

Summary
While it is generally accepted that anaerobic metabolism is required during infection, supporting experimental data have only been described in a limited number of studies. To provide additional evidence on the role of anaerobic metabolism in bacterial pathogens while invading mammalian hosts, we analysed the effect of the inactivation of FNR, the major regulatory protein involved in the adaptation to oxygen restrictive conditions, and of two of the FNR-regulated genes on the survival of Neisseria meningitidis serogroup B (MenB) in vivo. We found that fnr deletion resulted in more than 1 log reduction in the meningococcal capacity to proliferate both in infant rats and in mice. To identify which of the FNR-regulated genes were responsible for this attenuated phenotype, we defined the FNR regulon by combining DNA microarray analysis and FNR–DNA binding studies. Under oxygen-restricted conditions, FNR positively controlled the transcription of nine transcriptional units, the most upregulated of which were the two operons NMB0388-galM and mapA-pgmβ implicated in sugar metabolism and fermentation. When galM and mapA were knocked out, the mutants were attenuated by 2 and 3 logs respectively. As the operons are controlled by FNR, from these data we conclude that MenB survival in the host anatomical sites where oxygen is limiting is supported by sugar fermentation.
Introduction
During infection bacterial pathogens encounter host anatomical sites with low oxygen levels, a condition often exacerbated by pathological events such as ischaemia, oedema and inflammation, triggered by the infection process. The fact that most pathogens have anaerobic pathways capable of providing energy and eventually using nitrate and fumarate as final electron acceptors strongly suggests that anaerobic metabolism plays an important role in the infection process. However, while the interplay between oxygen availability and expression of virulence factors has been well documented in a number of reports (Leclerc et al., 1998; Landini and Zehnder, 2002; Krishnan et al., 2004), the experimental data supporting the requirement of anaerobic metabolism for in vivo infection are limited to relatively few studies. These studies include: the observation that the arcA and fumarate reductase genes were upregulated in Vibrio cholera isolated from stool samples of infected patients (Bina et al., 2003; Xu et al., 2003); the inability of Salmonella typhi and Mycobacterium bovis mutants defective in nitrate reductase activity to invade and proliferate in epithelial cells (Contreras et al., 1997) and to persist in organs of infected mice respectively (Fritz et al., 2002); the attenuation of virulence in the animal models of infection observed in Bordetella pertussis and Actinobacillus pleuropneumoniae mutants in which the fnr homologous genes, btr and hlyX, were deleted (Wood et al., 1998; Baltes et al., 2005). As FNR (Fumarate and Nitrate reductase Regulator protein) (Guest et al., 1996) is the main player in the metabolic switch from aerobic to anaerobic growth, these latter studies suggest that genes involved in anaerobic metabolism may be crucial for bacterial infection.
To further investigate on the link between anaerobic metabolism and bacterial infection and pathogenesis, we used the human pathogen Neisseria meningitidis as a model system. This microorganism is capable of adapting to different anatomical compartments of the host, including the nasopharyngeal mucosa, the bloodstream and the subarachnoid compartment (Nassif et al., 1999), sites where oxygen concentrations are expected to fluctuate. Therefore, it is expected that the organism is endowed with a fine-tuned regulation of expression of the aerobic and anaerobic metabolism genes and that mutations that impaired the capacity of the pathogen to cope with the oxygen-limiting conditions interfere with the infection process.
In the present article, we first show that, as it is the case for B. pertussis and A. pleuropneumoniae, mutations in the aerobic/anaerobic metabolism regulator fnr result in an attenuated phenotype. Furthermore, by whole genome transcription analysis we demonstrate that the mapA and galM genes, involved in sugar fermentation, are regulated by FNR and that their inactivation impairs the ability of N. meningitidis serogroup B (MenB) to proliferate in the mouse.
Results
FNR knockouts mutants are attenuated in vivo
To study the role of anaerobic metabolism in N. meningitidis pathogenesis, we first asked whether the deletion of the fnr gene affects N. meningitidis survival in vivo. The fnr gene was replaced with an erm cassette in two different N. meningitidis strains (MC58 and BZ232) to be used in an adult mouse model and an infant rat model (Wilks et al., 1998; Granoff et al., 2001) of N. meningitidis infection respectively. Under aerobic conditions, the growth rate of both MC-fnrKO and BZ-fnrKO mutants was comparable to the wild-type strains. This is consistent with the fact that FNR is inactive in the presence of oxygen (Khoroshilova et al., 1997). In contrast, when the mutants were cultured under oxygen restriction, the growth rate, as judged by viable cell counting, was reduced by twofold (data not shown). To analyse the effect of fnr deletion in vivo, 6-week-old mice were challenged with 107 colony-forming units (cfu) of either N. meningitidis MC-fnrKO or the isogenic wild-type strain. Eighteen hours after challenge, the animals were bled and bacteria counted by colony plating. While the wild type reached a bacteraemia of approximately 105 cfu ml−1, the number of viable bacteria of the MC-fnrKO deletion mutant was reduced by more than one order of magnitude (Table 1). A similar trend in the reduction of viable bacteria was obtained in the infant rat model using the BZ232 wild-type and BZ-fnrKO strains (Table 1).
Table 1.
Mouse and infant rat infection assays with wild-type and fnrKO strains.
| Mouse model | Rat model | ||||
|---|---|---|---|---|---|
| Bacterial strains | Challenge dose | Number of animals | Bacteraemiaa | Number of animals | Bacteraemiaa |
| MC58 wild type | 4.9 × 107 | 50 | 1.7 × 105* | nd | nd |
| MC-fnrKO | 5.1 × 107 | 50 | 9.8 × 103* | nd | nd |
| BZ232 wild type | 4.4 × 103 | nd | nd | 6 | 3.2 × 106** |
| BZ-fnrKO | 5.0 × 103 | nd | nd | 8 | 1.4 × 105** |
Values represent geometric means of the bacterial cfu ml−1 measured in each animal.
Four-day-old infant rats and 6-week-old BALB/c mice were infected with the indicated amount of bacteria. In the mouse infection model, animals were treated in five independent experiments, 10 mice per group. Bacteraemia was measured 18 h after intraperitoneal challenge.
P = 0.009.
P = 0.06.
The significance of differences in the levels of bacteraemia in repeated assays was estimated by two-tailed t-test.
nd, not determined.
Identification of the FNR regulon by DNA microarray analysis
To determine which of the FNR-regulated genes might be responsible for the attenuated phenotype of the strains carrying the fnr gene deletion, we utilized microarray analysis to define the FNR regulon of N. meningitidis. Global transcription profiling was performed on RNA samples extracted at different time points from both N. meningitidis MC58 and MC-fnrKO strains cultured in GC medium under oxygen-limiting conditions. Comparison of the gene expression profiles revealed that a group of 11 genes was upregulated by more than twofold in the wild-type strain and, in general, maintained an upregulated trend throughout the entire time-course of the analysis (Fig. 1A). The 11 genes were organized in nine putative transcriptional units. The two putative operons NMB0388-galM and mapA-pgmβ, whose operon organization was confirmed by reverse transcription polymerase chain reaction (RT-PCR) analysis (see Fig. S1 in Supplementary material), are involved in maltose catabolism (Qian et al., 1994) and intervene in the first steps of the degradation of simple sugars to form pyruvate. The aniA and the nosR genes, respectively, code for a nitrite reductase (AniA), which reduces nitrite to nitric oxide, and for a putative nitrous oxide reductase regulator that in Pseudomonas regulates the expression of nitrous oxide reductase (Arai et al., 2003). The NMB1805 and NMB1677 genes encode cytochrome c4 and cytochrome c5, respectively, belonging to the c-type cytochrome family. Finally, the last three genes of the group, NMB0363, NMB1806 and NMB1870, code for hypothetical proteins.
Fig. 1.
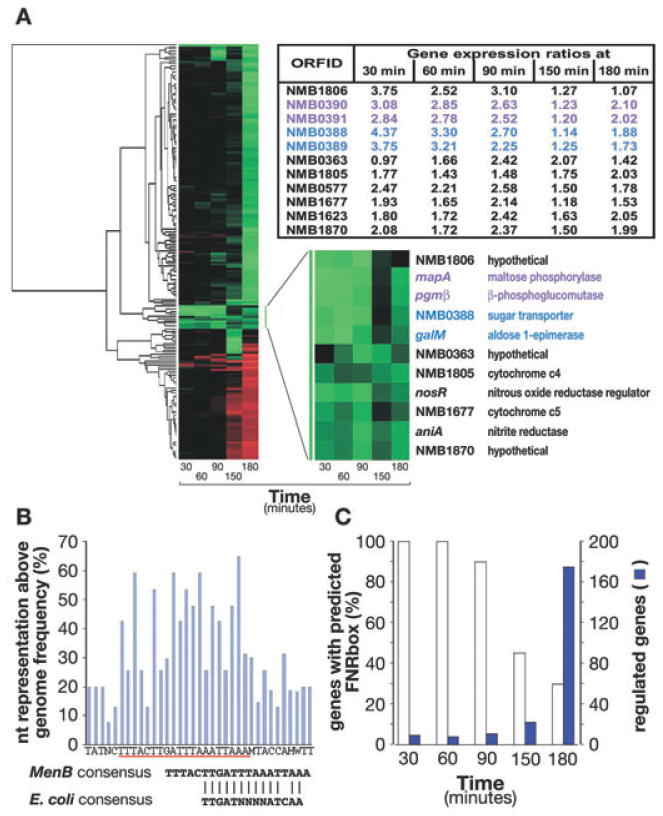
Transcription profile analysis of FNR-dependent genes and computational analysis of MenB FNR consensus sequence.
A. Hierarchical clustering of gene expression profiles of wild-type MC58 and MC-fnrKO strains grown under oxygen restriction compared by DNA microarray analysis. Red and green bars indicate down- and upregulated genes in the wild-type strain respectively. The zoomed panel represents the upregulated cluster of FNR-dependent genes whose expression ratios are shown in the table. Blue and purple identify genes belonging to the same transcriptional units.
B. Analysis of the FNR consensus sequence derived from the multiple sequence alignment of the upstream regions of the nine clustered upregulated transcriptional units. The graph reports the representation of each nucleotide of the derived consensus sequence above the MC58 genomic nucleotide frequency. The most conserved region of the derived consensus is highlighted in red. Pair-wise alignment of the derived MenB FNR consensus with the previously proposed E. coli FNR consensus is reported below.
C. Frequency of FNR-regulated genes carrying the FNR consensus sequence. The graph indicates the number of genes (in blue) regulated at different time points and the percentage of regulated genes (in white) with an FNR box in their promoter-proximal region.
After 3 h of growth under oxygen limitation, the difference in gene expression between the wild-type and mutant strains became much more pronounced. A total of 175 genes were differentially transcribed by more than twofold (Table S1 in Supplementary material), of which 126 were upregulated and 49 were downregulated. Although not further investigated, this phenomenon could be in part ascribable to the pleiotropic effect of prolonged oxygen starvation of the mutant strain, in which the major regulator of anaerobic metabolism is missing.
Confirmation of the FNR regulon by computational and biochemical analyses
To confirm the involvement of FNR in the regulation of the 11 genes identified by microarray analysis, we first scanned the promoter-proximal regions (400 nucleotides upstream from the respective start codons) of the nine transcriptional units belonging to the putative FNR cluster. In all nine transcriptional units, conserved motifs were found whose alignment generated the TTTACTTGATTTAAATTAAA consensus sequence (Fig. 1B and Fig. S2 in Supplementary material). This sequence includes a highly conserved TTGATT TAAATTAA sequence that differs by one nucleotide from both the Escherichia coli FNR box (TTGAT-N4-ATCAA) (Guest et al., 1996) and the FNR-binding sequence of the aniA gene of Neisseria gonorrhoeae, the only FNR box so far characterized in Neisseria (Lissenden et al., 2000). When the TTGATTTAAATTAA sequence was searched for in the upstream promoter regions of the 175 genes differentially expressed after 3 h of oxygen starvation (up to four-nucleotide differences accepted), it was found in approximately 30% of the genes (Fig. 1C). A comparable frequency was estimated by scanning all N. meningitidis annotated transcriptional units (Tettelin et al., 2000; data not shown). This indicates that the regulatory events directly controlled by FNR are likely to occur early after oxygen starvation and involve the nine clustered transcriptional units, all carrying an FNR consensus sequence. More complex regulatory pathways, not directly dependent on FNR, intervene after prolonged oxygen restriction.
To further support our hypothesis that the nine transcriptional units are under the direct control of FNR, we analysed the capacity of purified FNR to bind to the upstream promoter-proximal regions. Both the oxygen-resistant E. coli FNR D154A mutant protein (Ec FNR) (Bates et al., 1995) and the wild-type N. meningitidis FNR (Nm FNR) were utilized in electrophoretic mobility shift assay (EMSA) analysis. The Ec FNR was used to take advantage of its oxygen-resistant property so that these experiments could be carried out under normal laboratory conditions. Likewise, this allowed us to investigate the ability of the E. coli FNR to recognize N. meningitidis FNR binding sites. The Ec FNR was able to bind to five of the nine upstream regions, showing the highest affinity to the promoter-proximal regions of the aniA gene and the NMB0388-galM gene locus (Table S2 in Supplementary material). In contrast, the recombinant Nm FNR was capable of binding all nine upstream regions, although with an apparent lower affinity (Table S2). However, this was likely due to the partial inactivation of the highly oxygen sensitive Nm FNR, even if particular care was taken to run the experiments under anaerobic conditions. As examples, Ec FNR and Nm FNR binding to the promoter regions of aniA, NMB0388-galM and mapA-pgmβ transcriptional units is shown in Fig. 2. Nm FNR specifically interacted with all three upstream promoter regions when a protein concentration of 800 nM was used. In the case of the aniA fragment, Nm FNR interaction also occurred at 400 nM but the band shift was less pronounced, suggesting that the aniA region carries two FNR binding sites recognizing the regulatory protein with different affinities. Indeed, inspection of the DNA sequence of the fragment revealed the presence of two putative consensus FNR boxes located 253 nucleotides apart (data not shown). The fact that the Nm FNR–DNA complexes appeared like smears on gel is likely to be due to a partial inactivation of the regulatory protein during the electrophoresis run. In the case of Ec FNR, while no interaction was observed with the mapA-pgmβ promoter region, a strong binding to both aniA and NMB0388-galM fragments occurred at a protein concentration as low as 50 nM.
Fig. 2.
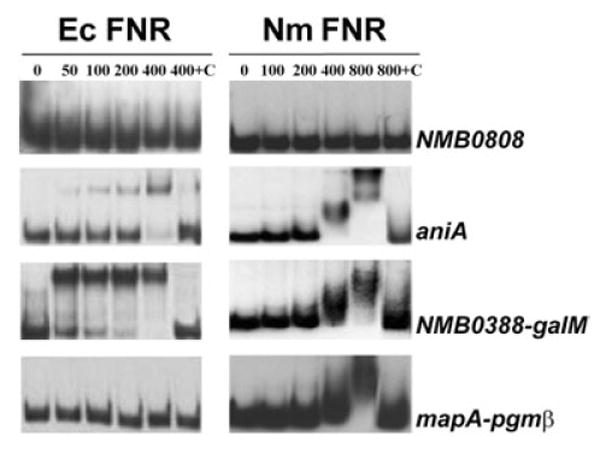
EMSA analysis of FNR-regulated genes. PCR-amplified upstream regions were incubated with different concentrations (nM) of purified Ec FNR and Nm FNR proteins and DNA protein binding was followed by gel retardation analysis in the presence of salmon sperm DNA. To assess FNR binding specificity, reactions containing a 10-fold molar excess of each specific unlabelled DNA region were also included (lanes 400+C and 800+C). NMB0808 was used as negative control as its expression was unaffected by FNR deletion and no FNR consensus sequence was identified in its promoter-proximal region.
The specificity of FNR binding to the upstream promoter regions was confirmed by EMSA competition experiments using both the Ec FNR and the Nm FNR and all the DNA fragments that showed affinity to the regulatory proteins. In these experiments, FNR binding to the labelled DNA fragments was followed in the presence of increasing amounts of corresponding unlabelled DNA. Under these conditions, 10-fold excess of unlabelled probes almost completely abolished the FNR binding to the labelled probes (Fig. 2 and data not shown).
Taken together, DNA microarray, bioinformatics and DNA binding studies consistently led to the conclusion that the nine transcriptional units are under the direct control of FNR.
galM and mapA mutants are attenuated in vivo
As galM and mapA-pgmβ are important genes for sugar metabolism under anaerobic conditions, we asked what role galM and mapA genes might have on meningococcal virulence.
To this aim, the genes were replaced with the erm cassette integrated in the chromosome in the opposite orientation with respect to original gene orientation (Fig. 3). RT-PCR was used to confirm each gene deletion. As expected, the replacement of galM and pgmβ genes resulted in the abrogation of the corresponding transcription products (Fig. 3, panels 2 and 3). We also analysed whether each gene inactivation had affected the transcription levels of the adjacent genes. As shown in Fig. 3, panel 1, galM deletion did not influence the transcription of the NMB0388 upstream gene, while marginally affected the transcription level of the mapA downstream gene (Fig. 3, panel 3). In the case of mapA gene replacement, the transcription levels of the downstream pgmβ gene and, marginally, of the upstream galM gene were both reduced. These secondary effects were likely due to the transcription activity of the erm promoter, which transcribes in the opposite direction with respect to the inactivated transcriptional units (Fig. 3).
Fig. 3.
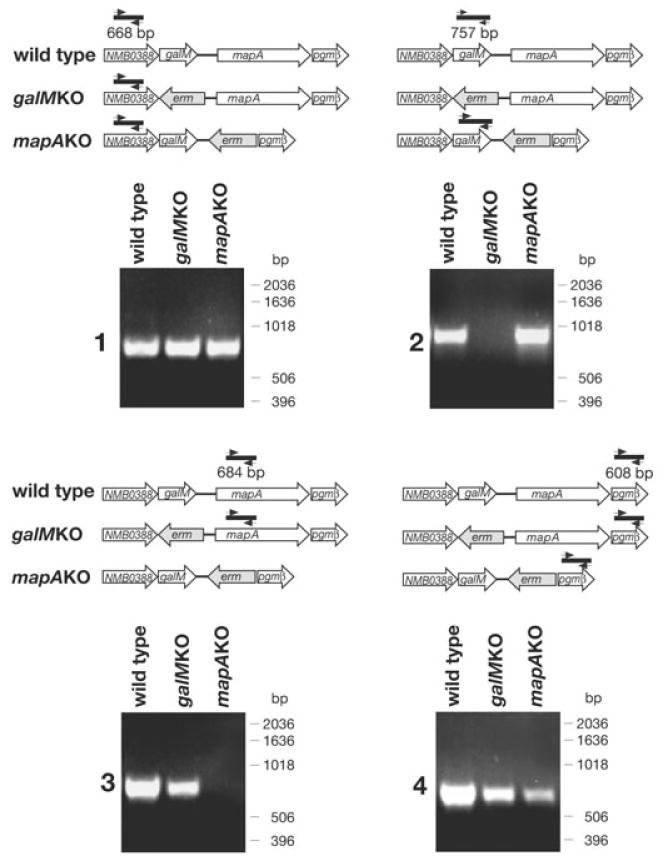
Transcription analysis of galM and mapA genes in wild-type and mutant strains. Total RNA from wild-type and mutant strains was reverse transcribed and the derived cDNAs were amplified using gene-specific primer pairs and analysed by agarose gel electrophoresis. All PCR reactions from non-reverse transcribed RNAs were negative (not shown). The genetic organization of the amplified regions is schematized above each corresponding gel. Arrows indicate the position of the primers used for the amplification reactions and bp numbers indicate the size of each PCR product. Molecular weight standards are given on the right side of each gel.
In conclusion, RT-PCR analyses confirmed each gene replacement. In the case of mapA inactivation, erm insertion resulted in a slight reduction in the mRNA levels of the neighbouring genes while galM gene substitution only marginally affected the transcription level of the mapA downstream gene. These effects should be taken into account for the correct interpretation of the subsequent phenotypic analyses.
Inactivation of both mapA and galM genes did not affect the growth kinetics of the two mutants in rich medium or in glucose-containing synthetic medium (Fig. S3A in Supplementary material). In contrast, both mutants showed an impaired capacity to grow in a chemically defined medium containing maltose as the main carbon source. This is consistent with the fact that the two genes are involved in maltose utilization (Fig. S3B in Supplementary material).
We next examined the ability of the individual N. meningitidis mutants to proliferate and survive following intraperitoneal injection in the mouse model of infection. As shown in Table 2, the levels of galMKO and mapAKO mutants in blood were reduced by 2 logs and 3 logs, respectively, as compared with the wild-type strain, suggesting that maltose utilization represents an important energy source for the in vivo growth of N. meningitidis.
Table 2.
Mouse infection assay with MC58 wild-type, galMKO and mapAKO strains.
| Bacterial strains | Challenge dose | Number of animals | Bacteraemiaa | Pb |
|---|---|---|---|---|
| MC58 wild type | 3.2 × 107 | 40 | 8.3 × 105 | |
| mapAKO | 3.6 × 107 | 40 | 3.9 × 102 | 2.4 × 10−7 |
| galMKO | 3.7 × 107 | 40 | 8.9 × 103 | 2.0 × 10−3 |
Values are geometric means of the bacterial cfu ml−1 measured in the blood of each animal.
The significance of difference in the level of bacteraemia was evaluated by two-tailed t-test.
Six-week-old BALB/c mice, 10 mice per group, were infected with the indicated amount of bacteria in four independent experiments. Bacteraemia was measured 18 h after intraperitoneal challenge.
Complementation of fnr, galM and mapA deletion mutants
To confirm that the attenuated phenotype of the mapAKO and galMKO mutants was specifically ascribable to the deleted genes, we complemented each mutant by inserting a functional copy of fnr, mapA and galM into the chromosome of MC-fnrKO, mapAKO and galMKO, respectively, between the convergent genes NMB1428 and NMB1429. From previous experience, we knew that gene insertions in this locus did not result in appreciable phenotypic effects (Grifantini et al., 2004). RT-PCR analysis of RNA samples obtained from the resulting three genetically complemented strains fnrKO_C, mapAKO_C and galMKO_C revealed that the transcription levels of fnr, mapA and galM in the three strains was comparable to the expression level in the wild-type strain (Fig. S4). We then analysed the phenotype of the fnrKO_C, mapAKO_C and galMKO_C strains in the mouse infection model. As shown in Table 3, the three strains were as virulent as the wild-type strain as judged by bacterial load in the blood of the challenged animals.
Table 3.
Mouse infection assay with MC58 wild-type and the complemented strains fnrKO_C, galMKO_C and mapAKO_C.
| Bacterial strains | Challenge dose | Number of animals | Bacteraemiaa | Pb |
|---|---|---|---|---|
| MC58 wild type | 1.7 × 107 | 20 | 6.3 × 105 | |
| fnrKO_C | 1.5 × 107 | 10 | 2.7 × 106 | 0.23 |
| mapAKO_C | 1.5 × 107 | 10 | 2.3 × 106 | 0.23 |
| galMKO_C | 1.5 × 107 | 10 | 2.3 × 106 | 0.47 |
Values are geometric means of the bacterial cfu ml−1 measured in the blood of each animal.
The significance of difference in the level of bacteraemia was evaluated by two-tailed t-test.
Six-week-old BALB/c mice, 10 mice per group, were infected with the indicated amount of bacteria. Bacteraemia was measured 18 h after intraperitoneal challenge.
These results indicate that maltose utilization may represent an important energy source for the in vivo growth of N. meningitidis.
Discussion
During infection, bacterial pathogens encounter anatomical compartments with low oxygen levels. In these types of environments, the ability of bacterial pathogens to activate the metabolic shift which allows them to cope with the oxygen-limited conditions is likely to be crucial for their proficient interactions with the host. To investigate on the interplay between anaerobic metabolism and bacterial infection, we have used N. meningitidis, a pathogen which appears to be particularly suitable for this kind of studies in that, during infection, it encounters different human compartments where oxygen partial pressure fluctuates quite substantially. In addition, two animal models exist, the infant rat and the adult mouse models (Wilks et al., 1998; Granoff et al., 2001) in which meningococci are administered intraperitoneally and bacterial infectivity is followed by measuring bacteraemia and animal survival. As oxygen-restricted conditions are encountered both in the peritoneum and in the blood, where oxygen is largely sequestered by haemoglobin, these models serve as a good system to examine how specific mutations in genes involved in the adaptation to oxygen restriction affect the virulence potential of N. meningitidis.
The first important observation of this study is that FNR inactivation attenuates the virulence of N. meningitidis in the two animal models utilized. Apart from the studies on the FNR homologues (Wood et al., 1998; Baltes et al., 2005), this is the first report that demonstrates a direct role of FNR in biological processes that ultimately lead to bacterial survival in vivo. FNR is the main regulatory protein responsible for the switch from aerobic to anaerobic metabolism in many bacteria (Guest et al., 1996). In E. coli, under anaerobic conditions, the FNR dimer carries two [4Fe-4S] clusters and acquires the capacity to interact with a sequence-specific, promoter-proximal DNA region (FNR box) (Guest et al., 1996). FNR binding results in the upregulation of a set of genes which are involved or linked to bacterial anaerobic functions by virtue of the fact that their expression is enhanced under oxygen limitation. Being FNR a regulatory protein, the observed attenuation of the fnr deletion mutant is most likely the result of an altered expression of some anaerobically activated genes. To select these genes, we defined the N. meningitidis FNR regulon, the second FNR regulon so far characterized in bacteria (Salmon et al., 2003; Kang et al., 2005).
The second important piece of information emerging from this study is that in the first 2 h of oxygen starvation, 11 genes, belonging to nine transcriptional units, were upregulated by FNR. The upstream promoter-proximal regions of all nine transcriptional units were able to bind a purified recombinant form of N. meningitidis FNR, as judged by gel retardation experiments. Inspection of the nucleotide sequences of these promoter regions allowed us to define a N. meningitidis FNR consensus box which appears to be very similar, but not identical to the E. coli FNR box (Guest et al., 1996). This is consistent with the fact that the oxygen-resistant E. coli FNR D154A mutant (Bates et al., 1995) was capable of binding only five of the nine upstream regions.
The N. meningitidis FNR regulon includes the aniA (nitrite reductase) and nosR (putative nitrous oxide regulator) genes. The aniA gene encodes a key gene in anaerobic respiration (Mellies et al., 1997; Householder et al., 1999) and catalyses the reduction of nitrite (NO2) to nitric oxide (NO), which is further reduced to nitrous oxide (N2O) by the sequential action of a FNR-independent nitric oxide reductase (norB) (Householder et al., 2000). Interestingly, from the present study, as well as from our previously published study (Grifantini et al., 2003), it appears that N. meningitidis exploits the two major regulatory proteins FNR and Fur to optimize the use of the denitrification pathway. On the one hand, FNR is utilized to upregulate nitrite respiration when oxygen is limiting; on the other hand, as nitrite reductase leads to the accumulation of toxic NO, the organism exploits the capacity of Fur to sense the oxidative stress imposed by NO accumulation and upregulates the expression of the nitric oxide reductase (NorB) (Delany et al., 2004). The most likely explanation why Fur also upregulates aniA is that N. meningitidis lives in anatomical compartments where both oxygen and iron are limiting. Therefore, the organism utilizes the same regulator (Fur) to simultaneously activate nitrite respiration, iron scavenging and detoxification from NO and iron overload.
The N. meningitidis FNR regulon also includes the two transcriptional units NMB0388-galM and mapA-pgmβ. NMB0388 encodes a putative sugar transporter, while the galM gene codes for an aldose 1-epimerase, an enzyme that catalyses the mutarotation of α and β anomers of esoses, such as glucose and galactose. As both anomers are needed in bacteria, aldose 1-epimerase allows the efficient utilization of lactose and galactose whose hydrolysis would otherwise generate only one anomer (Bouffard et al., 1994; Grossiord et al., 2003). MapA is a maltose phosphorylase catalysing the cleavage of maltose to glucose and β-glucose-1-phosphate; that is further converted into glucose 6-phosphate by a β-phosphoglucomutase, encoded by pgmβ (Fitting and Dourodoff, 1952; Arai et al., 2003).
The last two genes of the N. meningitidis FNR regulon with known function are NMB1805 and NMB1677, which code for cytocrome c4 and c5 respectively. In N. gonorrhoeae, FNR controls the expression of ccp, the cytochrome c peroxidase recently reported to be involved in protection of the organism from oxidative stress (Turner et al., 2003). As ccp homologues are not present in the MenB MC58 genome (Tettelin et al., 2000), it is tempting to speculate that NMB1805 and/or NMB1677 play an analogous protective role against reactive oxygen species.
The remaining three genes of the N. meningitidis FNR regulon belong to a hypothetical gene family. NMB0363 encodes a 45-amino-acid-long peptide, while the NMB1806 gene product is homologous to the gonococcal ORF1 and shows significant identity (about 48%) to hypothetical GTP-binding proteins present in several pathogens and sometimes involved in bacterial pathogenicity (van Putten et al., 1998; Zusman et al., 2004). Finally, NMB1870 encodes the surface-associated lipoprotein GNA1870, a recently discovered antigen proposed as an anti-MenB vaccine candidate (Masignani et al., 2003).
The third relevant observation of this study is that FNR contributes to the virulence of N. meningitidis through the modulation of the expression of at least two transcriptional units, NMB0388-galM and mapA-pgmβ. These genes are involved in maltose catabolism (Qian et al., 1994) and intervene in the first steps of the degradation of simple sugars to form pyruvate. Inactivation of each of these transcriptional units results in the attenuation of N. meningitidis in the mouse. In particular, the galM and mapA knockout mutants were reduced in their ability to survive in the blood by two and three orders of magnitude respectively. The observation that these FNR-dependent genes are important for the survival/proliferation of N. meningitidis in the bloodstream of the animals strongly suggests that in this environment where oxygen is limiting the pathogen requires the sugar fermentation pathways for both carbon and energy source.
The possible role of aniA in N. meningitidis infection deserves our last comment. According to our microarray data, FNR upregulates this gene by more than twofold in oxygen-restricted conditions. Considering that AniA plays a key role in anaerobic respiration, and that the AniA-mediated denitrification pathway and oxygen contribute similarly to neisseral respiration in microaerobic conditions (Rock et al., 2005), one might expect that the observed in vivo attenuated phenotype of the fnr-deletion mutant is partially ascribable to the insufficient expression of this gene. While we are investigating on this attractive possibility (which seems to be supported by our preliminary data), it has to be pointed out that, based on the genome sequence analysis, N. meningitidis seems to have an incomplete denitrification pathway, in that homologous sequences to known N2O reductases, which further convert nitrous oxide to dinitrogen, have not been identified (Anjum et al., 2002). This implies that in N. meningitidis, less energy is derived from denitrification as compared with bacteria having a complete pathway and would explain why N. meningitidis is classified as a strict aerobic organism (Rock et al., 2005). Therefore, the contribution of FNR in aniA upregulation might not necessarily influence N. meningitidis virulence.
Experimental procedures
Bacterial strains, cultures and reagents
Unless differently stated, MenB strains were grown on GC broth or GC agar plates (BD Biosciences) supplemented with 4 g l−1 glucose, 0.1 g l−1 glutamine, 2.2 mg l−1 cocarboxylase at 37°C in 5% CO2. When necessary, erythromycin (5 μg ml−1) was added to the GC medium. Bacteria were inoculated with colonies taken from freshly grown aerobic plates and incubated aerobically to logarithmic phase (OD600 0.4) in GC broth and harvested. For aerobic growth, bacteria were resuspended in fresh medium, and 5 ml of aliquots was incubated aerobically at 37°C in 15 ml tubes, shaken at 160 r.p.m. For oxygen-restricted growth, bacteria were resuspended in degassed GC medium and 15 ml of aliquots was incubated in 15 ml sealed tubes. When specified, MenB strains were grown aerobically in a chemically defined medium (La Scolea and Young, 1974), containing starch (0.1%) in combination with either glucose (0.75%) or maltose (0.75%) as main carbon sources.
Escherichia coli DH5α or BL21 (DE3) was grown aerobically in Luria–Broth (LB) medium (Difco) at 37°C. When appropriate, ampicillin (100 μg ml−1), erythromycin (200 μg ml−1) and isopropyl-beta-d-galactopyranoside (IPTG, 0.5 mM) were added to the medium. Unless specified, all chemicals used in this study were purchased from Sigma. Restriction enzymes and DNA modification enzymes were from New England Biolabs.
Mutant preparation and RT-PCR analysis
fnr, mapA, galM null mutants were produced by replacing their entire coding sequences with an erythromycin resistance cassette (erm) (Trieu-Cuot et al., 1990). The 500 bp upstream and downstream regions of the gene to be deleted were amplified from MC58 genomic DNA and cloned into plasmid pT7.7 (Amersham Biosciences) on either side of the erm gene. The newly derived plasmids were linearized and used for allelic exchange in MenB. The resulting mutants, namely MC-fnrKO, BZ-fnrKO, mapAKO and galMKO, were verified by PCR of genomic DNA with specific primers.
Complementation of the MC-fnrKO, mapAKO and galMKO deletion mutants was achieved by insertion of the deleted genes (the amplified fragments included the coding regions only) into a non-coding chromosomal location between the two converging ORFs NMB1428 and NMB1429, under the control of the erythromycin resistance gene promoter. This was obtained by cloning the erythromycin resistance gene promoter (Trieu-Cuot et al., 1990) along with and directly upstream of the coding sequence of genes fnr, galM and mapA into the pSl-ComCmr vector (Grifantini et al., 2004). The newly generated plasmids were used to transform MC-fnrKO, mapAKO and galMKO deletion mutants, selecting for chloramphenicol resistance, and the resulting strains fnrKO_C, mapAKO_C and galMKO_C were verified by PCR with specific primers.
For RT-PCR analysis, total RNA (2 μg) was reverse transcribed with random primers and Superscript II Reverse Transcriptase (Invitrogen). The resulting cDNAs were amplified using primer pairs specific for fnr, NMB0388, mapA, galM, pgmβ. RNA-containing PCR reactions, in which the reverse transcriptase step was omitted, were used as controls for DNA contamination. Genomic DNA was amplified as positive control with the same primer sets.
Microarray procedures, hybridization and analysis
Microarray comparisons were performed on wild-type MC58 and MC-fnrKO strains grown in oxygen restriction for 3 h. At time points, total bacterial RNA was extracted from three independent cultures of each sample by using the RNeasy kit (Qiagen, Valencia, CA), pooled and used for cDNA labelling. The hybridization probe was constituted by a mixture of the differently labelled cDNA derived from MC58 and MC-fnrKO. Probe hybridization, washing, slide scanning and statistical and clustering analyses were performed as previously described (Grifantini et al., 2003). Genes whose expression ratios changed above twofold and had P-values < 0.01 were considered up- or downregulated.
Computational analysis of FNR binding site
The 400 bp upstream regions of all potential transcriptional units (defined as a cluster of adjacent genes having inter-genic region < 50 bp in length) present in the meningococcal genome were scanned for the presence of conserved sequences using the findpatterns program of the Wisconsin Package (Accelrys, San Diego), under default parameters.
Purification of FNR proteins from MenB and E. coli and FNR binding studies
The MenB fnr gene was PCR-amplified from MC58 chromosomal DNA by using specific primers annealing at the 5′ and 3′ ends of the gene. E. coli D154A fnr gene was obtained by PCR from E. coli BL21 (DE3) chromosomal DNA. For amplification, two primer pairs were opportunely designed so as to amplify the gene in two partially overlapping fragments, containing the A154 codon within the overlapping region, that were subsequently fused by PCR. The resulting PCR product was verified by DNA sequencing.
Both E. coli D154A mutant FNR (Ec FNR) and MenB FNR (Nm FNR) proteins were expressed as C-terminal His-tagged fusions by cloning their corresponding amplified genes into pET21b+ vector (Novagen), using the E. coli BL21 (DE3) strain as the recipient.
Expression of Nm FNR was obtained by growing the recombinant E. coli strain in an anaerobic chamber at 25°C. All subsequent steps were carried out as previously described (Lazazzera et al., 1996). Purification of the His-tagged Nm FNR was performed by metal ion affinity chromatography (IMAC) (Amersham Biosciences) in the presence of 1.7 mM sodium dithionite. Expression and purification of Ec FNR was performed aerobically, as described (Ziegelhoffer and Kiley, 1995).
For EMSA on PCR-amplified fragments, 0.025 pmoles of 32P-ATP-end-labelled promoter fragments were incubated with purified FNR proteins, at a protein dimer concentration ranging from 0 to 800 nM, in the presence of 100 ng of salmon sperm DNA as a specific competitor. For competition experiments, the 32P-ATP-end-labelled promoter fragments were incubated with Ec FNR (400 nM) and Nm FNR (800 nM) in the presence of variable amounts of each unlabelled DNA fragments. FNR binding was assessed as described (Ziegelhoffer and Kiley, 1995; Lazazzera et al., 1996)
Animal models
For the mouse model, bacteria were grown overnight on brain–heart infusion (BHI) broth agar plates, then harvested in BHI broth and the number of bacteria was estimated by measuring the OD600 of the bacterial suspension. Bacteria were harvested by centrifugation, resuspended in BHI medium supplemented with 1% Iron Dextran (w/v) at the desired concentration and 100 μl of suspension was injected in each animal. Six-week-old BALB/c female mice (Charles River laboratories), 10 mice per group, were infected intraperitoneally with 107 bacteria of wild-type MC58 or its isogenic mutant strains. Bacterial concentration was confirmed by colony plating. Bacteraemia was assessed by colony plating at 18 h, collecting blood from a tail vein bleed.
The infant rat model was performed as previously described (Granoff et al., 2001). In brief, bacteria were grown to log-phase in Mueller-Hinton (Sigma, St Louis, MO) supplemented with 0.25% glucose, washed and resupended at the desired concentration in PBS-1%BSA. Four-day-old infant pups from litters of outbred Wistar rats (Charles River, Hollister, CA) were randomly redistributed to the nursing mothers. Groups of six to eight animals were challenged intraperitoneally with bacteria of wild-type BZ232 or its isogenic BZ-fnrKO strain. Eighteen hours after the bacterial challenge, blood specimens were obtained by cardiac puncture and blood aliquots were plated onto chocolate agar for viable counting. Standard errors were evaluated by dividing the standard deviation values by the squared root value of number of animals. In the different experiments, standard errors did not vary by more than 1 log the values of the geometrical mean. Statistical difference among groups was evaluated by two-tailed Student's t-test. The infant rat studies were approved by the CHORI institutional committee responsible for overseeing experiments in animals.
Supplementary Material
The following supplementary material is available for this article online:
This material is available as part of the online article from http://www.blackwell-synergy.com
Fig. S1. RT-PCR analysis of NMB0388-galM and mapA-pgmβ operons.
Fig. S2. Multiple sequence alignment of upstream regions of EMSA-positive genes.
Fig. S3. Role of mapA and pgmβ genes in maltose metabolism.
Fig. S4. Transcription analysis of fnr, galM and mapA genes in wild-type and complemented strains.
Table S2. FNR binding to the upstream regions of the nine FNR-regulated transcriptional units.
Table S1. Expression ratios of genes found differentially expressed in MC58 wild type versus MC-fnrko mutant under oxygen restriction.
Acknowledgments
We wish to thank Rino Rappuoli for the useful discussions and the critical reading of the manuscript. We are also grateful to Antonietta Maiorino for her expert secretarial assistance and to Giorgio Corsi for artwork. This work was supported by Public Health Service Grant RO1 AI46464 from the National Institute of Allergy and Infectious Diseases, NIH, and NIH training grant T32-HL007951 (J.A.W.).
References
- Anjum MF, Stevanin TM, Read RC, Moir JW. Nitric oxide metabolism in Neisseria meningitidis. J Bacteriol. 2002;84:2987–2993. doi: 10.1128/JB.184.11.2987-2993.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai H, Mizutani M, Igarashi Y. Transcriptional regulation of the nos genes for nitrous oxide reductase in Pseudomonas aeruginosa. Microbiol. 2003;149:29–36. doi: 10.1099/mic.0.25936-0. [DOI] [PubMed] [Google Scholar]
- Baltes N, N'diaye M, Jacobsen ID, Maas A, Buettner FF, Gerlach GF. Deletion of the anaerobic regulator HlyX causes reduced colonization and persistence of Actinobacillus pleuropneumoniae in the porcine respiratory tract. Infect Immun. 2005;73:4614–4619. doi: 10.1128/IAI.73.8.4614-4619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DM, Lazazzera BA, Kiley PJ. Characterization of FNR* mutant proteins indicates two distinct mechanisms for altering oxygen regulation of the Escherichia coli transcription factor FNR. J Bacteriol. 1995;177:3972–3978. doi: 10.1128/jb.177.14.3972-3978.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc Natl Acad Sci USA. 2003;100:2801–2806. doi: 10.1073/pnas.2628026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouffard GG, Rudd KE, Adhya SL. Dependence of lactose metabolism upon mutarotase encoded in the gal operon in Escherichia coli. J Mol Biol. 1994;244:269–278. doi: 10.1006/jmbi.1994.1728. [DOI] [PubMed] [Google Scholar]
- Contreras I, Toro CS, Troncoso G, Mora GC. Salmonella typhi mutants defective in anaerobic respiration are impaired in their ability to replicate within epithelial cells. Microbiol. 1997;43:2665–2672. doi: 10.1099/00221287-143-8-2665. [DOI] [PubMed] [Google Scholar]
- Delany I, Rappuoli R, Scarlato V. Fur functions as an activator and as a repressor of putative virulence genes in Neisseria meningitidis. Mol Microbiol. 2004;52:1081–1090. doi: 10.1111/j.1365-2958.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- Fitting C, Dourodoff MJ. Phosphorolysis of maltose by enzyme preparation from Neisseria meningitidis. J Biol Chem. 1952;199:153–163. [PubMed] [Google Scholar]
- Fritz C, Maass S, Kret A, Bange FC. Dependence of Mycobacterium bovis BCG on anaerobic nitrate reductase for persistence is tissue specific. Infect Immun. 2002;1:286–291. doi: 10.1128/IAI.70.1.286-291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granoff DM, Moe GR, Giuliani MM, Abu-Bodie J, Santini L, Brunelli B, et al. A novel mimetic antigen eliciting protective antibody to Neisseria meningitidis. J Immunol. 2001;167:6487–6496. doi: 10.4049/jimmunol.167.11.6487. [DOI] [PubMed] [Google Scholar]
- Grifantini R, Sebastian S, Frigimelica E, Draghi M, Bartolini E, Muzzi A, et al. Identification of iron-activated and -repressed Fur-dependent genes by transcriptome analysis of Neisseria meningitidis group B. Proc Natl Acad Sci USA. 2003;100:9542–9547. doi: 10.1073/pnas.1033001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifantini R, Frigimelica E, Delany I, Bartolini E, Giovinazzi S, Balloni S, et al. Characterization of a novel Neisseria meningitidis Fur and iron-regulated operon required for protection from oxidative stress: utility of DNA microarray in the assignment of the biological role of hypothetical genes. Mol Microbiol. 2004;54:962–979. doi: 10.1111/j.1365-2958.2004.04315.x. [DOI] [PubMed] [Google Scholar]
- Grossiord BP, Luesink EJ, Vaughan EE, Arnaud A, de Vos WM. Characterization, expression, and mutation of the Lactococcus lactis galPMKTE genes, involved in galactose utilization via the Leloir pathway. J Bacteriol. 2003;85:870–878. doi: 10.1128/JB.185.3.870-878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest JR, Green J, Irvine AS, Spiro S. The FNR modulon and FNR-regulated gene expression. In: Lin ECC, Lynch AS, editors. Regulation of Gene Expression in Escherichia coli. New York, NY: Champman and Hall; 1996. pp. 317–342. [Google Scholar]
- Householder TC, Belli WA, Lissenden S, Cole JA, Clark VL. Cis- and trans-acting elements involved in regulation of aniA, the gene encoding the major anaerobically induced outer membrane protein in Neisseria gonorrhoeae. J Bacteriol. 1999;8:54–55. doi: 10.1128/jb.181.2.541-551.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Householder TC, Fozo EM, Cardinale JA, Clark VL. Gonococcal nitric oxide reductase is encoded by a single gene, norB, which is required for anaerobic growth and is induced by nitric oxide. Infect Immun. 2000;68:524–526. doi: 10.1128/iai.68.9.5241-5246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Weber KD, Qiu Y, Kiley PJ, Blattner FR. Genome-wide expression analysis indicates that FNR of Escherichia coli K-12 regulates a large number of genes of unknown function. J Bacteriol. 2005;187:1135–1160. doi: 10.1128/JB.187.3.1135-1160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoroshilova N, Popescu C, Munck E, Beinert H, Kiley PJ. Iron-sulfur cluster disassembly in the FNR protein of Escherichia coli by O2: [4Fe-4S] to [2Fe-2S] conversion with loss of biological activity. Proc Natl Acad Sci USA. 1997;94:6087–6092. doi: 10.1073/pnas.94.12.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan HH, Ghosh A, Paul K, Chowdhury R. Effect of anaerobiosis on expression of virulence factors in Vibrio cholerae. Infect Immun. 2004;72:3961–3967. doi: 10.1128/IAI.72.7.3961-3967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scolea LJ, Jr, Young FE. Development of a defined minimal medium for the growth of Neisseria gonorrhoeae. Appl Microbiol. 1974;28:70–76. doi: 10.1128/am.28.1.70-76.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landini P, Zehnder AJ. The global regulatory hns gene negatively affects adhesion to solid surfaces by anaerobically grown Escherichia coli by modulating expression of flagellar genes and lipopolysaccharide production. J Bacteriol. 2002;184:1522–1529. doi: 10.1128/JB.184.6.1522-1529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazazzera BA, Beinert H, Khoroshilova N, Kennedy MC, Kiley PJ. DNA binding and dimerization of the Fe-S-containing FNR protein from Escherichia coli are regulated by oxygen. J Biol Chem. 1996;271:2762–2768. doi: 10.1074/jbc.271.5.2762. [DOI] [PubMed] [Google Scholar]
- Leclerc GJ, Tartera C, Metcalf ES. Environmental regulation of Salmonella typhi invasion-defective mutants. Infect Immun. 1998;66:682–691. doi: 10.1128/iai.66.2.682-691.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissenden S, Mohan S, Overton T, Regan T, Crooke H, Cardinale JA, et al. Identification of transcription activators that regulate gonococcal adaptation from aerobic to anaerobic or oxygen-limited growth. Mol Microbiol. 2000;37:839–855. doi: 10.1046/j.1365-2958.2000.02050.x. [DOI] [PubMed] [Google Scholar]
- Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. 2003;197:789–799. doi: 10.1084/jem.20021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellies J, Jose J, Meyer TF. The Neisseria gonorrhoeae gene aniA encodes an inducible nitrite reductase. Mol Gen Genet. 1997;256:525–532. doi: 10.1007/s004380050597. [DOI] [PubMed] [Google Scholar]
- Nassif X, Pujol C, Morand P, Eugene E. Interactions of pathogenic Neisseria with host cells. Is it possible to assemble the puzzle? Mol Microbiol. 1999;32:1124–1132. doi: 10.1046/j.1365-2958.1999.01416.x. [DOI] [PubMed] [Google Scholar]
- van Putten JP, Duensing TD, Carlson J. Gonococcal invasion of epithelial cells driven by P.IA, a bacterial ion channel with GTP binding properties. J Exp Med. 1998;88:941–952. doi: 10.1084/jem.188.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian N, Stanley GA, Hahn-Hagerdal B, Radstrom P. Purification and characterization of two phosphoglucomutases from Lactococcus lactis subsp. lactis and their regulation in maltose- and glucose-utilizing cells. J Bacteriol. 1994;176:5304–5311. doi: 10.1128/jb.176.17.5304-5311.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JD, Mahnane MR, Anjum MF, Shaw JG, Read RC, Moir JWB. The pathogen Neisseria meningitidis requires oxygen, but supplements growth by denitrification. Nitrite, nitric oxide and oxygen control respiratory flux at genetic and metabolic levels. Mol Microbiol. 2005;58:800–809. doi: 10.1111/j.1365-2958.2005.04866.x. [DOI] [PubMed] [Google Scholar]
- Salmon K, Hung SP, Mekjian K, Baldi P, Hatfield GW, Gunsalus RP. Global gene expression profiling in Escherichia coli K2. The effects of oxygen availability and FNR. J Biol Chem. 2003;278:29837–29855. doi: 10.1074/jbc.M213060200. [DOI] [PubMed] [Google Scholar]
- Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, et al. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science. 2000;287:1809–1815. doi: 10.1126/science.287.5459.1809. [DOI] [PubMed] [Google Scholar]
- Trieu-Cuot P, Poyart-Salmeron C, Carlier C, Courvalin P. Nucleotide sequence of the erythromycin resistance gene of the conjugative transposon Tn1545. Nucleic Acids Res. 1990;8:3660. doi: 10.1093/nar/18.12.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner S, Reid E, Smith H, Cole J. A none cytocrome c peroxidase from Neisseria gonorrhoeae: a lipoprotein from a gram-negative bacterium. Biochem J. 2003;373:865–873. doi: 10.1042/BJ20030088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks KE, Dunn KL, Farrant JL, Reddin KM, Gorringe AR, Langford PR, Kroll JS. Periplasmic superoxide dismutase in meningococcal pathogenicity. Infect Immun. 1998;66:213–217. doi: 10.1128/iai.66.1.213-217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood GE, Khelef N, Guiso N, Friedman RL. Identification of Btr-regulated genes using a titration assay. Search for a role for this transcriptional regulator in the growth and virulence of Bordetella pertussis. Gene. 1998;209:51–58. doi: 10.1016/s0378-1119(98)00031-6. [DOI] [PubMed] [Google Scholar]
- Xu Q, Dziejman M, Mekalanos JJ. Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc Natl Acad Sci USA. 2003;100:1286–1291. doi: 10.1073/pnas.0337479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelhoffer EC, Kiley PJ. In vitro analysis of a constitutively active mutant form of the Escherichia coli global transcription factor FNR. J Mol Biol. 1995;245:35–36. doi: 10.1006/jmbi.1994.0029. [DOI] [PubMed] [Google Scholar]
- Zusman T, Feldman M, Halperin E, Segal G. Characterization of the icmH and icmF genes required for Legionella pneumophila intracellular growth, genes that are present in many bacteria associated with eukaryotic cells. Infect Immun. 2004;72:3398–3409. doi: 10.1128/IAI.72.6.3398-3409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. RT-PCR analysis of NMB0388-galM and mapA-pgmβ operons.
Fig. S2. Multiple sequence alignment of upstream regions of EMSA-positive genes.
Fig. S3. Role of mapA and pgmβ genes in maltose metabolism.
Fig. S4. Transcription analysis of fnr, galM and mapA genes in wild-type and complemented strains.
Table S2. FNR binding to the upstream regions of the nine FNR-regulated transcriptional units.
Table S1. Expression ratios of genes found differentially expressed in MC58 wild type versus MC-fnrko mutant under oxygen restriction.