiments on SUR2B peptides (containing residues 1308–1399 of SUR2B) with and without the S1351A and/or S1387A mutation. The peptides were fused to MBP and were expressed in bacteria. After purification with the amylose affinity column, these peptides were subjected to in vitro phosphorylation in the presence of the catalytic subunit of PKA and 32P-γ-labeled-ATP. Strong phosphorylation was seen in the WT peptide and the peptide containing the S1351A mutation. The peptides with the S1387A mutation either alone or jointly with the S1351A mutation failed to be phosphorylated (Fig. 6A).
Fig. 6.
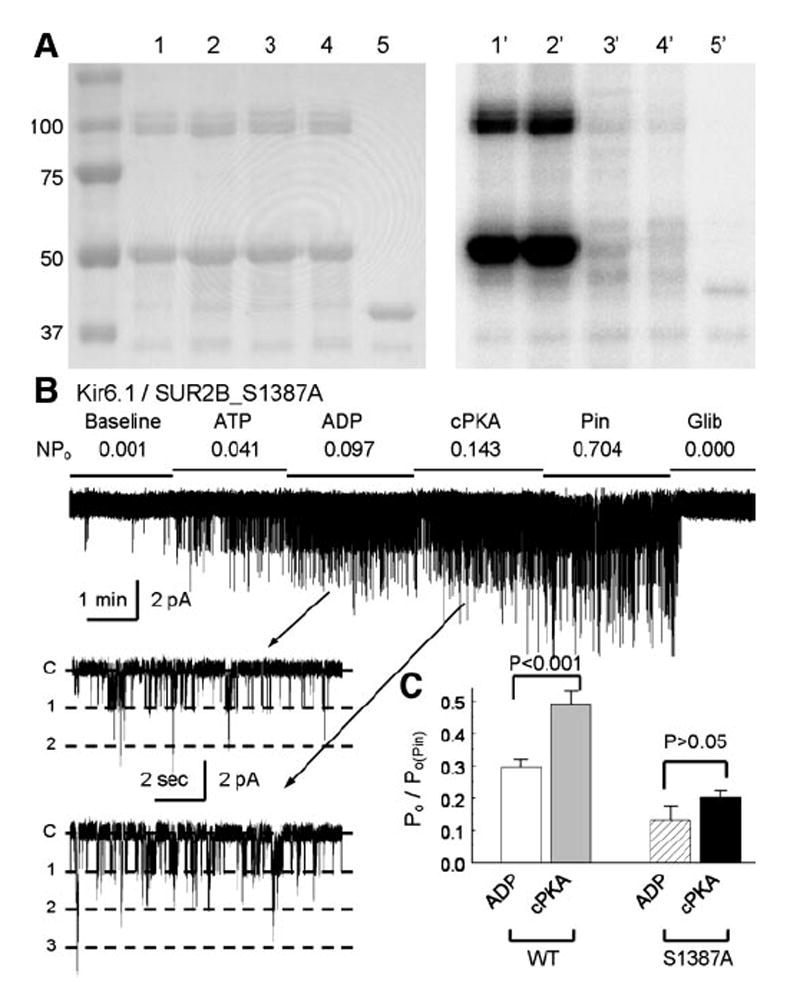
Characterization of Ser1387 in PKA phosphorylation. A: short peptide in SUR2B (residues 1308–1399) was fused to C-terminus of MBP. Site-specific mutations of S1351A and/or S1387A were created on the peptide. After purification with amylose-affinitive beads, correct peptides were revealed in SDS-PAGE gel (left). A 50-kDa band is seen in lanes 1–4 containing WT, S1351A, S1387A, and S1351A/S1387A double mutations, respectively. Lane 5, MBP protein. Another band of ~100 kDa is also seen, suggesting dimerization of the fusion peptides. Autoradiograph with 32P-γ-labeled ATP after an 8-h exposure (right) showed positive labeling of the WT and S1351A but not the S1387A, and S1351A/S1387A peptides and the control MBP. B and C: compared with the WT channel, the S1387A mutant was slightly activated by the catalytic subunit of PKA (n = 5). Note that the S1387A currents with cPKA exposure were smaller than the baseline level of the WT channel.
In excised patches, the S1387A channel had a rather low baseline activity. Channel activity increased with an exposure to 0.5 mM ADP and 1.0 mM ATP. Under this condition, the channel activity was slightly stimulated by the catalytic subunit of PKA (100 U/ml) (Fig. 6, B and C). Although the S1387A mutation did not completely eliminate the channel activation, the channel activity remained lower than the basal activity of the WT channel (Fig. 6C). These results are consistent with our observations in whole cell recordings, indicating that the Ser1387 is likely to be a PKA site (Table 1 and Fig. 5A).
Activation of vascular KATP channels by isoproterenol
In acutely dissociated VSMs obtained from rat mesenteric arteries, inward K+ currents were activated with an exposure to 100 nM isoproterenol (Fig. 7A). The isoproterenol-activated currents averaged 35.4 ± 7.3% (n = 6) of the total currents activated by 10 μM pinacidil and showed a nearly identical pattern to the Kir6.1/SUR2B currents expressed in HEK cells (Fig. 7, A and B). The same concentration of isoproterenol failed to produce significant current activation when the pipette solution contained PKI5–24 (Fig. 7B). These results, which are consistent with those obtained from the Kir6.1/SUR2B channel, suggest that the VMS-endogenous KATP channels are activated by PKA phosphorylation.
Fig. 7.
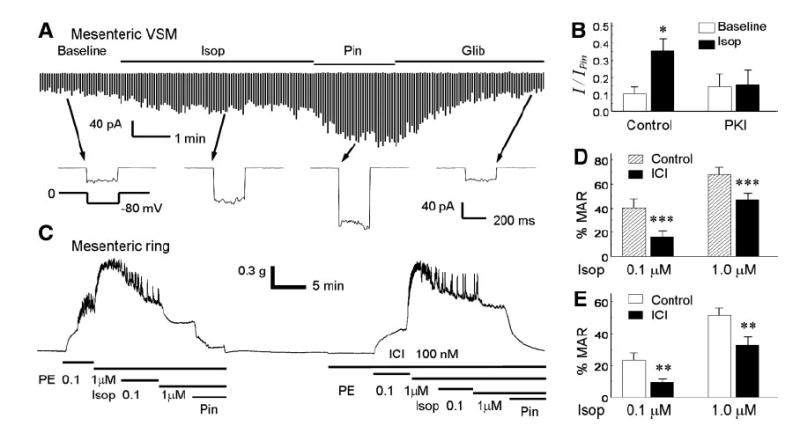
Effects of isoproterenol on vascular smooth muscles. A: whole-cell currents were recorded from a vascular smooth myocyte (VSM) acutely dissociated from the mesenteric artery. Exposure to 100 nM isoproterenol activated the currents that were further activated by 10 μM pinacidil. Bottom: current traces recorded with a single voltage protocol. B: isopoterenol-activated currents were not seen in the presence of PKI5–24 (PKI, 10 μM) in the pipette solution. C: in a mesenteric ring, phenylephrine (PE) produced vasoconstriction that was reversed by isoproterenol. Such vasorelaxation effects were markedly attenuated in the presence of a β2-AR antagonist ICI-118551 (ICI). Note that pinacidil can completely relax the mesenteric ring. D: summary of vasorelaxation effects of isoproterenol on the PE-induced vasoconstriction with and without ICI on endothelium-intact rings (n = 7). E: similar vasorelaxation was observed in endothelium-denuded rings (n = 6). **P < 0.01; ***P < 0.001.
In endothelium-intact mesenteric rings, phenylephrine produced vasoconstriction. Such vasoconstriction could be dose-dependently reversed by isoproterenol and was completely eliminated by pinacidil (Fig. 7C). The vasorelaxation effect of isoproterenol was significantly attenuated with a pretreatment with the β2-AR antagonist ICI-118551 (100 nM) (Fig. 7, C and D). A similar phenomenon was found in endothelium-denuded mesenteric rings (Fig. 7E). These results suggest that the isoproterenol-induced vasodilation involves β2-AR and requires the activation of VSM-endogenous KATP channels, consistent with our observations in the heterologous expression system and acutely dissociated VSMs.
DISCUSSION
Our results have shown that the Kir6.1/SUR2B channel is a downstream target of β2-ARs. The channel regulation results from activation of the Gs-adenylyl cyclase-cAMP-PKA pathway. Two serine residues in the NBD2 are critical for the channel activation by isoproterenol and forskolin. One of the residues, indeed, can be phosphorylated by PKA in vitro. The PKA stimulation seems to underscore the activation of VSM-endogenous KATP channels and relaxation of mesenteric arteries by isoproterenol.
The Kir6.1/SUR2B channel activation by isoproterenol is mediated by the β2-ARs-Gs-adenylyl cyclase-cAMP-PKA pathway. The β2-AR involvement is consistent with existing experimental evidence showing that the β2-ARs are expressed in vascular smooth muscles, β2-AR antagonism affects vascular tones, and β2-AR gene targeting causes disruption of vascular regulation and hypertension (6, 11-14, 20). In agreement with previous findings in cell-endogenous KATP channels (5, 45, 47), the Kir6.1/SUR2B channel activation by isoproterenol relies on PKA activity, as PKA inhibitors, RP-cAMP, and PKI5–24, block the channel activation. Activation of adenylyl cyclase is necessary, since intracellular dialysis of cAMP and activation of the adenylyl cyclase by forskolin augment the Kir6.1/SUR2B currents to the same extent as isoproterenol. The β2-ARs can be phosphorylated by PKA leading to a switch to Gi cascade (9). Our results suggest that Gi is not a key player in the KATP channel activation by isoproterenol, as the channel activation remains following Gi inhibition by a pretreatment of the cells with pertussis toxin. The involvement of the Gs-adenylyl cyclase-cAMP-PKA intracellular signaling system for Kir6.1/SUR2B channel activation is consistent with several previous reports on vascular endogenous KATP channels (5, 14, 20, 23, 31, 45, 47). Beside the PKA system, the exchange proteins directly activated by cAMP (Epacs) have been reported to mediate the inhibitory effects of cAMP on the pancreatic KATP isoform (22, 25). Glucagon-like peptide-1 raises cAMP concentrations that initiate binding of Epacs to SUR1 and inhibit the Kir6.2/SUR1 channel (22). Such a PKA-independent effect of cAMP does not seem to play a significant role in the activation of vascular KATP channel, as the channel activation is abolished by PKA inhibitors, as well as mutation of the PKA site in SUR2B.
A previous study has shown that the Kir6.1/SUR2B channel is modulated by PKA, and the channel activation was due to direct phosphorylation of the channel proteins at three sites (one in Kir6.1 and two in SUR2B) (33). We have examined these residues in the present study. However, our results suggest that these three residues do not seem to play a role in the Kir6.1/SUR2B channel activation by PKA in the presence of physiological levels of nucleotides, as mutations of these residues did not show significant effect on the channel activation by isoproterenol and forskolin. The different observations are probably due to experimental conditions. The Kir6.1/ SUR2B currents were recorded in the presence of 0.5 mM UDP in the study by Quinn et al. (33) compared with 0.5 mM ADP in the present study. Because the Kir6.1/SUR2B channel is strongly activated by UDP, forskolin only increased the whole cell current amplitude by ~50% in their study (33) by ~500% in the present study.
In the present study, we have systematically mutated all 11 consensus PKA sites in the Kir6.1 subunit. Ten of them do not seem to be functionally phosphorylated by PKA, as mutations to nonphosphorylatable residues do not affect the channel activation by forskolin. The role of the other residue Thr190 remains uncertain, as channels with a mutation at this position were nonfunctional (42; see also Fig. 4D).
In the SUR2B subunit, our systematic mutational analysis revealed two serine residues, that is, Ser1351 and Ser1387, that are both located in the NBD2. Mutation of either one abrogates Kir6.1/SUR2B channel activation by isoproterenol and forskolin. Our in vitro phosphorylation study in a purified fusion peptide of SUR2B shows that mutation of Ser1351 does not affect phosphorylation by PKA. A straightforward explanation of the results is that the Ser1351 may be involved in ADP binding on the Walker-A motif. Its mutations thus affect ADP binding as well as the consequence of PKA phosphorylation of another residue(s). It is also possible that the isolated peptide may have lost its normal folding and failed to be phosphorylated in vitro. Interestingly, a corresponding serine is also found in SUR1 (Ser1387); its mutation (SUR1_S1387F) and deletion (SUR1_ΔS1387) have been found in patients with congenital hyperinsulinism (1, 41, 43). In the current study, a replacement of this serine residue (SUR2B_S1351) with either nonpolar alanine or polar asparigine causes disruption of the channel activation by PKA stimulation. Therefore, Ser1351 is an important site for channel regulation, although it does not seem to be phosphorylated by PKA.
Why doesn’t the mutation of corresponding residue in NBD1 (Ser710) affect the channel activation by PKA? This may be related to the difference in the function of NBD1 and NBD2. The NBD1 in SURs hosts a Mg2+-independent high-affinity nucleotide-binding domain, while the nucleotide-binding domain in NBD2 is Mg2+ dependent and has low affinity (27). Thus mutation of the serine residue in NBD1 may have little effect on nucleotide binding and channel activity.
Ser1387 is a phosphorylation site critical for the channel regulation by PKA, as shown in our pharmacological studies, mutagenesis analysis, in vitro phosphorylation assay, and direct exposure to cPKA. This is a novel finding compared with previous studies in PKA regulation on KATP channels. Lin et al. (26) and Beguin et al. (2) have found that PKA activates Kir6.2/SUR1 through phosphorylation of the Kir6.2 subunit (Ser224 and Ser372, respectively). SUR1 subunit has also been proposed as a target of PKA. Beguin et al. (2) reported that a human-specific residue on SUR1 (Ser1571) has basal level phosphorylation. Light et al. (25) reported that the Kir6.2/ SUR1 channel is inhibited by the glucagon-like peptide through PKA phosphorylation at Ser1448 of the SUR1 subunit. Both PKA sites on SUR1 subunit are located in the NBD2, in which ADP binding takes place (27). Thus, it is possible that PKA phosphorylation affects the ADP sensitivity and thus the channel activity (25). These two sites are SUR1 specific, as the corresponding sites are not phosphorylatable residues or not in a consensus PKA sequence in SUR2B. In contrast, our newly identified PKA phosphorylation site Ser1387 in SUR2B is conserved among species and in all three SURs (Fig. 8A). It is of interest to know whether such a site plays a role in other isoforms of KATP channels.
Fig. 8.
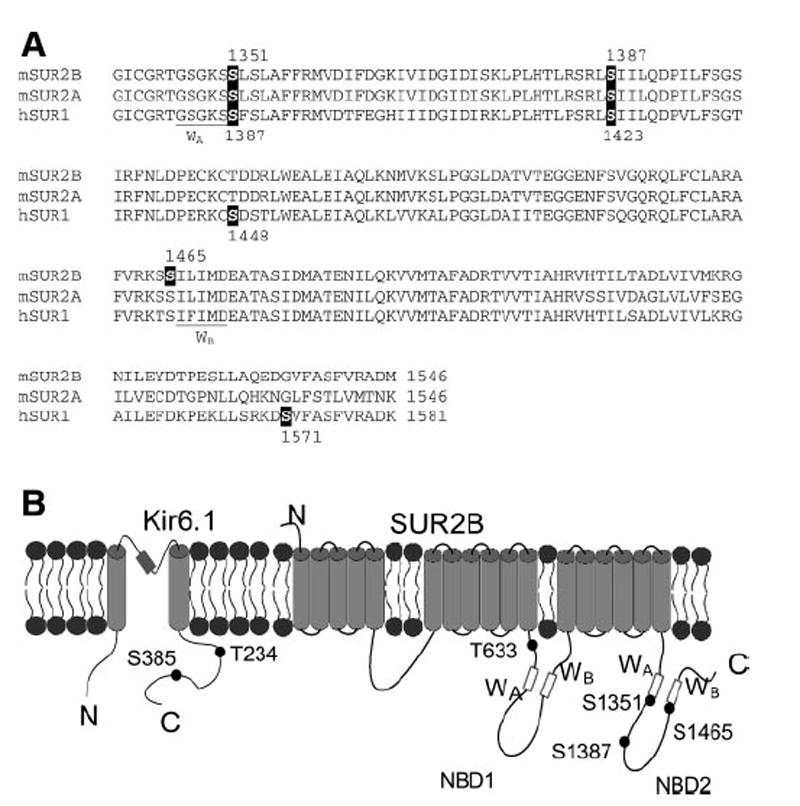
The sites important for PKA regulation on KATP channels. A: alignment of NBD2 of mouse SUR2A, SUR2B (residue 1339-end), and human SUR1 (residue 1376-end). The nucleotide binding Walker A (WA) and Walker B (WB) are underlined. The S1351 that is found important for PKA activation and PKA phosphorylation site (S1387) in SUR2B, and the conserved sites in SUR2A and SUR1 are highlighted in reverse form. The previously suggested PKA site (S1465) in SUR2B and sites (S1448 and S1571) in SUR1 are also highlighted. B: schematic representation of Kir6.1 and SUR2B. The nucleotide binding domain 1 (NBD1) and 2 (NBD2) and Walker A (WA) and Walker B (WB) and N, C terminus are illustrated. The relative positions of important sites (Kir6.1_Thr234, Ser385 and SUR2B_Thr633, Ser1351, Ser1387, and Ser1465) are marked. Note the Kir6.1_Thr234 and Ser385 are also the corresponding sites of Kir6.2_Thr224 and Ser372).
It should be noted that Ser1387 cannot explain the full effects of PKA as cPKA moderately activates the Kir6.1/ SUR2B_S1387A channel in inside-out configuration (Fig. 6B). This may be due to the presence of other unconventional PKA sites that were not detected by our PKA consensus sequence screening. Besides, the possibility of phosphorylation on Ser1351 cannot be excluded, as the isolated peptide may differ in protein folding, losing the capability to be phosphorylated. In addition, there may be other adaptor proteins and scaffolds absent in the in vitro phosphorylation assay, such as A-kinase anchoring proteins (15) and caveolae (37) that are important for PKA modulation of native vascular KATP channels.
In conclusion, our results indicate that the Kir6.1/SUR2B channel is a downstream effector of β2 receptors. The channel activation involves Gs, adenylyl cyclase, cAMP, and PKA. Two serine residues (Ser1351 and Ser1387) in the SUR2B subunit are important for PKA activation, and the channel protein is likely phosphorylated at Ser1387. The demonstration of an effector protein of β2-ARs and intracellular signaling cascades may allow for the creation of therapeutical modalities by targeting these molecules and their regulation and controlling vascular tones more effectively.
Acknowledgments
We are grateful to Dr. Susumu Seino and Yoshihisa Kurachi for their gifts of the Kir6.1 and SUR2B cDNAs, respectively. We also thank Nga Ta, Anetta Pool, and Morium Chowdhury for their technical assistance.
GRANTS This work was supported by the National Institutes of Health Grant HL-067890 and Georgia State University Research Program Enhancement Fund.
References
- 1.Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- 2.Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human KATP channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 1999;18:4722–4732. doi: 10.1093/emboj/18.17.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 4.Cao K, Tang GH, Hu DH, Wang R. Molecular basis of ATP-sensitive K+ channels in rat vascular smooth muscles. Biochem Biophys Res Commun. 2002;296:463–469. doi: 10.1016/s0006-291x(02)00892-6. [DOI] [PubMed] [Google Scholar]
- 5.Chang HY. The involvement of ATP-sensitive potassium channels in beta2-adrenoceptor agonist-induced vasodilatation on rat diaphragmatic microcirculation. Br J Pharmacol. 1997;121:1024–1030. doi: 10.1038/sj.bjp.0701192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the beta 2 adrenergic receptor gene. J Biol Chem. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- 7.Chutkow WA, Pu JL, Wheeler MT, Wada T, Makielski JC, Burant CF, McNally EM. Episodic coronary artery vasospasm and hypertension develop in the absence of Sur2 KATP channels. J Clin Invest. 2002;110:203–208. doi: 10.1172/JCI15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui Y, Tran S, Tinker A, Clapp LH. The molecular composition of KATP channels in human pulmonary artery smooth muscle cells and their modulation by groWTh. Am J Respir Cell Mol Biol. 2002;26:135–143. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- 9.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 10.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science. 2001;293:98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 11.Dishy V, Landau R, Sofowora GG, Xie HG, Smiley RM, Kim RB, Byrne DW, Wood AJJ, Stein CM. beta2-Adrenoceptor Thr164IIe polymorphism is associated with markedly decreased vasodilator and increased vasoconstrictor sensitivity in vivo. Pharmacogenetics. 2004;14:517–522. doi: 10.1097/01.fpc.0000114763.78957.ec. [DOI] [PubMed] [Google Scholar]
- 12.Ge D, Huang J, He J, Li B, Duan X, Chen R, Gu D. beta2-Adrenergic receptor gene variations associated with stage-2 hypertension in northern Han Chinese. Ann Hum Genet. 2005;69:36–44. doi: 10.1046/j.1529-8817.2003.00093.x. [DOI] [PubMed] [Google Scholar]
- 13.Goto K, Fujii K, Abe I. Impaired beta-adrenergic hyperpolarization in arteries from prehypertensive spontaneously hypertensive rats. Hypertension. 2001;37:609–613. doi: 10.1161/01.hyp.37.2.609. [DOI] [PubMed] [Google Scholar]
- 14.Guimaraes S, Moura D. Vascular adrenoceptors: An update. Pharmacol Rev. 2001;53:319–356. [PubMed] [Google Scholar]
- 15.Hayabuchi Y, Dart C, Standen NB. Evidence for involvement of A-kinase anchoring protein in activation of rat arterial KATP channels by protein kinase A. J Physiol London. 2001;536:421–427. doi: 10.1111/j.1469-7793.2001.0421c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hein TW, Zhang CH, Wang W, Kuo L. Heterogeneous beta2-adrenoceptor expression and dilation in coronary arterioles across the left ventricular wall. Circulation. 2004;110:2708–2712. doi: 10.1161/01.CIR.0000134962.22830.CF. [DOI] [PubMed] [Google Scholar]
- 17.Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005;33:W226–W229. doi: 10.1093/nar/gki471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jansen-Olesen I, Mortensen CH, El Bariaki N, Ploug KB. Characterization of KATP -channels in rat basilar and middle cerebral arteries: Studies of vasomotor responses and mRNA expression. Eur J Pharmacol. 2005;523:109–118. doi: 10.1016/j.ejphar.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 20.Johnson M. Molecular mechanisms of beta2-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117:18–24. doi: 10.1016/j.jaci.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Kane GC, Lam CF, O’Cochlain F, Hodgson DM, Reyes S, Liu XK, Miki T, Seino S, Katusic ZS, Terzic A. Gene knockout of the KCNJ8-encoded Kir6.1 KATP channel imparts fatal susceptibility to endotoxemia. FASEB J. 2006;20:2271–2280. doi: 10.1096/fj.06-6349com. [DOI] [PubMed] [Google Scholar]
- 22.Kang GX, Chepurny OG, Malester B, Rindler MJ, Rehmann H, Bos JL, Schwede F, Coetzee WA, Holz GG. cAMP sensor Epac as a determinant of ATP-sensitive potassium channel activity in human pancreatic beta cells and rat INS-1 cells. J Physiol London. 2006;573:595–609. doi: 10.1113/jphysiol.2006.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleppisch T, Nelson MT. Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase. Proc Natl Acad Sci USA. 1995;92:12441–12445. doi: 10.1073/pnas.92.26.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Wu J, Jiang C. Differential expression of Kir6.1 and SUR2B mRNAs in the vasculature of various tissues in rats. J Membr Biol. 2003;196:61–69. doi: 10.1007/s00232-003-0625-z. [DOI] [PubMed] [Google Scholar]
- 25.Light PE, Fox JEM, Riedel MJ, Wheeler MB. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Mol Endocrinol. 2002;16:2135–2144. doi: 10.1210/me.2002-0084. [DOI] [PubMed] [Google Scholar]
- 26.Lin YF, Jan YN, Jan LY. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. EMBO J. 2000;19:942–955. doi: 10.1093/emboj/19.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuo M, Kimura Y, Ueda K. KATP channel interaction with adenine nucleotides. J Mol Cell Cardiol. 2005;38:907–916. doi: 10.1016/j.yjmcc.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 28.Miki T, Suzuki M, Shibasaki T, Uemura H, Sato T, Yamaguchi K, Koseki H, Iwanaga T, Nakaya H, Seino S. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- 29.Ming Z, Parent R, Lavallee M. beta2-Adrenergic dilation of resistance coronary vessels involves KATP channels and nitric oxide in conscious dogs. Circulation. 1997;95:1568–1576. doi: 10.1161/01.cir.95.6.1568. [DOI] [PubMed] [Google Scholar]
- 30.Morrissey A, Rosner E, Lanning J, Parachuru L, Dhar CP, Han S, Lopez G, Tong X, Yoshida H, Nakamura TY, Artman M, Giblin JP, Tinker A, Coetzee WA. Immunolocalization of KATP channel subunits in mouse and rat cardiac myocytes and the coronary vasculature. BMC Physiol. 2005;5:1. doi: 10.1186/1472-6793-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quayle JM, Bonev AD, Brayden JE, Nelson MT. Calcitonin-gene-related peptide activated ATP-sensitive K+ currents in rabbit arterial smooth-muscle via protein-kinase-A. J Physiol London. 1994;475:9–13. doi: 10.1113/jphysiol.1994.sp020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 33.Quinn KV, Giblin JP, Tinker A. Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ Res. 2004;94:1359–1366. doi: 10.1161/01.RES.0000128513.34817.c4. [DOI] [PubMed] [Google Scholar]
- 34.Randall MD, Mcculloch AI. The involvement of ATP-sensitive potassium channels in beta-adrenoceptor-mediated vasorelaxation in the rat isolated mesenteric arterial bed. Br J Pharmacol. 1995;115:607–612. doi: 10.1111/j.1476-5381.1995.tb14975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Repunte VP, Nakamura H, Fujita A, Horio Y, Findlay I, Pott L, Kurachi Y. Extracellular links in Kir subunits control the unitary conductance of SUR/Kir6.0 ion channels. EMBO J. 1999;18:3317–3324. doi: 10.1093/emboj/18.12.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenblum WI. ATP-sensitive potassium channels in the cerebral circulation. Stroke. 2003;34:1547–1552. doi: 10.1161/01.STR.0000070425.98202.B5. [DOI] [PubMed] [Google Scholar]
- 37.Sampson LJ, Hayabuchi Y, Standen NB, Dart C. Caveolae localize protein kinase A signaling to arterial ATP-sensitive potassium channels. Circ Res. 2004;95:1012–1018. doi: 10.1161/01.RES.0000148634.47095.ab. [DOI] [PubMed] [Google Scholar]
- 38.Satoh E, Yamada M, Kondo C, Repunte VP, Horio Y, Iijima T, Kurachi Y. Intracellular nucleotide-mediated gating of SUR/Kir6.0 complex potassium channels expressed in a mammalian cell line and its modification by pinacidil. J Physiol London. 1998;511:663–674. doi: 10.1111/j.1469-7793.1998.663bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt JM, Stork PJS. beta2-Adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein Rap1 and the serine/threonine kinase B-Raf. J Biol Chem. 2000;275:25342–25350. doi: 10.1074/jbc.M003213200. [DOI] [PubMed] [Google Scholar]
- 40.Sheridan BC, McIntyre RC, Meldrum DR, Fullerton DA. KATP channels contribute to beta- and adenosine receptor-mediated pulmonary vasorelaxation. Am J Physiol Lung Cell Molec Physiol. 1997;17:L950–L956. doi: 10.1152/ajplung.1997.273.5.L950. [DOI] [PubMed] [Google Scholar]
- 41.Stanley CA, Thornton PS, Ganguly A, MacMullen C, Underwood P, Bhatia P, Steinkrauss L, Wanner L, Kaye R, Ruchelli E, Suchi M, Adzick NS. Preoperative evaluation of infants with focal or diffuse congenital hyperinsulinism by intravenous acute insulin response tests and selective pancreatic arterial calcium stimulation. J Clin Endocrinol Metab. 2004;89:288–296. doi: 10.1210/jc.2003-030965. [DOI] [PubMed] [Google Scholar]
- 42.Thorneloe KS, Maruyama Y, Malcolm AT, Light PE, Walsh MP, Cole WC. Protein kinase C modulation of recombinant ATP-sensitive K+ channels composed of Kir6.1 and/or Kir62 expressed with SUR2B. J Physiol London. 2002;541:65–80. doi: 10.1113/jphysiol.2002.018101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thornton PS, MacMullen C, Ganguly A, Ruchelli E, Steinkrauss L, Crane A, Aguilar-Bryan L, Stanley CA. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes. 2003;52:2403–2410. doi: 10.2337/diabetes.52.9.2403. [DOI] [PubMed] [Google Scholar]
- 44.Wang XR, Wu JP, Li L, Chen FX, Wang RP, Jiang C. Hypercapnic acidosis activates KATP channels in vascular smooth muscles. Circ Res. 2003;92:1225–1232. doi: 10.1161/01.RES.0000075601.95738.6D. [DOI] [PubMed] [Google Scholar]
- 45.Wellman GC, Quayle JM, Standen NB. ATP-sensitive K+ channel activation by calcitonin gene-related peptide and protein kinase A in pig coronary arterial smooth muscle. J Physiol London. 1998;507:117–129. doi: 10.1111/j.1469-7793.1998.117bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamada M, Isomoto S, Matsumoto S, Kondo C, Shindo T, Horio Y, Kurachi Y. Sulphonylurea receptor 2B Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J Physiol London. 1997;499:715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye D, Zhou W, Lee HC. Activation of rat mesenteric arterial KATP channels by 11, 12-epoxyeicosatrienoic acid. Am J Physio Heart Circ Physiol. 2005;288:H358–H364. doi: 10.1152/ajpheart.00423.2004. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Bolton TB. Activation by intracellular GDP, metabolic inhibition and pinacidil of A glibenclamide-sensitive K-channel in smooth-muscle cells of rat mesenteric-artery. Br J Pharmacol. 1995;114:662–672. doi: 10.1111/j.1476-5381.1995.tb17190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang HL, Bolton TB. Two types of ATP-sensitive potassium channels in rat portal vein smooth muscle cells. Br J Pharmacol. 1996;118:105–114. doi: 10.1111/j.1476-5381.1996.tb15372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]