

Abstract
Minute tissue samples or single cells increasingly provide the starting material for gene expression profiling, which often requires RNA amplification. Although much effort has been put into optimizing amplification protocols, the relative abundance of RNA templates in the amplified product is frequently biased. We applied a T7 polymerase-based technique to amplify RNA from two tissues of a cichlid fish and compared expression levels of unamplified and amplified RNA on a cDNA microarray. Amplification bias was generally minor and comprised features that were lost (1.3%) or gained (2.5%) through amplification and features that were scored as regulated before but unregulated after amplification (4.2%) or vice versa (19.5%). We examined ten sequence-specific properties and found that GC content, folding energy, hairpin length and number, and lengths of poly-A and poly-T stretches significantly affected RNA amplification. We conclude that, if RNA amplification is used in gene expression studies, preceding experiments controlling for amplification bias should be performed.
Keywords: RNA amplification bias, GC content, poly-A, poly-T, hairpin, folding energy, T7 polymerase
Introduction
Profiling gene expression on a genomic scale using microarray technology has become an important tool in uncovering the molecular basis of many biological processes [1]. To be successful, however, this approach requires RNA of sufficient quantity and quality, which is often a limiting factor. This is especially true for mRNA extracted from tissue sections or even single cells, by methods such as micropunches [2], laser capture microdissection [3; 4; 5], microaspiration [5] and fluorescence- activated cell sorting [6]. To overcome this problem researchers frequently amplify the RNA.
To date, there are two major amplification techniques commonly in use that can also be applied in combination [7; 8]: 1) exponential amplification of cDNA by polymerase chain reaction (PCR) [9] 2) and linear RNA amplification by in vitro transcription (various modifications of the original “Eberwine method” presented by van Gelder and co-workers [10]). Although both approaches have some drawbacks [11], linear amplification is generally favored ([11; 12; 13] but see [14]). Many studies have focused on optimizing these amplification strategies [15; 16; 17; 18; 19], and the performance of multiple protocols has been evaluated by several groups using quantitative real-time PCR [20; 21] or microarray analysis [22; 23; 24; 25; 26].
It has become clear that all amplification procedures introduce some bias, i.e. RNA species may become over- or under-represented in the amplified RNA compared with the starting RNA [7; 14; 27]. However, identifying mRNAs that are prone to amplification bias due to specific sequence properties is crucial in order to avoid potentially erroneous results. The bias introduced by differentially amplifying various RNA species has commonly been measured on cDNA- or oligonucleotide-based microarrays as the difference in signal intensity or ratio between amplified and non-amplified RNA. However, only few studies have explored how sequence and/or structure-specific properties of RNA species might affect differential amplification [28; 29]. Non-random loss in transcripts due to linear RNA amplification of rat biopsy material was reported by van Haaften et al. [29] who analyzed gene expression profiles of both unamplified and amplified RNA on the Affymetrix platform. When compared to hybridization signals of unamplified RNA, 21% of reporters were undetectable on arrays with amplified RNA. In contrast, only 3% of reporters scored as absent in unamplified RNA were counted present after amplification. Sequences of reporters that disappeared after amplification had a significantly higher GC-content, and significantly more and longer hairpins than those present before and after amplification. Notably, the widely used Affymetrix oligo-array platform requires even unamplified RNA to go through one round of in vitro transcription and thus amplification during the labeling procedure.
However, the evaluation of amplification bias in gene expression studies that utilize oligonucleotide-array platforms may not be directly applicable to cDNA microarrays. In addition to vast differences in the labeling procedures, only one set of probes is hybridized to microarrays containing short (< 100bp) oligonucleotides while two probes labeled with different fluorescent dyes are competitively hybridized to cDNA arrays containing double-stranded DNA (usually several hundred bp and up to several kb long). Moreover, the peculiarities of each method require the application of specifically developed software for processing expression intensities. Wadenbäck et al. [28] compared two amplification techniques by hybridizing amplified RNA to a pine tree cDNA microarray, and assessed the relative bias each method introduced. They showed that linear amplification by T7 RNA polymerase, when compared with PCR-based RNA amplification, yielded transcripts with a greater range in lengths and greater estimated mean length as well as greater variation of expression levels; average GC-content, however, was lower.
Applying a linear amplification protocol based on in vitro transcription [16] (Figure 1) and using a cDNA microarray platform, we show here that certain RNA sequence properties significantly bias amplification efficiency. We compare gene expression levels of total RNA with amplified RNA (Figure 2) from two very different tissue types, brain and muscle, of the African cichlid fish Astatotilapia burtoni. We use an expanded custom-built microarray [30] that contains more than 17,700 features, derived from several tissues/organs. We then determine whether sequence properties (e.g., GC-content, tandem repeats, hairpin-structure, poly-A and poly-T stretches, etc.) (Table 1) explain the differences in over- or underrepresentation of transcripts due to the amplification procedure. Specifically, we focus on two major categories: A) features on the array that were absent in the unamplified samples but appeared after amplification as well as features that were present in the unamplified samples but disappeared after amplification; B) features that gave different regulation signals before and after amplification.
Figure 1.

Flowchart of linear RNA amplification with the ExpressArt® mRNA Amplification Kit Nano version (AmpTech GmbH). mRNA is reverse transcribed with an anchored oligo(dT)-primer (without T7-promoter). In an attempt to minimize 3'-bias, double stranded cDNA is produced with a “Box-random-trinucleotide primer”, which preferentially binds near the 3'-ends of nucleic acid molecules. In the first round of amplification, a T7-promoter/oligo(dT) primer binds in reverse orientation to the single stranded cDNA with the Box sequence tag at the 3'end. This double stranded cDNA is then used as a template for in vitro transcription, which generates antisense oriented RNA. The antisense oriented RNA in turn is used as a template for cDNA synthesis and second round amplification.
Figure 2.
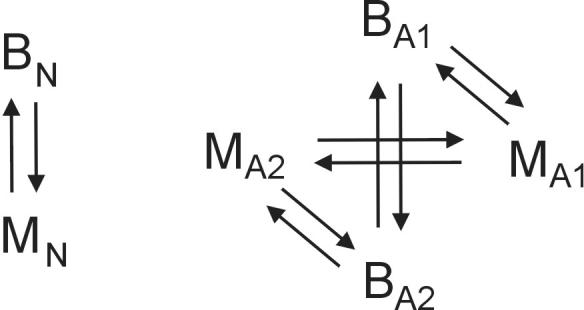
Experimental design of microarray hybridization. Each comparison was done in duplicate using dye-swap (total of 10 slides). BN, non-amplified brain; MN, non-amplified muscle; B1A, B2A, amplified brain, technical replicate 1 and 2; M1A, M2A, amplified muscle, technical replicate 1 and 2.
Table 1.
Sequence parameters and algorithms used in the present analysis.
| Sequence characteristics |
Computer program | Specifications |
|---|---|---|
| GC-content | Perl script | Relative GC-content [%] |
| Tandem repeats |
Tandem Repeat Finder (Benson 1999) |
Default parameters, score threshold 50, unit length ≥ 2 nt; Analyzed for presence/absence |
| Poly-A stretches |
Tandem Repeat Finder (Benson 1999) |
Default parameters, score threshold 50, unit=mono-nucleotide “A” ≥ 25 [nt] |
| Poly-T stretches |
Tandem Repeat Finder (Benson 1999) |
Default parameters, score threshold 50, unit=mono-nucleotide “T” ≥ 25 [nt] |
| Folding energy | RNAfold (Hofacker 2003) | Default parameters [Kcal/mol] |
| Normalized folding energy (Z-score) |
RNAfold (Hofacker 2003) | Z-score: each sequence was shuffled 1000 times by maintaining the di-nucleotide frequency to avoid composition biases, and the folding energy was calculated. The average of resulting energies was subtracted from the folding energy of the real sequence and divided by the standard deviation. |
| Number of hairpins |
RNAfold (Hofacker 2003) | Number of hairpins were determined from RNAfold output |
| Maximum length of hairpins |
RNAfold (Hofacker 2003) | Length of the longest hairpin was extracted from RNAfold output [nt] |
| GC-skew | Perl script | (G+C)/(G−C) (Perna & Kocher 1995) |
| AT-skew | Perl script | (A+T)/(A−T) (Perna & Kocher 1995) |
Results
Amplification bias measured from expression intensities
The overall amplification bias as inferred from correlations between log2 ratios of expression intensities of unamplified and amplified RNA and, between amplified replicates, was small and comparable to results of one-round-amplification of other linear amplification methods [31]. Log2 ratios co-varied strongly when we compared expression data obtained from unamplified and amplified RNA (Pearson correlation coefficient r = 0.78; Figure 3a) and even more so when we compared two technical replicates of RNA amplification (r = 0.96; Figure 3b).
Figure 3.

(a) Correlation between log2 transformed ratios of hybridization intensities of non-amplified RNA versus amplified RNA. (calculated mean of 2 slides total RNA and 4 slides amplified RNA; only B/M comparisons, no B/B or M/M). Dark blue, regulated before and after amplification; pink, regulated before, unregulated after amplification; yellow, unregulated before, regulated after amplification; light blue, unregulated before and after amplification. (b) Correlation between ratios of log2 transformed hybridization intensities of replicated amplified RNA (B/M).
Reliability of amplified samples to detect gene expression differences
We found that 1.3% (219) of array features called present in the non-amplified set disappeared after amplification, while 2.5% (425) called absent in the non-amplified set appeared after amplification (Table 2). 18.1% (3058) of features were differentially expressed between brain and muscle in the unamplified samples compared to 33.4% (5643) features in the amplified samples. 13.9% (2353) of array features were differentially expressed in both unamplified and amplified samples, while 4.2% (705) of features were exclusive to unamplifed and 19.5% (3290) of features exclusive to the amplified set. Of the features which were differentially expressed in both sets, 98.73% (2323) were concordant while only 1.27% (30) showed an inversion in regulation between non-amplified and amplified samples. Plots of the log-ratio of hybridization intensities (M) versus their mean log-expression (A) [32] illustrate that features, which were exclusively regulated in either the unamplified or amplified sample, were not restricted to low intensity spots but rather covered the entire intensity range (Figure 4).
Table 2.
Percent of total number of features and sequence-specific properties (see Table 1 for definitions) statistically tested for being correlated with biased RNA amplification (see heatmap in Figure 6). Mean and standard deviation (SD) are given for features meeting the criteria in bold. Where applicable, the test statistics t (t-test) and Z (Mann-Whitney test) are listed with corresponding p-values. Number and percent of features with tandem repeats are listed with Pearson Chi-Square test statistics and p-values.
| GC-Content | Tandem repeats | ||||||
|---|---|---|---|---|---|---|---|
| % | Mean (± SD) | (t) or (Z) | p | N | Pearson Chi- Square |
p | |
|
Present before and after amplification |
93.4 | 43.9 (±7.95%) | 983 (11.27%) |
||||
|
Appeared after amplification vs. present in both |
2.5 | 45.7 (±7.73%) | −3.108 (Z) |
0.002 | 19 (8.72%) | 1.397 | 0.237 |
|
Disappeared after amplification vs. present in both |
1.3 | 42.2 (±7.79%) | −2.541 (Z) |
0.011 | 14 (8.92%) | 0.858 | 0.354 |
|
No change in regulation after amplification |
69.5 | 43.7 (±7.93%) | 706 (12.91%) |
||||
|
Gained regulation signal after amplification vs. no change in regulation |
19.5 | 43.9 (±7.88%) | 0.977 (t) | 0.329 | 212 (13.05%) |
0.018 | 0.893 |
|
Lost regulation signal after amplification vs. no change in regulation |
4.2 | 47.3 (±8.08%) | −8.733 (Z) |
0.000 | 36 (9.52%) | 2.910 | 0.088 |
| Length polyA | Length polyT | ||||||
| % | Mean (± SD) | (t) or (Z) | p | Mean (± SD) |
(t) or (Z) | p | |
|
Present before and after amplification |
93.4 | 45.6 (±30.10nt) | 44.4 (±29.20nt) |
||||
|
Appeared after amplification vs. present in both |
2.5 | 28.0 (±2.71nt) | −10.595 (t) |
0.000 | 35.1 (±17.02nt) |
−2.573 (t) | 0.017 |
|
Disappeared after amplification vs. present in both |
1.3 | 40.1 (±24.00nt) | −0.690 (t) | 0.490 | 41.9 (±36.39nt) |
−0.434 (t) | 0.664 |
|
No change in regulation after amplification |
69.5 | 47.0 (±30.67nt) | 45.4 (±29.92nt) |
||||
|
Gained regulation signal after amplification vs. no change in regulation |
19.5 | 41.6 (±27.63nt) | −2.421 (Z) |
0.015 | 41.6 (±27.40nt) |
−2.254 (t) | 0.025 |
|
Lost regulation signal after amplification vs. no change in regulation |
4.2 | 42.0 (±23.83nt) | −0.584 (t) | 0.559 | 36.1 (±15.32nt) |
−3.175 (t) | 0.003 |
| Folding energy | Z-score folding energy | ||||||
| % | Mean (± SD) | (t) or (Z) | p | Mean (± SD) |
(t) or (Z) | p | |
|
Present before and after amplification |
93.4 | −149.6 (±68.27Kcal/mol) |
−0.21 (±1.295) |
||||
|
Appeared after amplification vs. present in both |
2.5 | −127.6 (±61.29Kcal/mol) |
−4.577 (Z) | 0.000 | −0.13 (±1.216) |
0.908 (t) | 0.365 |
|
Disappeared after amplification vs. present in both |
1.3 | −147.4 (±62.00Kcal/mol) |
0.443 (t) | 0.659 | −0.47 (±1.579) |
−2.083 (t) | 0.039 |
|
No change in regulation after amplification |
69.5 | −151.4 (±69.54Kcal/mol) |
−0.2 (±1.30) |
||||
|
Gained regulation signal after amplification vs. no change in regulation |
19.5 | −150.4 (±65.71Kcal/mol) |
0.573 (t) | 0.567 | −0.20 (±1.249) |
0.558 (t) | 0.577 |
|
Lost regulation signal after amplification vs. no change in regulation |
4.2 | −118.2 (±54.40Kcal/mol) |
−9.299 (Z) | 0.000 | −0.12 (±1.391) |
−2.273 (Z) | 0.023 |
| Number hairpins | Length hairpins | ||||||
| % | Mean (± SD) | (t) or (Z) | p | Mean (± SD) |
(t) or (Z) | p | |
|
Present before and after amplification |
93.4 | 33.0 (±14.78) | 10.6 (±3.99nt) |
||||
|
Appeared after amplification vs. present in both |
2.5 | 28.5 (±13.52) | −4.378 (Z) |
0.000 | 9.7 (±3.10nt) |
−4.445 (Z) | 0.000 |
|
Disappeared after amplification vs. present in both |
1.3 | 33.4 (±14.23) | 0.367 (t) | 0.714 | 11.4 (±5.47nt) |
1.795 (t) | 0.075 |
|
No change in regulation after amplification |
69.5 | 33.3 (±14.88) | 10.7 (±4.05nt) |
||||
|
Gained regulation signal after amplification vs. no change in regulation |
19.5 | 33.5 (±14.60) | 0.611 (t) | 0.541 | 10.6 (±3.87nt) |
−0.576 (t) | 0.565 |
|
Lost regulation signal after amplification vs. no change in regulation |
4.2 | 25.4 (±12.42) | −10.352 (Z) |
0.000 | 9.7 (±3.07nt) |
−6.506 (Z) | 0.000 |
| GC-skew | AT-skew | ||||||
| % | Mean (± SD) | (t) or (Z) | p | Mean (± SD) |
(t) or (Z) | p | |
|
Present before and after amplification |
93.4 | 0.00 (±0.134) | 0.02 (±0.157) |
||||
|
Appeared after amplification vs. present in both |
2.5 | 0.01 (±0.115) |
0.179 (t) | 0.858 | 0.01 (±0.136) | −0.980 (t) | 0.328 |
|
Disappeared after amplification vs. present in both |
1.3 | 0.02 (±0.176) | 0.999 (t) | 0.319 | −0.01 (±0.137) |
−2.088 (t) | 0.038 |
|
No change in regulation after amplification |
69.5 | 0.00 (±0.136) | 0.02 (±0.159) |
||||
|
Gained regulation signal after amplification vs. no change in regulation |
19.5 | 0.01 (±0.131) | 0.376 (t) | 0.707 | 0.02 (±0.151) |
−0.401 (t) | 0.688 |
|
Lost regulation signal after amplification vs. no change in regulation |
4.2 | 0.01 (±0.127) |
1.060 (t) | 0.289 | 0.03 (±0.158) |
1.131 (t) | 0.258 |
Figure 4.
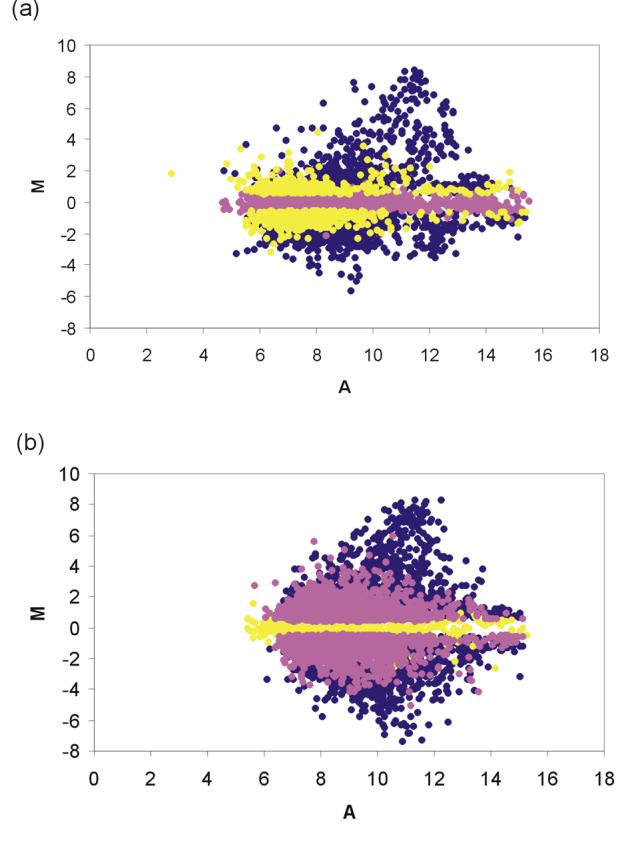
Log-ratio of hybridization intensities of brain RNA and muscle RNA (M) versus their mean log-intensities (A) for (a) unamplified RNA and (b) amplified RNA. M = log2I1 − log2I2 and A = (log2I1 + log2I2)/2, where I1 is the mean intensity signal of brain RNA and I2 is the mean intensity signal of muscle RNA. Dark blue, features regulated both in unamplified and amplified samples; yellow, regulated exclusively in unamplified samples; pink, regulated exclusively in amplified samples.
Amplification bias is linked to sequence-specific properties
Both categories tested (array features scored as present that were either gained or lost after amplification; and features that gave different regulation signals before and after amplification) revealed a bias in amplification that depended on sequence-specific properties of the RNA species (Table 2 and Figure 5).
Figure 5.
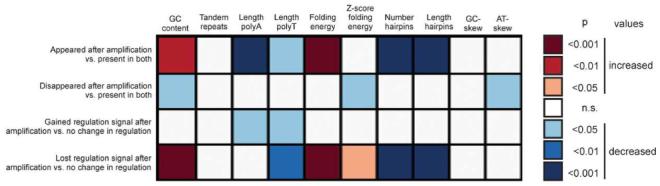
Heatmap presentation of significance values for the sequence properties that influence amplification efficiency as determined by genes that were gained/lost as either present or regulated after amplification. Red (blue) colors indicate that sequences that were gained/lost had increased (decreased) values in the respective sequence parameter. For detailed information see Table 2.
For example, features on the array that were absent before and present after amplification were characterized by a significantly higher GC-content (45.7±7.7%) than features that were present before and absent after amplification (42.2±7.8%) as well as those that were present on both array types (43.9±8.0%) (Figure 6). Furthermore, when tested against features that showed no change in regulation (GC content: 43.7±7.9%), we found a significant difference in GC-content for features where we gained regulation after amplification (43.9±7.88%), but not in those where we lost regulation after amplification (47.3±8.08%).
Figure 6.
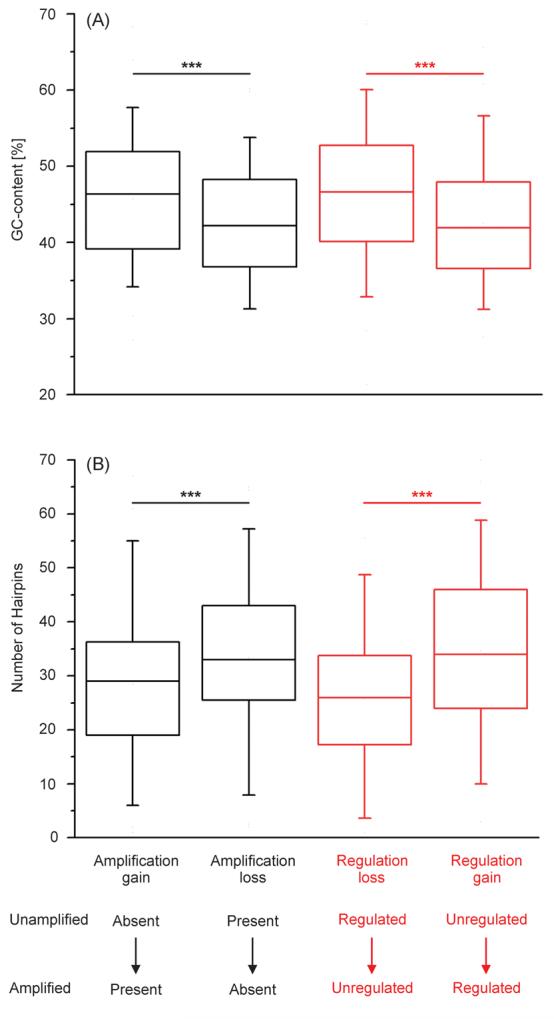
Box and whisker plot showing (A) GC-content and (B) predicted number of hairpins for sequences where we detected amplification bias. Error bars represent 95% confidence intervals. Asterisks indicate significance level < 0.001.
This analysis assumes that the GC content of each EST is representative of that of the full length RNA species that is subject to the amplification procedure. However, to date only ∼ 30 complete coding regions have been cloned and sequenced for A. burtoni, even though a whole genome sequencing project is underway (see URL http://www.genome.gov/11007951). In the absence of large numbers of known full-length RNAs, we decided to compare the GC contents of individual ESTs to the contigs they cluster into using the contigs contained by the DFCI GeneIndex for A. burtoni and built from the ESTs present on our array (see URL http://compbio.dfci.harvard.edu/tgi/cgi-bin/tgi/gimain.pl?gudb=a_burtoni). These contigs are on average 200 bp longer than the individual ESTs. We find that the GC content of 98.2% ESTs that clustered into contigs is not significantly different (even at a liberal cutoff of p<0.01) from that of the contigs themselves.
Furthermore, we found that features which appeared after amplification had significantly shorter poly-A and poly-T stretches, higher folding energy, as well as less and shorter hairpins (Table 2, Figure 5 and 6). Features that disappeared after amplification had a significantly lower normalized folding energy and lower AT-skew. Features that were gained as regulated after amplification had significantly shorter poly-A and poly-T stretches than those that showed no change in regulation. Features for which we lost regulation signal after amplification had significantly shorter poly-T stretches, significantly higher folding energy and normalized folding energy, as well as significantly fewer and shorter hairpins. In none of the categories tested we found significant differences in tandem repeats and GC-skew.
Discussion
Minimally invasive methods and new technological advances such as fine needle biopsies, laser capture microdissection and microaspiration provide the basis for selective small-scale tissue extractions as they are used for example, in early cancer diagnostics and neurobiological research [2; 5; 6; 33]. However, these techniques frequently yield only minute amounts of starting material for gene expression profiling, which renders RNA amplification absolutely indispensable. A major concern with any amplification procedure is the faithful retention of the relative transcript abundance and, thus, it is not surprising that in the last decade, much effort has been invested into developing reliable and efficient ways of amplifying RNA [17].
In the classic “Eberwine method” [10], from which all linear amplification protocols were derived, mRNA is amplified with T7-polymerase in one, two or even three rounds of amplification. An alternative strategy that can efficiently amplify samples containing far fewer molecules than needed for linear amplification, is PCR with DNA polymerase. Both methods have their strengths and weaknesses and, depending on the amount and sequence properties of the template, might be the method of choice under one condition, but sub-optimal under another condition. If there is enough starting material available, linear amplification methods are generally favored, mainly because DNA polymerases tend to mis-incorporate bases and generate shorter transcripts than RNA polymerases ([11; 12; 13; 28] but see [14]). In our study, we used a commercially available linear amplification kit, based on the method by Baugh et al. [16], that was developed in an attempt to maximize the length of the original RNA templates.
A bias introduced in the course of an amplification process can be measured in a number of different ways of which quantitative real-time PCR [20; 21] and microarray analysis [22; 23; 24; 25; 26] are two commonly used methods. Most studies have restricted their analysis to a comparison between expression intensity values of non-amplified versus amplified RNA. Only recently have scientists started to take into account sequence-specific properties that might be prone to be amplified in a biased fashion [28; 29]. Our study presents a comprehensive analysis of RNA amplification bias associated with certain nucleotide strand characteristics such as GC-content, length of poly-A stretches, folding energy, number and length of hairpins, etc. (see Tables 1 and 2). We measured differences in gene expression by competitively hybridizing two tissue types from the cichlid fish Astatotilapia burtoni to a custom-built cDNA microarray. The use of cDNA libraries of expressed sequence tags (ESTs) to construct DNA microarrays is a good and often the only way for performing large-scale gene expression studies in non-traditional model systems, for which no genome has yet been sequenced [30; 34].
Our results show that the linear T7-based amplification method used in this study produced RNA that remained a representative sample of the starting material. Overall, we observe reliable gene expression patterns for amplified samples that are comparable to results of one-round amplifications in a previous survey [31]. However, we also detect bias in expression intensities between non-amplified and amplified RNA (Figure 3a). By hybridizing either T7- or PCR- amplified RNA to cDNA microarrays, Wadenbäck et al. [28] demonstrated differential amplification efficiencies measured as the percentage of detectable spots or signals above background (88%, T7; 71%, PCR). Amplification bias was also reported by van Haaften et al. [29] who observed considerable loss of signals (21%) on an Affymetrix oligoarray after T7-amplification. Their proportion of features called present before but absent after amplification was thus almost 20-fold higher than what we found in our own study (1.3%). For oligoarray experiments, loosing such a high number of signals due to RNA amplification seems not unusual [8] and may in part be due to the greater demand of target quantities [7] and the nature of the amplification procedure. Interestingly, the relative proportions of features that appeared after amplification were comparable, with 3% [29] and 2.5% (this study), respectively. Statistical analyses of correlations between gene expression patterns and sequence-specific properties revealed non-random amplification bias in all three studies including ours [28; 29].
Wadenbäck et al. [28] found for pine that amplification performance by T7 RNA polymerase appeared to depend on three different sequence properties: the mean GC-content was lower, the range of sequence lengths, the estimated mean range as well as the variation of expression levels were greater after PCR amplification. In our study, those transcripts for which we gained a detection signal after linear amplification were shown to have a significantly higher GC-content (45.7±7.73%) compared to those that were present in both the unamplified and amplified sample (43.9±7.95%) as well as those that disappeared after amplification (42.2±7.79%). This result is in sharp contrast with the findings of van Haaften et al. [29] who found just the opposite pattern. They compared unamplified and linear-T7-based amplified RNA derived from biopsies of rat cardiac tissue by means of hybridization to Affymetrix GeneChip gene arrays, which are utilizing short oligonucleotides as targets. In their experiment, reporters that disappeared after amplification had a significantly higher GC-content (53.7±4.0%) compared to those that remained detectable (47.8±5.5%). In reference to a previous study by Arezi et al. [35], this observation led them to conclude that higher affinity bonds between nucleotides G and C as compared to A and T were responsible for the inferior performance of the DNA polymerase during amplification. Arezi et al. [35] indeed discovered a difficulty for several PCR enzymes to efficiently amplify sequences with high GC-content, but the T7-polymerase was not included in their analysis. Because of the intriguingly similar codon distribution patterns of phages and their respective host genomes (e.g., T7: 48% GC content; E. coli host: 49% GC content), it has been suggested that such enzymes transcribe or replicate efficiently only in a narrow window of GC-content [36]. It follows then that, even if the same enzyme and amplification protocol is used, the magnitude of amplification bias observed for one species might not be predictive of the biases observed in other study organisms that have different base compositions. Mean GC-content is generally higher in the gene rich regions of homeothermic genomes than in those of poikilotherms, such as fish and reptiles [37]. Zhang et al. [38] calculated a mean GC-content of 51.8±6.0% for the coding region of Rattus norwegicus genes based on ∼6000 genes. The average GC-content of Astatotilapia burtoni is 43.9±8.0%, which is consistent with findings in other teleosts (e.g. stickleback, average GC-content ∼ 42%; Grimwood et al. unpublished data). Given these differences in base composition, it is therefore not surprising that our results for GC-content are inverse to those of van Haaften et al. [29]. Our results suggest that both relatively high (53.7±4.0%; [29]) and relatively low GC-content (42.2±7.8%) is sub-optimal for the performance of the T7-polymerase and that GC-content in-between those “extremes” may allow fairly unbiased amplification. As mentioned, the average GC-content of the T7 phage is 48% [36] and falls right into the range where amplification did not result in a loss of signal in either rat or cichlid fish. Polacek et al. [20], using cDNA arrays, analyzed transcripts expressed in human endothelial cells and compared the number of regulated genes that were exclusively found in either the unamplified or the T7-based linearly amplified sample with those common to both samples. When we calculated the mean GC-content of the published sequences available from this study [20] (≤ 1000 nt of the 3'end), we found an increased GC-content for genes exclusively regulated in the unamplified data set compared with those only regulated in the amplified sample or those that were regulated in both samples (data not shown). This finding is consistent with the results of our study, where we found features that were exclusively regulated in the unamplified sample to have a significantly higher GC-content than features of the other two categories.
Although GC-content may considerably affect the efficiency of the amplification process it is certainly not the only factor that does so. Van Haaften et al. [29] already showed that sequences with high predicted numbers and lengths of hairpins were less likely to be amplified by the T7-polymerase than control sequences. Our study corroborates their results and gives further insights in the role other sequence properties such as poly-A and poly-T stretches play in the amplification procedure. We find that sequences with significantly shorter poly-A and poly-T stretches, fewer and shorter hairpins, or higher folding energies were preferentially amplified. Long stretches of mononucleotides can serve as alternative priming sites for oligonucleotides during cDNA synthesis and can entail the generation of two or more truncated products instead of one complete sequence [39]. Moreover, they often pose an obstacle for enzymes, especially when they are occupied by an internally primed oligonucleotide, resulting in the abortion of strand extension [39]. The latter is also more likely to happen, the more hairpins a sequence has and the longer the hairpins are. Several of these sequence properties are thus non-independent but rather related to each other. High stability is generally reflected by a low folding energy due to the formation of secondary structures. We also find that features that disappeared after amplification had a significantly lower Z-score of the folding energy compared to those detectable in both sets. The low Z-score indicates that the molecules are more stable than random sequences with the same composition and maintained di-nucleotide frequency.
RNA-amplification results in an overall increase of template molecules. A greater number of microarray features may then be detected above threshold if the amplified RNA is labeled at a higher efficiency than the unamplified RNA. Alternatively, an increase in the number of detectable features would be observed if the amplified sample included less material that would contribute to background. These hypotheses are reflected in the results of previous studies [20; 40] as well as in our observations of a higher percentage of features that were scored as unregulated before but as regulated after amplification (19.5%) than vice versa (4.2%). Importantly, these signals were not restricted to low intensity (i.e., variable) spots but instead covered the whole intensity range (Figure 4). However, given that the sample size (number of arrays hybridized) was smaller in the experiment using unamplified RNA, the number of regulated array features may be underestimated due to decreased statistical power (when compared with the amplified RNA experiment). The features that appeared unregulated before but regulated after amplification were characterized by significantly shorter poly-A and poly-T stretches while the features that appeared regulated before but unregulated after amplification had a significantly higher GC content, shorter poly-T stretches, higher folding energy, higher Z-scores of folding energy, as well as fewer and shorter hairpins as compared to those for which no change in regulation was observed before and after amplification. These sequence properties were thus shown to hamper efficient RNA amplification.
In many cases, amplification of small amounts of RNA is absolutely necessary for analyzing gene expression of particular tissue samples. With the linear amplification method we used in this study we show that the amplified RNA overall represents a valid sample of the original template. However, one has to be aware of the fact that some RNAs are prone to be multiplied in a non-random fashion due to certain sequence properties. We hypothesize that this bias may depend on the sequence characteristics that the enzyme utilized in the amplification procedure has been adapted to in the course of evolution. Unamplified and amplified samples should therefore never directly be compared.
Many variations of the linear RNA amplification protocol are in use, and it is not possible at this point to infer how and to what extent variations in a given protocol affect the nature of RNA amplification with respect to certain sequence properties. As more studies become available that relate sequence characteristics to different amplification protocols, it may become possible to assign generalized sequence –based “risk factors” to RNA species. Until then, experiments that utilize RNA amplification for microarray hybridization should ideally be accompanied by a control that compares unamplified and amplified RNA expression patterns in order to identify sequences that are prone to be affected by amplification bias.
Materials and Methods
RNA isolation
RNA was isolated from muscle (M) and whole brain (B) of one lab bred territorial male East African cichlid fish, Astatotilapia burtoni. The tissue samples that had originally been stored in RNAlater were homogenized in 500μl TRIZOL (Invitrogen) and were processed according to the manufacturer's instruction. Pellets were re-dissolved in RNA Storage Solution (Ambion). Quality of RNA and absence of genomic DNA was assessed on a BioAnalyzer 2100 (Agilent) using the Agilent Total RNA Nano Chip assay. Clear 28S and 18S ribosomal bands and a high ratio of 28S versus 18S rRNA indicated that the extractions met the criteria for downstream genetic analysis. Quantity and purity (OD260/280 and OD260/230) of RNA was evaluated by using a NanoDrop ND-1000 spectrophotometer.
Specifications of RNA amplification kit
Amplification strategies that are based on in vitro transcription with T7 RNA-polymerase are generally prone to lose sequence information of the 5'end [41]. This is mainly due to 1) the alignment of the T7-promoter/oligo(dT) primer and the transcription start at the 3' poly(A)-tail of the original mRNA, and 2) the use of random hexamers during the second cDNA synthesis step, causing reduction of fragment length. Based on the protocol by Baugh et al. [16], the ExpressArt® mRNA amplification kit (AmpTech GmbH) facilitates the generation of almost full-length molecules. Compared to other protocols, the main improvement is the use of a Box-random-trinucleotide primer, which preferentially binds near the 3' ends of the fragments and, together with the subsequent annealing of a T7-promoter/oligo(dT) primer, generates double stranded template for in vitro transcription with defined sequences at both ends (Figure 1).
RNA amplification
Five hundred ng of each sample were used in replicate as template for two rounds of linear amplification using the ExpressArt mRNA amplification kit (nano version, AmpTech GmBH) with the following modifications. For first round amplification: In place of the First Strand cDNA Synthesis Mix 1, was 0.75ul 10x Primer A, 0.5ul dNTPs, 0.75ul DEPC H2O. In place of First Strand cDNA Sythesis Mix 2, was 2μl 5x RT Buffer, 0.5μl RNase Inhibitor, 0.5μl RT Enzyme, 3μl DEPC H2O. In place of RNase Mix 3, was 0.5μl RNase. In place of Second Strand cDNA Sythesis Mix 4 was 0.5μl 10x Primer B, 0.5μl dNTPs, 3ul 5x Extender Buffer, 10μl DEPC H2O. In place of Extender Enzyme A Mix 5 was 0.5μl Extender Enzyme A. In place of Primer Erase Mix 6 was 0.5μl Primer Erase. In place of Primer C was 1.0μl 10x Primer C. In place of Extender Enzyme B Mix 7 was 0.5μl Extender Enzyme B. Quantity of amplified RNA was measured with a NanoDrop ND-1000 spectrophotometer. Assuming that 1-5% of total RNA correspond to poly (A)+ RNA, the first round of amplification yielded an approximately 1000 to 4000 fold increase while the second round yielded an approximately 200 fold increase of poly (A)+ equivalents. These amplification factors correlate well with the expected values given by the manufacturer of the amplification kit for 500 ng input material in both the first and second round of amplification. The size distribution of second-round amplified RNA was measured using a Nano Chip assay on the Agilent Bioanalzyer. RNA fragments peaked at approximately 500 bp and ranged up to 2000 bp.
Microarray Hybridization
Two μg of unamplified RNA (M/B) and 1μg of amplified RNA (M/B; two replicates; M/M, B/B), each adjusted to a total volume of 14.5μl, were mixed with 1μl of Primer Solution (poly-T, unamplified; poly-A, amplified) and incubated for 10min at 70°C and 10min at 4°C. cDNA synthesis was achieved by combining 5.6μl 5x reaction buffer, 2.8μl 0.1M DTT, 0.75μl 50x amino-allyl-dUTP/dNTP mix (2.5mM each dATP, dCTP, dGTP; 1.5 mM dTTP; 10 mM Amino Allyl dUTP), 2μl (200U/μl) SuperScript II (Invitrogen) and 2.8μl DEPC water, then incubating the reaction at 42°C for 2 hours. Subsequently, samples were hydrolyzed and purified [30] prior to being labeled with Cy3 or Cy5 of the CyDye Post-labeling reactive Dye Pack (Amersham) and incubated at room temperature for 1 hour in the dark. Each sample was coupled twice, once to Cy3 and once to Cy5 to control for bias due to dye-specific fluorescence properties. Unincorporated primers and dye were removed by applying the labeled cDNAs to Qiagen PCR columns and by performing several washing steps. Amplified samples were then divided in half to take part in two separate hybridization reactions (see experimental design, Figure 2). Two samples labeled with different dyes were pooled for competitive hybridization and further concentrated and filtered. After adjusting the volume to 50μl with DEPC water, 6μl 20x SSC, 3μl Poly (dA) Poly (dT), 0.96μl 1M HEPES (pH 7.5) and 0.6μl 0.1M DTT were added. The reaction was filtered and 0.9μl 10% SDS were added. Fifty μl of each probe mixture were applied to a custom-built A. burtoni cDNA array [30], containing both a brain specific library as well as a library from several other tissues. The array consists of more than 17,700 features (including several controls) with 5% sequence redundancy. Hybridization was performed under a coverslip submerged in a humidified chamber (Telechem) at 65°C for 15 to 16 hours in the dark. Slides were rinsed at room temperature, first in 0.6x SSC, 0.025% SDS, 0.001M DTT, and then in 0.05x SSC, 0.001M DTT, before they were centrifuged until dry. The slides were kept in the dark until they were scanned using an Axon 4000B scanner (Molecular Devices) with the software package Genepix 5.0 (Molecular Devices).
Data normalization and analysis
Scanned array images were visually inspected to adjust grids to spots deviating from the standard pattern and to exclude features with hybridization artifacts from further analysis. After this first filtering process the data were imported into R (v2.3.1) and analyzed with the LIMMA (LInear Models for MicroArray data; [42]) software package, and spots with intensity values less than 2 standard deviations above local background were eliminated. The two-color microarray data were normalized [43] by applying a within array print-tip loess normalization. Given that the array consisted of PCR-products originating from two different cDNA libraries normalization was performed for each group separately. Differential expression was determined using intensity ratios in a linear modeling and empirical Bayes analysis of gene expression levels (LIMMA t-test; [44]), and were subsequently log2-transformed to render up- and down-regulated values of the same scale and comparable.
Overall bias of RNA amplification was assessed by comparing the log2 ratios of hybridization intensities for each feature in the amplified sample arrays to the corresponding values in the unamplified arrays. Reproducibility of the amplification procedure was evaluated by comparing the mean log2 ratio of intensity values of the two technical replicates B/M (dye reversal). Bias introduced by amplification due to particular sequence properties of RNA molecules was tested for features that A) were present/absent before and absent/present after amplification, respectively; B) were unregulated/regulated before and regulated/unregulated after amplification, respectively. All ESTs for which sequence information was available (∼54%) were analyzed for the following characteristics (see Table 1): 1) GC-content, 2) tandem repeats, 3) poly-A stretches, 4) poly-T stretches, 5) folding energy, 6) folding energy normalized against a permuted set (Z-score), 7) number of hairpins, 8) maximum length of hairpins, 9) GC-skew, 10) AT-skew. Statistical analyses were performed for the two different bias categories A) and B) as mentioned above, and each of the sequence characteristics separately with the statistical package SPSS version 10.0 (SPSS 1999). Depending on whether the data fit a normal distribution, which was evaluated with the Kolmogorov-Smirnov test, a t-test or nonparametric Mann-Whitney test was applied.
Acknowledgements
We thank Jiangwen Zhang (Harvard University) for assistance with the implementation of batch normalization, members of the Hofmann laboratory for discussions, Celeste and Michael Kidd (University of Texas at Austin) and two anonymous reviewers for valuable comments on the manuscript. This work was supported by an Erwin-Schrödinger postdoctoral fellowship provided by the FWF Austrian Science Fund (Nina Duftner), NIGMS grant GM068763, the Bauer Center for Genomics Research at Harvard University and the Institute for Cellular and Molecular Biology at the University of Texas at Austin (Hans A. Hofmann). Matthieu Legendre is supported by the NIH center grant GM068763 to the Harvard Center for Modular Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoheisel JD. Microarray technology: beyond transcript profiling and genotype analysis. Nature. 2006;7:200–210. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- 2.Xiao XQ, Grove KL, Lau SY, McWeeney S, Smith SM. Deoxyribonucleic acid microarray analysis of gene expression pattern in the arcuate nucleus/ventromedial nucleus of hypothalamus during lactation. Endocrinology. 2005;146:4391–4398. doi: 10.1210/en.2005-0561. [DOI] [PubMed] [Google Scholar]
- 3.Emmert-Buck E, Bonner RF, Smith PDC, F. R, Zhuang Z, Goldstein S, Weiss RA, Liotta LA. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 4.Bonner RF, Emmert-Buck E, Cole K, Pohida T, Chuaqui R, Goldstein S, Liotta LA. Laser capture microdissection: molecular analysis of tissue. Science. 1997;278:1481–1483. doi: 10.1126/science.278.5342.1481. [DOI] [PubMed] [Google Scholar]
- 5.Ginsberg SD, Elarova I, Ruben M, Tan F, Counts SE, Eberwine JH, Trojanowski JQ, Hemby SE, Mufson EJ, Che S. Single-cell gene expression analysis: implications for neurodegenerative and neuropsychiatric disorders. Neurochem. Res. 2004;29:1053–1064. doi: 10.1023/b:nere.0000023593.77052.f7. [DOI] [PubMed] [Google Scholar]
- 6.Singh S, Bhattacherjee V, Mukhopadhyay P, Worth CA, Wellhausen SR, Warner CP, Greene RM, Pisano MM. Fluorescence-activated cell sorting of EGFP-labeled neural crest cells from murine embryonic craniofacial tissue. J. Biomed. Biotechnol. 2005;3:232–237. doi: 10.1155/JBB.2005.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji W, Zhou W, Gregg K, Lindpaintner K, Davis S, Davis S. A method for gene expression analysis by oligonucleotide arrays from minute biological materials. Anal. Biochem. 2004;331:329–339. doi: 10.1016/j.ab.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 8.Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, Ueda HR, Saitou M. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006;34:e42. doi: 10.1093/nar/gkl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dulac C, Axel R. A novel family of genes encoding putative pheromone receptors in mammals. Cell. 1995;83:195–206. doi: 10.1016/0092-8674(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 10.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nygaard V, Hovig E. Options available for profiling small samples: a review of sample amplification technology when combined with microarray profiling. Nucleic Acids Res. 2006;34:996–1014. doi: 10.1093/nar/gkj499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lockhart DJ, Winzeler E. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- 13.Gustincich S, Contini M, Gariboldi M, Puopolo M, Kadota K, Bono H, LeMieux J, Walsh P, Carninci P, Hayashizaki Y, Okazaki Y, Raviola E. Gene discovery in genetically labeled single dopaminergic neurons of the retina. Proc. Natl. Acad. Sci. USA. 2004;101:5069–5074. doi: 10.1073/pnas.0400913101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iscove NN, Barbara M, Gu M, Gibson M, Modi C, Winegarden N. Representation is faithfully preserved in global cDNA amplified exponentially from sub-picogram quantities of mRNA. Nat. Biotechnol. 2002;20:940–943. doi: 10.1038/nbt729. [DOI] [PubMed] [Google Scholar]
- 15.Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM. High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 16.Baugh LR, Hill AA, Brown EL, Hunter CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res. 2001;29:e29. doi: 10.1093/nar/29.5.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang E. RNA amplification for successful gene profiling analysis. J. Transl. Med. 2005;5:28. doi: 10.1186/1479-5876-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Upson JJ, Stoyanova R, Cooper HS, Patriotis C, Ross EA, Boman B, Clapper ML, Knudson AG, Bellacosa A. Optimized procedures for microarray analysis of histological specimens processed by laser capture microdissection. J. Cell. Physiol. 2004;201:366–373. doi: 10.1002/jcp.20073. [DOI] [PubMed] [Google Scholar]
- 19.Ohara R, Kikuno RF, Kitamura H, Ohara O. cDNA library construction froma small amount of RNA: adaptor-ligation approach for two-round cRNA amplification using T7 and SP6 RNA polymerases. BioTechniques. 2005;38:451–458. doi: 10.2144/05383RR01. [DOI] [PubMed] [Google Scholar]
- 20.Polacek DC, Passerini AG, Shi C, Francesco NM, Manduchi E, Grant GR, Powell S, Bischof H, Winkler H, Stoeckert CJJ, Davies PF. Fidelity and enhanced sensitivity of differential transcription profiles following linear amplification of nanogram amounts of endothelial mRNA. Physiol. Genomics. 2003;13:147–156. doi: 10.1152/physiolgenomics.00173.2002. [DOI] [PubMed] [Google Scholar]
- 21.Thelen P, Burfeind P, Grzmil M, Voigt S, Ringert R-H, Hemmerlein B. cDNA microarray analysis with amplified RNA after isolation of intact cellular RNA from neoplastic and non-neoplastic prostate tissue separated by laser microdissections. I. J. Oncol. 2004;24:1085–1092. [PubMed] [Google Scholar]
- 22.Nygaard V, Løland A, Holden M, Langaas M, Rue H, Liu F, Myklebost O, Fodstad Ø, Hovig E, Smith-Sørensen B. Effects of mRNA amplification on gene expression ratios in cDNA experiments estimated by analysis of variance. BMC Genomics. 2003;4:11. doi: 10.1186/1471-2164-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park JY, Kim SY, Lee JH, Song J, Noh JH, Lee SH, Park WS, Yoo NJ, Lee JY, Nam SW. Application of amplified RNA and evaluation of cRNA targets for spotted-oligonucleotide microarray. Biochem. Biophys. Res. Commun. 2004;325:1346–1352. doi: 10.1016/j.bbrc.2004.10.151. [DOI] [PubMed] [Google Scholar]
- 24.Schneider J, Buneß A, Huber W, Volz J, Kioschis P, Hafner M, Poustka A, Sültmann H. Systematic analysis of T7 RNA polymerase based in vitro linear RNA amplification for use in microarray experiments. BMC Genomics. 2004;5:29. doi: 10.1186/1471-2164-5-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han J, Lee H, Nguyen NY, Beaucage SL, Puri RK. Novel multiple 5′-amino-modified primer for DNA microarrays. Genomics. 2005;86:252–258. doi: 10.1016/j.ygeno.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Schindler H, Wiese A, Auer J, Burtscher H. cRNA target preparation for microarrays: Comparison of gene expression profiles generated with different amplification procedures. Anal. Biochem. 2005;344:92–101. doi: 10.1016/j.ab.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Klur S, Toy K, Williams MP, Certa U. Evaluation of procedures for amplification of small-size samples for hybridization on microarrrays. Genomics. 2004;83 doi: 10.1016/j.ygeno.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Wadenbäck J, Clapham DH, Craig D, Sederoff R, Peter GF, von Arnold S, Egertsdotter U. Comparison of standard exponential and linear techniques to amplify small cDNA samples for microarrays. BMC Genomics. 2005;6:61. doi: 10.1186/1471-2164-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Haaften RIM, Schroen B, Janssen BJA, van Erk A, Debets JJM, Smeets HJM, Smits JFM, van den Wijngaard A, Pinto YM, Evelo CTA. Biologically relevant effects of mRNA amplification on gene expression profiles. BMC Bioinformatics. 2006;7:200. doi: 10.1186/1471-2105-7-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renn SCP, Aubin-Horth N, Hofmann HA. Biological meaningful expression profiling across species using heterologous hybridization to a cDNA microarray. BMC Genomics. 2004;5:42. doi: 10.1186/1471-2164-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marko NF, Frank B, Quackenbush J, Lee NH. A robust method for the amplification of RNA in the sense orientation. BMC Genomics. 2005;6:27. doi: 10.1186/1471-2164-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dudoit S, Yang YH, Callow MJ, Speed TP. Statistical methods for identifying differentially expressed genes in replicated cDNA expression experiments. Statist. Sinica. 2002;12:111–140. [Google Scholar]
- 33.Whiteley GR. Proteomic patterns for cancer diagnosis - promise and challenges. Mol. BioSyst. 2006;2:358–363. doi: 10.1039/b607260g. [DOI] [PubMed] [Google Scholar]
- 34.Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression withinand among natural populations. Nat. Genet. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- 35.Arezi B, Xing W, Sorge JA, Hogrefe HH. Amplification efficiency of thermostable DNA polymerases. Anal. Biochem. 2003;321:226–235. doi: 10.1016/s0003-2697(03)00465-2. [DOI] [PubMed] [Google Scholar]
- 36.Kunisawa T, Kanaya S, Kutter E. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA Res. 1998;5:319–326. doi: 10.1093/dnares/5.6.319. [DOI] [PubMed] [Google Scholar]
- 37.Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Kasif S, Cantor CR, Broude NE. GC/AT-content spikes as genomic punctuation marks. Proc. Natl. Acad. Sci. USA. 2004;101:16855–16860. doi: 10.1073/pnas.0407821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nam DK, Lee S, Zhou G, Cao X, Wang C, Clark T, Chen J, Rowley JD, Wang SM. Oligo(dT) primer generates a high frequency of truncated cDNAs through internal poly(A) priming during reverse transcription. Proc. Natl. Acad. Sci. USA. 2002;99:6152–6156. doi: 10.1073/pnas.092140899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu L, Wang J, Baggerly K, Wang H, Fuller GN, Hamilton SR, Coombes KR, Zhang W. Obtaining reliable information from minute amounts of RNA using cDNA microarrays. BMC Genomics. 2002;3:16. doi: 10.1186/1471-2164-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kenzelmann M, Klären R, Hergenhahn M, Bonrouhi M, Groene H-J, Schmid W, Schuetz G. High-accuracy amplification of nanogram total RNA amounts for gene profiling. Genomics. 2004;83:550–558. doi: 10.1016/j.ygeno.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 42.Smyth GK. Limma: linear models and empirical models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and computational biology solutions using R and Bioconductor. Springer; New York: 2005. pp. 397–420. [Google Scholar]
- 43.Smyth GK, Speed TP. Normalization of cDNA microarray data. Methods. 2003;31:265–273. doi: 10.1016/s1046-2023(03)00155-5. [DOI] [PubMed] [Google Scholar]
- 44.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]