

Abstract
Membrane protein function within the membrane interstices is achieved by mechanisms that are not typically available to water-soluble proteins. The whole balance of molecular interactions that stabilize three-dimensional structure in the membrane environment is different from that in an aqueous environment. As a result interhelical interactions are often dominated by non-specific van der Waals interactions permitting dynamics and conformational heterogeneity in these interfaces. Here, solid-state NMR data of the transmembrane domain of the M2 protein from influenza A virus are used to exemplify such conformational plasticity in a tetrameric helical bundle. Such data lead to very high resolution structural restraints that can identify both subtle and substantial structural differences associated with various states of the protein. Spectra from samples using two different preparation protocols, samples prepared in the presence and absence of amantadine, and spectra as a function of pH are used to illustrate conformational plasticity.
Keywords: M2 channel, Influenza A virus, Conformational plasticity, PISEMA, Solid-state NMR, Membrane protein
1. Introduction
It is becoming apparent that membrane proteins typically have multiple conformational states that require conformational transitions and large amplitude molecular motions to perform their function. Here, we present an example of this conformational plasticity, the M2 proton channel domain from influenza A virus, a tetrameric protein with a single transmembrane helix contributed from each monomer. Solid-state NMR spectroscopy of uniformly aligned samples in planar liquid–crystalline lipid bilayers is used to characterize structural details of the transmembrane domain that has been shown to function as a proton channel and can be blocked by the anti-viral drug, amantadine. The molecular structures discussed here have been previously published, but new structural data extend the range of sample conditions to illustrate the conformational heterogeneity and plasticity of this protein.
Membrane proteins are fundamentally very different from water-soluble proteins. Their amino acid composition is substantially different [1]. Charged residues and amide side-chain residues are more common in water-soluble α-helices than transmembrane α-helices by nearly a factor of three, while hydrophobic and weakly hydrophilic residues are more common in membrane proteins than water-soluble proteins by 50%. In addition, these residues have a distribution profile as a function of depth into the lipid bilayer [2]. The reduced diversity in side-chain functionality results in a reduced capacity to support electrostatic intramolecular and intermolecular interactions. Consequently, the whole balance of molecular interactions is altered for the stabilization of membrane protein structure. The distribution profile for the amino acids parallels gradients in dielectric constant [3], water concentration and fluidity [4] in the membrane environment. This anisotropy of the lipid bilayers is used by cells to generate the vital electrical, chemical, and mechanical potentials, which are derived from or lead to phenomena, such as membrane thinning and hydrophobic mismatch, curvature frustration, charge polarization and lateral force gradients [5–7]. These phenomena affect membrane protein structure, orientation, dynamics and function.
Engelman and Steitz [8] recognized more than 25 years ago that the folding of α-helices within the membrane was going to be energetically problematical and hypothesized that these secondary structural elements would fold at the bilayer surface followed by insertion into the membrane [9,10]. In this fashion the hydrogen bonds are formed in an environment that has considerable water, a known catalyst for hydrogen bond rearrangements [11–14]. Computational evidence has been building for this folding hypothesis [15,16], however, this mechanism raises many interesting questions; among them is the issue that helical folding is occurring in a different environment than that for the native protein—i.e., the bilayer surface versus the bilayer interior. Since structural stability is dependent on the protein's environment, it is possible that these helices are kinetically trapped states in the bilayer interstices. Such kinetically trapped conformations have been observed for peptides in non-protic organic solvents [14] and in lipid bilayers [17]. Conformations that are crosslinked by hydrogen bonds in a low dielectric environment may become trapped due to the strength of the interaction in that environment and the lack of a catalyst to facilitate its rearrangement. In part, the lack of water in the membrane interior and the necessity for water in folding α-helices provides an explanation for Engelman's hypothesis. In addition, it may explain why aqueous access to the amide backbone in membrane proteins is quite variable; while most of the amide protons of lac permease [18] and M2 protein [19] can be readily exchanged, the amides in the transmembrane region of diacylglycerol kinase are extremely resistant to H–D exchange [20]. The overall result is that helices are structurally very stable when water is scarce [21] and often very uniform in local structure (torsion angles varying by as little as ±6° [22]).
For many membrane proteins the very high concentration of aliphatic side-chains in the bilayer interstices generates a largely greasy interface between transmembrane helices. The result is that these helix–helix interactions are dominated by van der Waals and other weak electrostatic interactions. Where hydrogen bonding is rare the non-specificity of these interfacial interactions may readily permit dynamics and conformational transitions between low energy functional states. Indeed, there has been considerable discussion in the literature of the rough potential energy surfaces for proteins, in general [23] and membrane proteins appear to support a particularly rough energy potential [24,25] as opposed to a single deep potential energy minimum at the bottom of a smooth folding funnel. There is a large literature that documents almost indiscriminate chemical crosslinking between helices in polytopic membrane proteins provided that the reaction times are lengthy [26]. Moreover, hydrogen–deuterium exchange experiments on the full-length M2 protein have shown that rotational excursions occur about the helical axis exposing the normally lipid facing amides to an aqueous pore [19]. Furthermore, there is a growing literature that has characterized multiple functional and structural states of membrane proteins involving different packing arrangements for α-helices [27,28].
M2 protein is a proton channel and an important drug target for influenza virus. The protein is a 96-residue polypeptide with a single transmembrane helix (residues 26–43). The channel forms as a result of tetramerization [29–31] and the proton conductance is acid activated below pH 6.5 [32–34], a process that is thought to result when three of the four His-37 residues become charged [35]. The anti-viral drugs, amantadine and rimantadine that block conductance of both the full-length protein and the isolated transmembrane domain (M2-TMD) [36–39] bind to both the activated and inactivated states without breaking the time averaged tetrameric symmetry [40] and are therefore thought to bind on the symmetry axis. Recently, the tetramerization of the isolated transmembrane domain has been elegantly confirmed by observing 19F dipolar interactions in a CODEX solid-state NMR experiment [41,42]. The structure of this domain at high pH has been solved [43,44] and also in the presence of amantadine [40]. Additional structural studies of the transmembrane domain have been performed using cysteine mutagenesis [45], analytical ultracentrifugation [46], and UV resonance Raman [47]. These results and those from many molecular dynamics studies [48–51] have generated insights into the multiple functional states, structures and dynamics of this protein. Solid-state NMR structural data as a function of pH and as a function of sample preparation protocol have recently been obtained and will be presented here for the first time.
Solid-state NMR is rapidly developing as a major structural technology for the characterization of membrane proteins [52–56]. This methodology requires neither that the sample be a solid nor that it has rapid isotropic motions, as is required by X-ray crystallography and solution NMR, respectively. Solid-state NMR experiments can be performed on liquid–crystalline lipid bilayer preparations—the most native-like membrane protein environment used by the front-line technologies for structural characterization today. The structural restraints of interest here are obtained as orientational restraints from uniformly aligned planar liquid–crystalline bilayer samples. In PISEMA experiments [57] where the anisotropic 15N chemical shift is correlated with the 15N–1H dipolar interaction, resonance patterns are often observed for α-helical structures, known as PISA wheels [58,59]. The position, shape and size of the PISA wheel within the spectrum independently characterize the tilt angle of helices relative to the bilayer normal. There are now 10 unique membrane protein structures characterized by solid-state NMR with coordinates deposited in the Protein Data Bank. Applications extend from peptide characterizations [41,60–62] to full-length proteins [54,55,63–65]. Numerous advances in sample preparation [54–56] and NMR probe technology [66,67] are leading to the increased numbers, size, and complexity of 3D structural characterizations in liquid–crystalline lipid bilayer environments.
2. Materials and methods
2.1. Peptide synthesis and sample preparation
The transmembrane peptide of M2 protein (S22SAPLVVAASIIGILHLILWILARL46) was synthesized by either standard Fmoc chemical synthesis [68] or by biosynthesis through a maltose binding protein fusion expression system as described by Hu et al. [62]. High purity was achieved for the chemically synthesized peptide as previously described [68]. For the biosynthetic preparation the cleavage reaction (Tobacco Etch Virus protease), which results in an additional three residues (SNA) on the amino terminus of the peptide was stopped by the addition of trichloroacetic acid (TCA) at a concentration of 6% and the precipitate was collected by centrifugation. After washing the pellet twice with water to remove residual TCA, the protein was lyophilized in a vacuum centrifuge. 20 ml methanol/l culture was added and gently mixed for several hours at room temperature. To remove the undissolved protein (MBP and TEV), the solution was centrifuged at 13,000×g for 20 min, and the supernatant was collected. The peptide was then lyophilized in a vacuum centrifuge and stored at −20 °C.
Two different protocols were used to prepare the M2-TMD samples (Fig. 1), both are initiated by codissolving peptide and dimyristoyl-phosphatidylcholine (DMPC) in trifluoroethanol. In one case the organic solution is spread directly on glass slides (organic protocol) prior to the evaporation of the solvent and hydration to ∼50% by weight to form the planar bilayers [44]. In this case the molar ratio of peptide to lipid is 1:16. Ratios up to 1:30 have shown no substantial difference in the spectra [69]. In the second case the organic solvent is evaporated and liposomes are prepared (liposome protocol) in bulk solution prior to ultracentrifugation, spreading on the glass slides, and dehydration/rehydration [40]. For this latter protocol a molar ratio of peptide to lipid of 1:50 was used although no significant variation in the spectra have been observed over a broad range of molar ratios. The formation of the liposomes containing the M2 peptide was prepared by suspension in a phosphate buffer (50 mM, pH 8.0) or acetate buffer (50 mM, pH 5.0). The proteoliposomes were bath sonicated and dialyzed against buffer for 1 day prior to ultracentrifugation at 196,000×g. The pelleted proteoliposomes were used to prepare aligned samples on glass slides by dehydrating the sample in a ∼70% relative humidity environment followed by rehydration in a 96% relative humidity environment resulting in ∼50% by weight water in the sample, before sealing the samples in square glass tubing. The pH of the samples was characterized from the supernatant of the ultracentrifugation run [35]. Amantadine was added by dialysis at 10 mM concentration in the presence of buffer.
Fig. 1.
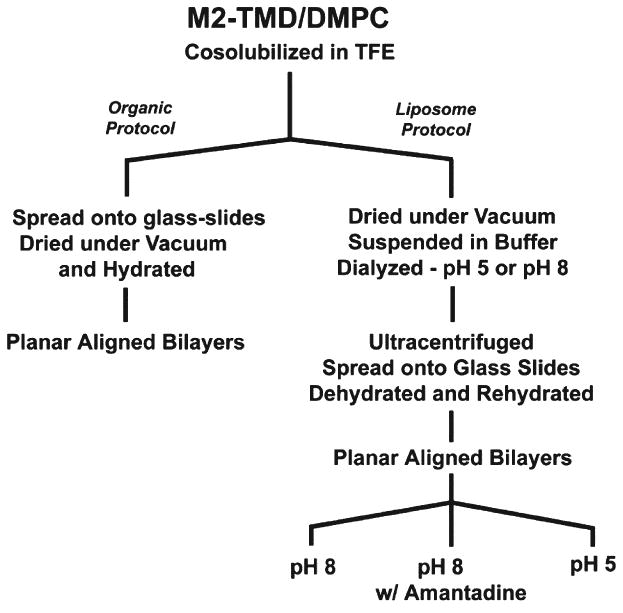
Flow chart summarizing the sample preparation protocols for the lipid bilayer preparations of M2-TMD. Details of the protocols are presented in the Materials and methods section.
While early studies were performed using the organic solvent protocol, the liposome protocol has been used in recent years for all of our M2-TMD studies as it more closely follows the protocols we use for full-length membrane proteins. Dialysis provides an opportunity to buffer the sample and to accurately measure its pH. In addition it provides an opportunity to dialyze out residual organic solvent.
2.2. Solid-state NMR experiments
The solid-state NMR data that are described here for the first time (as opposed to data previously published) were all performed on a Bruker DRX 600 NMR wide bore spectrometer using an NHMFL Low-E double resonance probe. Samples were observed at 303 K using a substantial flow of air for maintaining the sample temperature and yet we found that optimal spectra were obtained with a lengthy 6-s recycle delay—the result of residual sample heating. For the PISEMA [57] experimental setup, typically a 90° pulse of 6.5-μs and an 800-μs contact time were used for cross-polarization with a 40-kHz spin-lock field. The same RF field was also used during the Lee–Goldberg spin exchange at the magic angle and 65-kHz decoupling field was applied for proton decoupling with the SPINAL decoupling sequence [70]. 32 t1 increments were obtained, zero-filled to 256 points in the t1 dimension before Fourier transformation. A concentrated solution of NH4NO3 was used as the external frequency reference, defined as 26 ppm relative to liquid ammonia for all 15N spectra.
3. Results
Solid-state PISEMA spectra of M2-TMD aligned samples from organic and liposomal preparations are compared in Fig. 2. These spectra are all obtained above the gel to liquid–crystalline phase transition temperature. The spectrum from the organic protocol (red, [43]) is actually a superposition of a set of spectra from single and multiple site 15N-labeled samples. The spectrum from the liposomal protocol (blue) uses a biosynthetic uniformly 15N-labeled peptide. The observed anisotropic resonance frequencies are a consequence of the orientationally dependent 15N chemical shift interaction and the 15N–1H dipolar interaction. The uniformly 15N-labeled sample represents 11 additional backbone resonances and the likelihood of 3 side-chain resonances compared to the superimposed specific site labeled spectra (Fig. 2A). Not all of these resonances are likely to be in the spectral range displayed and a few may possess isotropic motions and very low intensity. The spectral comparison in Fig. 2B shows significant shifts in the resonance frequencies and changes in linewidth. For a nearly ideal α-helical structure, a pattern of resonances, known as a PISA wheel [58,59], is observed in the PISEMA spectra. The position, size, and shape of the PISA wheel are very sensitive to the tilt angle of the helix relative to the bilayer normal. Theoretical PISA wheels based on φ/ψ torsion angles of −60°, −45°, respectively [71], are superimposed on the spectra in Fig. 2. The experimental data from both preparations clearly show the presence of PISA wheels. The tilt angle from the organic protocol (Fig. 2A) is fit by a PISA wheel reflecting a 38° tilt angle, while that from the liposomal protocol is tilted at 32° (Fig. 2C). To indicate the error range for the helical tilt, additional PISA wheels are displayed in Fig. 2C at ±3° showing that the error in this characterization is less than ±3°.
Fig. 2.
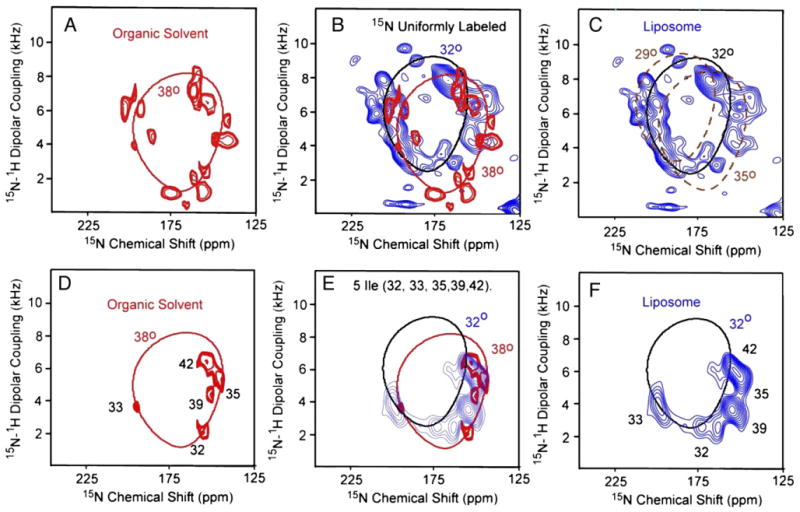
Two-dimensional 1H–15N PISEMA spectra of M2-TMD in DMPC bilayers above the gel to liquid–crystalline phase transition temperature prepared by either the organic protocol (A and D; red) or liposomal protocol (C and F; blue). The spectra from these two protocols are superimposed in panels B and E. The PISEMA spectrum (A) from the organic solvent preparation is a superimposed set of spectra from single and multiple site labeled samples representing 65% of the backbone amide sites [43]. The backbone sites that are not labeled are residues 23, 24, 31, 34, 37, 44, 45, and 46 — most of which are in the hydrophilic terminal regions and may give rise to isotropically averaged signals. The PISEMA spectrum in C is from a uniformly 15N-labeled sample that has 3 extra residues at the amino terminus [62]. The PISEMA spectra in panels D–F are from 5-site 15N isoleucine-labeled samples. The spectrum in panel D is adapted from Wang et al. [59].
The PISEMA spectra and assignments (Fig. 2D and F) of the 5-site 15N isoleucine-labeled samples are compared in Fig. 2E. The frequencies of the isoleucine resonances are only slightly different for the two preparations. These highly reproducible spectra show that both conformations are in a transmembrane configuration and that the tilt of these two stable conformations differs by only 6°. The rotational orientation of the helices differs by no more than 10° judged from the 5-site isoleucine-labeled samples, implying that the same transmembrane configuration is present. The resonance linewidths in the PISEMA spectra from the organic solvent preparation are significantly narrower than that prepared from liposomes suggesting less low-frequency dynamics or conformational heterogeneity in the organic preparation.
Fig. 3 displays the PISEMA spectra of uniformly 15N-labeled (A and C) and 5 site 15N isoleucine (D and F)-labeled M2-TMD in the presence (red; C, F) and the absence (blue; A and D) of amantadine at pH 8.0. Amantadine is a prescription anti-viral drug known to bind and block the M2 proton channel. The uniformly labeled peptides were biosynthesized while the isoleucine-labeled peptides were chemically synthesized. All of the samples were prepared using the liposomal protocol. The effects of amantadine binding on the resonance frequencies and line-widths in the PISEMA spectra are illustrated in Fig. 3B and E. The PISEMA spectra in the presence of amantadine show substantially narrower resonance linewidths compared to the spectra in its absence. The reduced linewidth suggests that the amplitude of low-frequency motions has been substantially reduced or that conformational heterogeneity has been significantly reduced upon amantadine binding [72].
Fig. 3.
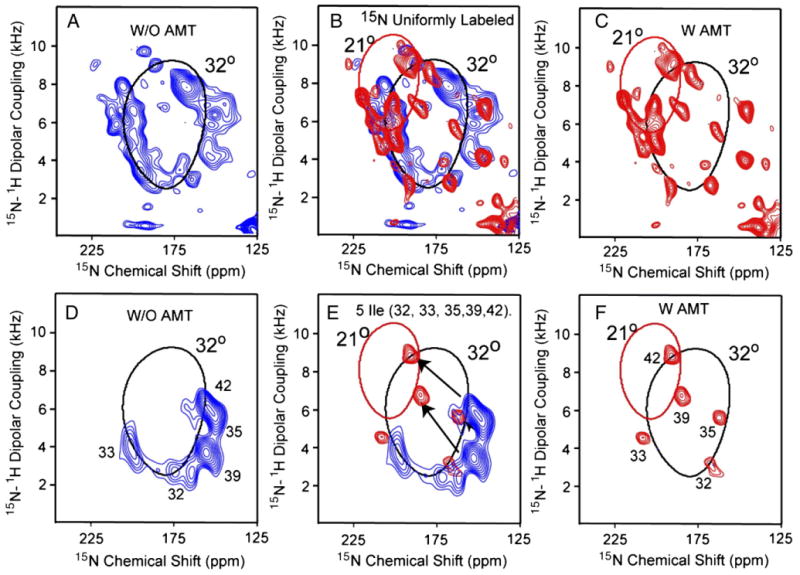
Two-dimensional 1H/15N PISEMA spectra of uniformly 15N-labeled M2-TMD (A–C) and 5-site 15N isoleucine-labeled (D–F) in aligned DMPC planar bilayers at pH 8.0 and 303 K in the absence of amantadine (A and D; red—same as spectra in Fig. 2C and F), in its presence (C and F; blue [40]) and where the spectra are superimposed (B and E). All samples were prepared by the liposomal protocol.
Previously the assignments of the resonances for both Fig. 3C and F using single site and multiple site labeled M2-TMD in the presence of amantadine have been reported [40]. The resonances from Ile 32, 33, and 35 show only subtle changes in chemical shift and dipolar coupling, while Ile 39 and 42 in the C-terminal portion of the transmembrane helix show dramatic chemical shift and dipolar coupling changes. Indeed, these resonances shift to another PISA wheel, which corresponds to a 21° tilt angle, instead of the 32° PISA wheel that characterizes the N-terminal region of the helix. Consequently, the M2-TMD helix develops an 11° kink in the presence of amantadine. The dipolar wave analysis [40] clearly shows that there is no significant change (≤±10°) in the phase of the wave at the kink site, consequently the rotational orientation remains the same with or without the kink and amantadine.
Fig. 4 displays the PISEMA spectra of uniformly 15N-labeled M2-TMD at pH 5.0 (red) and pH 8.0 (blue). The 1D chemical shift spectra shown in Fig. 4B are slices through the dipolar dimension at 7.28 kHz. The signal distribution around the wheel is quite similar and consequently there appears to be no or very little change in helix tilt upon lowering the pH from 8.0 to 5.0. While the pH 8 liposomal sample shows much broader resonances in the absence of amantadine than in its presence, as described above, here in the absence of amantadine and at pH 5 the linewidths are even broader. Indeed, upon lowering the pH the linewidth is observed to more than double. To sort out the resonance overlap in these PISEMA spectra of uniformly 15N-labeled M2-TMD, an 15N-labeled Gly34 sample was prepared to monitor the effect of pH and amantadine on a single resonance. One- and two-dimensional 15N spectra are shown in Fig. 5 at pH 8.0 (black; A, D, E), pH 5.0 (blue; B, E), and pH 8.0 in the presence of 10 mM amantadine (red; C, D). At pH 8.0, it has been noted earlier that, on average, the resonance linewidth narrows by a factor of two in the presence of amantadine and that small changes in the chemical shift and dipolar coupling frequencies are observed in the turn of the helix (e.g., Ile 32, 33, and 35) that includes Gly34. Here, a discernable shift of 14 ppm and 0.8 kHz is observed upon binding of amantadine to the channel at pH 8.0. The line shape in both the 1D and 2D spectra suggests the possibility of conformational heterogeneity. This is particularly interesting for the data in the absence of amantadine at pH 8, which suggest that the amantadine-bound conformational state is sampled even in the absence of amantadine. In fact, in Fig. 3E the Ile35 resonance in the absence of amantadine appears to show a small population of the amantadine-bound conformation, further supporting the Gly34 observation. Moreover, in the absence of amantadine a change in pH results in a dramatic broadening of the Gly34 resonance both in 1D and 2D spectra and in both the chemical shift and dipolar dimension. Once again, these are indications of conformational exchange between two or more conformational states.
Fig. 4.
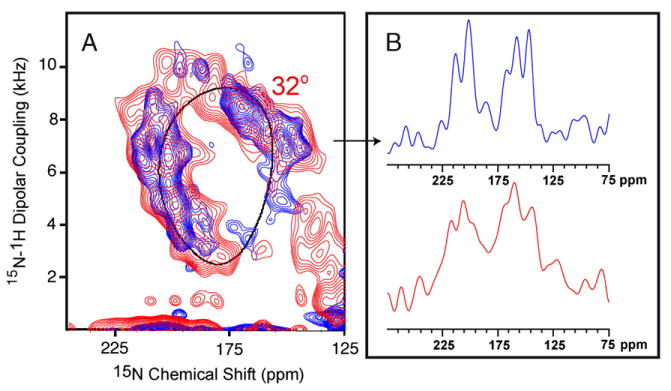
(A) Two-dimensional 1H/15N PISEMA spectra of uniformly 15N-labeled M2-TMD in aligned DMPC planar bilayers at 303 K and pH 5.0 (red) and pH 8.0 (blue—same as spectra in Fig. 2C). (A) One-dimensional chemical shift slices through the dipolar dimension at 7.28 kHz at pH 5.0 (red) and pH 8.0 (blue). All samples were prepared by the liposomal protocol.
Fig. 5.
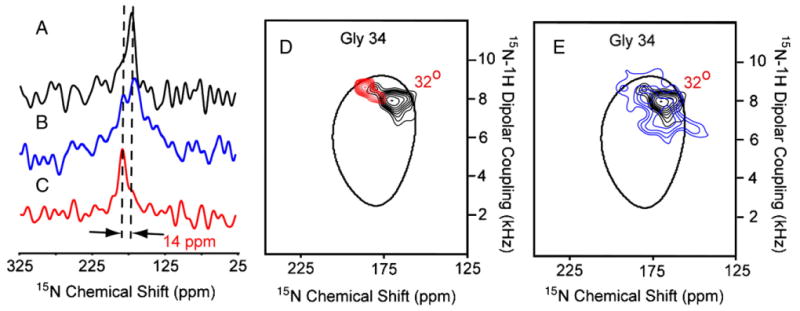
15N spectra of 15N Gly-34 labeled M2-TMD in aligned DMPC bilayers at 303 K prepared by the liposomal protocol. (A) One-dimensional spectra at pH 8.0 (black), (B) pH 5.0 (blue), (C) and at pH 8.0 (red) in the presence of 10 mM amantadine. (D and E) Superimposed two-dimensional 1H/15N PISEMA spectra at pH 8.0 (black), pH 5.0 (blue), pH 8.0 (red) in the presence of 10 mM amantadine in aligned DMPC planar bilayers. For the sake of simplicity the 32° PISA wheel is the same as that drawn in the previous figures even though it is known that the 15N chemical shift tensor for glycine would dictate that this wheel should be shifted by 5–7 ppm towards 0 ppm.
4. Discussion
The PISEMA data are a very sensitive probe of molecular structure, it is however not linearly sensitive, due to the P2cosθ dependency of molecular orientation on the spin interaction frequency. Consequently, variations in orientation of spin interaction tensors with respect to the magnetic field will induce a greater change in frequency than other orientations. Recently, a manuscript was published that described theoretical lineshapes that would be expected for different molecular orientations to the magnetic field and the linewidth based on orientational disorder in both the dipolar and chemical shift dimensions varied substantially [73]. For instance, an N–H vector that made an angle of 30±2° with respect to the bilayer normal and the magnetic field would have a range of dipolar coupling 2.5 times that for an N–H vector that made an angle of 10±2°. Importantly, the observed linewidths in the dipolar dimension of PISEMA spectra are typically much less than 1 kHz generating a structural restraint that has an error bar that is typically less than ±2°.
PISA wheels are also very sensitive probes of α-helical structure. Since local structural variation can shift resonance frequencies, the pattern of resonances can become blurred by the scatter in the resonances induced by variations in the φ/ψ torsion angles. It has recently been shown [22] that for the pH 8 M2-TMD resonance pattern without amantadine bound that the maximum variation in backbone torsion angles is only ±4°. Even though these resonances are relatively broad the uniformity of the helical structure can be precisely determined. This level of precision is higher than any other structural approach for membrane proteins and it is being achieved for proteins in a liquid–crystalline lipid bilayer environment.
PISEMA data and PISA wheel analyses have been presented here for several M2-TMD sample conditions. Firstly, there are significant spectral changes that occur with changes in the sample protocol. Previously, solvent history-dependent conformational states were extensively characterized for gramicidin A by solid-state NMR [74], CD [75], HPLC [76,77], Raman and Infrared spectroscopy [78]. It was also shown that conformational states with different hydrogen bonding patterns could be kinetically trapped in a membrane environment [17]. Here, with M2-TMD the conformations arising from the organic solvent and liposomal protocols appear to have the same hydrogen bonding pattern and only a modest change in helical tilt. There is no clear evidence for a helix kink or for changes in the backbone torsion angles, emphasizing that the helices in these two structures appear to be essentially rigid rods with weak, relatively non-specific interactions between the rods that are influenced by the protocol. Consequently, we suspect that this does not appear to be a situation where there is a kinetically trapped conformational state, but potentially a bound residual organic solvent molecule from the organic protocol that has modified the weak set of interactions between the helices. If so, this conformational change is an artifact, but it emphasizes that subtle changes in the environment can give rise to reproducible structural modifications and that the potential energy surface is broad, shallow, and rough [25]. Considering the complexity of the native membrane environment and the ability of cells to modulate these environments this result may also be biologically relevant.
A more dramatic conformational change is observed when amantadine binds to M2-TMD at pH 8. Importantly, the time-averaged tetrameric symmetry is maintained indicating that interactions of the amantidine must be fourfold averaged and therefore, amantadine does not appear to bind specifically to one monomer either at the channel mouth or to the exterior of the channel as has been suggested [79] based on electrophysiological data. The observed kink in the transmembrane helices appears to originate in the turn of the helix that includes the glycine residue. In water soluble proteins glycine is often a helix breaker, but for membrane proteins the situation is more complex. Glycine is a very common amino acid in transmembrane helices [1] and apparently, the stability of the intrahelical hydrogen bonding in a membrane environment is sufficient to counter the destabilizing effect of this residue [22]. However, glycine kink sites have been observed in other proteins such as KvAP where it facilitates the structural transition between two functional states [80]. Because of this potential for glycine to be part of a uniform helix as well as a kink site, glycine residues can be referred to as ‘pro-kink’ sites when they are observed in uniform helices. The relatively large change in structure induced upon amantadine binding is yet again suggestive of the weak interactions between helices and the relatively delicate balance of helix stability about a glycine residue.
Ligand binding has resulted in sharper resonances reflecting either reduced dynamics or reduced conformational heterogeneity, this is typical of ligand binding interactions, but it is surprising here, since amantadine is a hydrocarbon cage structure with an amino group undergoing time-shared interactions with four monomers. In other words, how does a largely hydrophobic ligand with little capacity for specific interactions reduce dynamics and/or conformational heterogeneity? This invites the intriguing possibility that it is not the ligand per se that causes these effects, but the displacement of water by the ligand. Water in the pore of this structure has the potential to destabilize hydrogen bonds and the weak electrostatic interactions between helices and when the water is displaced this potential is also displaced.
The PISEMA data at low pH have only recently been obtainable. Previously, spectra showed very poor sensitivity, but with the advent of low-electric field NMR probes [67] the heat deposited in the bilayer samples has been reduced by more than an order of magnitude and consequently, it is now possible to have a stable and reproducible sample. Fig. 5 shows 1D and PISEMA spectra of pH 5 and 8 samples in the absence of amantadine and of a pH 8 sample in the presence of amantadine. The resonance linewidths are substantially broader at pH 5 than those in the spectra at pH 8. While broad linewidths in M2-TMD spectra have previously been correlated with efficient relaxation and low-frequency dynamics [72], here there is strong evidence from the single site Gly-34 spectra and also from the Ile35 spectrum that conformational heterogeneity contributes significantly to the linewidths. However, differences of 10 to 20 ppm in anisotropic chemical shift and of 1 to 2 kHz in dipolar coupling are consistent with a small range of structural variation, if it is assumed that the glycine amide remains hydrogen bonded. The fact that heterogeneously broadened line shapes are observed and are consistent with small structural changes continues to suggest a rough potential energy surface [23] and structural differences dependent on non-specific interactions with modest potential energy barriers between conformational states. Indeed, the characterization of low-frequency dynamics and conformational heterogeneity only differs by the rate constant for inter-conversion, i.e., the magnitude of the potential energy barrier between the conformational states as described by a potential energy surface. Since high aliphatic content and low hydrophilic content are typical of transmembrane regions of membrane proteins it can be anticipated that multiple conformational states, interconversion between states and low-frequency dynamics will all be frequent properties of membrane proteins.
Interestingly, there is some evidence in the form of a weak signal for the amantadine-bound conformation (Fig. 5D) in two samples that have never been exposed to amantadine and some evidence for the amantadine-free state when there is a high enough concentration to saturate more than 99% of the sites. This hints at the interesting possibility that amantadine simply shifts a conformational equilibrium between two states that are sampled in its absence. This is interesting because we know that the amantadine-bound state does not conduct protons and if this state is sampled in the absence of amantadine then this could be a mechanism for opening and closing the channel in the absence of amantadine.
The plasticity of membrane proteins is becoming evident through the identification of multiple functional, structural, and dynamic states, as well as a fundamental understanding of the molecular interactions responsible for stabilizing these states. The M2 proton channel is known to have pH activated and inactivated states, as well as open and blocked states. Here, we have documented these conformational states that appear to primarily differ in helix packing interactions although one transition appears to be facilitated by a glycine residue in the midst of a transmembrane helix. Importantly, dynamics vary substantially among these states and it is interesting to note that the amantadine blocked state (i.e., least functional state) is the most rigid of the structures, while the pH 5 state is both the most dynamic and heterogeneous of the states, as well as the state with highest H+ conductance.
While some membrane proteins have evolved mechanisms to have a relatively rigid structure, such as those proteins that have a GxxxG sequence of amino acids, it appears that the plasticity of membrane protein structures has frequently been put to functional use. Such use is likely to be observed repeatedly in this class of proteins and consequently the task for membrane protein structural biologists is not just the characterization of one structure, but rather the characterization of a set of structures for each protein leading to a description of the potential energy surface. Furthermore, the weak interhelical interactions that are largely responsible for the plasticity of these proteins also result in the potential for these proteins to be easily distorted in sample preparation protocols and therefore great care must be taken to ensure that structures are not distorted by using a poor membrane mimetic environment or harsh protocol conditions.
Acknowledgments
The authors are grateful to the staff of the National High Magnetic Field Laboratory NMR facility (A. Blue) and the staff of Bioanalytical Synthesis and Services Laboratory (H. Hendricks and U. Goli) for their expertise, assistance and maintenance of instruments essential for this work. The authors also thank P. Gor'kov and W. Brey for their development of Low-E static Probes used in these experiments. This work was supported by the National Institute of Health, AI 23007. The experiments were largely performed at the National High Magnetic Field Laboratory, supported by Cooperative Agreement (DMR-0084173) and the state of Florida.
References
- 1.Eilers M, Patel AB, Liu W, Smith SO. Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys J. 2002;82:2720–2736. doi: 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White SH, von Heijne G. Transmembrane helices before, during and after insertion. Curr Opin Struct Biol. 2005;15:378–386. doi: 10.1016/j.sbi.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 3.White SH, Wiener MC. Determination of the structure of fluid lipid bilayer membranes. In: DiSalvo EA, Simon SA, editors. Permeability and Stability of Lipid Bilayers. CRC Press; Boca Raton: 1994. pp. 1–19. [Google Scholar]
- 4.Marrink SJ, Berkowitz M. Water and membranes. In: DiSalvo EA, Simon SA, editors. Permeability and Stability of Lipid Bilayers. CRC Press; Boca Raton: 1994. pp. 21–48. [Google Scholar]
- 5.Cantor RS. Lipid composition and the lateral pressure profile in bilayers. Biophys J. 1999;76:2625–2639. doi: 10.1016/S0006-3495(99)77415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Botelho AV, Gibson NJ, Thurmond RL, Wang Y, Brown MF. Conformational energetics of rhodopsin modulated by nonlamellar forming lipids. Biochemistry. 2002;41:6354–6368. doi: 10.1021/bi011995g. [DOI] [PubMed] [Google Scholar]
- 7.Sachs JN, Engelman DM. Introduction to the membrane protein reviews: the interplay of structure, dynamics and environment in membrane protein function. Ann Rev Biochem. 2006;75:707–712. doi: 10.1146/annurev.biochem.75.110105.142336. [DOI] [PubMed] [Google Scholar]
- 8.Engelman DM, Steitz TA. The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell. 1981;23:411–422. doi: 10.1016/0092-8674(81)90136-7. [DOI] [PubMed] [Google Scholar]
- 9.Popot JL, Engelman DM. Membrane protein folding and oligomerization: the two-stage model. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 10.Engelman DM. Crossing the hydrophobic barrier: insertion of membrane proteins. Science. 1996;274:1850–1851. doi: 10.1126/science.274.5294.1850. [DOI] [PubMed] [Google Scholar]
- 11.Sheinerman FB, Brooks CL., III Molecular picture of folding of a small ab protein. Proc Natl Acad Sci U S A. 1998;95:1562–1567. doi: 10.1073/pnas.95.4.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson GW, Cho CH. Role of hydration water in protein unfolding. Biophys J. 1999;77:3311–3318. doi: 10.1016/S0006-3495(99)77162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitter J. The temperature dependence of integral molecular motions in hydrated and dry α-amylase: the role of hydration water in the dynamical transition of proteins. Biophys J. 1999;76:1034–1042. doi: 10.1016/S0006-3495(99)77268-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu F, Cross TA. Water: a “foldase” that catalyzes hydrogen bond exchange in polypeptide conformational rearrangements. Proc Natl Acad Sci U S A. 1999;96:9057–9061. doi: 10.1073/pnas.96.16.9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Im W, Brooks CL., III De novo folding of membrane proteins: an exploration of the structure and NMR properties of the FD coat protein. J Mol Biol. 2004;337:513–519. doi: 10.1016/j.jmb.2004.01.045. [DOI] [PubMed] [Google Scholar]
- 16.Im W, Brooks CL., III Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc Natl Acad Sci U S A. 2005;102:6771–6776. doi: 10.1073/pnas.0408135102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arumugam S, Pascal S, North CL, Hu W, Lee KC, Cotten M, Ketchem RR, Xu F, Brenneman M, Kovacs F, Tian F, Wang A, Huo S, Cross TA. Conformational trapping in a membrane environment: a regulatory mechanism for protein activity? Proc Natl Acad Sci U S A. 1996;93:5872–5876. doi: 10.1073/pnas.93.12.5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patzlaff JS, Moeller JA, Barry BA, Brooker RJ. Fourier transform infrared analysis of purified lactose permease: a monodisperse lactose permease preparation is stably folded, α-helical, and highly accessible to deuterium exchange. Biochemistry. 1998;37:15363–15375. doi: 10.1021/bi981142x. [DOI] [PubMed] [Google Scholar]
- 19.Tian C, Gao FP, Pinto LH, Lamb RA, Cross TA. Initial structure and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci. 2003;12:2597–2605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oxenoid K, Kim HJ, Jacob J, Sonnichsen FD, Sanders CR. NMR Assignments for a helical 40 kDa membrane protein. J Am Chem Soc. 2004;126:5048–5049. doi: 10.1021/ja049916m. [DOI] [PubMed] [Google Scholar]
- 21.Janovjak H, Kedrov A, Cisneros DA, Sapra KT, Struckmeier J, Muller DJ. Imaging and detecting molecular interactions of single transmembrane proteins. Neurobiol Aging. 2006;27:546–561. doi: 10.1016/j.neurobiolaging.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 22.Page R, Li C, Hu J, Gao FP, Cross TA. Lipid bilayers: an essential environment for the understanding of membrane proteins. Magn Reson Chem. doi: 10.1002/mrc.2077. in press. [DOI] [PubMed] [Google Scholar]
- 23.James LC, Tawfik DS. Conformational diversity and protein evolution—a 60 year old hypothesis revisited. TRENDS Biochem Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 24.Mitra A, Tascione R, Auerbach A, Licht S. Plasticity of acetylcholine receptor gating motions via rate–energy relationships. Biophys J. 2005;89:3071–3078. doi: 10.1529/biophysj.105.068783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 26.Hunt JF, Earnest TN, Bousche O, Kalghatgi K, Reilly K, Horvath C, Rothschild KJ, Engelman DM. A biophysical study of integral membrane protein folding. Biochemistry. 1997;36:15156–15176. doi: 10.1021/bi970146j. [DOI] [PubMed] [Google Scholar]
- 27.Spencer RH, Rees DC. The α-helix and the organization and gating of channels. Annu Rev Biophys Biomol Struct. 2002;31:207–233. doi: 10.1146/annurev.biophys.31.082901.134329. [DOI] [PubMed] [Google Scholar]
- 28.Cordero-Morales JF, Guello LG, Zhao Y, Jogini V, Cortes DM, Roux B, Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat Struct Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- 29.Holsinger LJ, Lamb RA. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology. 1991;183:32–43. doi: 10.1016/0042-6822(91)90115-r. [DOI] [PubMed] [Google Scholar]
- 30.Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: evidence that it forms a tetrameric channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaguchi T, Tu Q, Pinto LH, Lamb RA. The active oligomeric state of the minimalistic influenza virus M2 ion channel is a tetramer. Proc Natl Acad Sci U S A. 1997;94:5000–5005. doi: 10.1073/pnas.94.10.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou N, Hay AJ. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol. 1996;494(Pt 2):329–336. doi: 10.1113/jphysiol.1996.sp021495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimbo K, Brassard DL, Lamb RA, Pinto LH. Ion selectivity and activation of the M2 ion channel of influenza virus. Biophys J. 1996;70:1335–1346. doi: 10.1016/S0006-3495(96)79690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH. Permeation and activation of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:31038–31050. doi: 10.1074/jbc.M003663200. [DOI] [PubMed] [Google Scholar]
- 35.Hu J, Fu R, Nishimura K, Zhang L, Zhu HX, Busath DD, Vijayvergiya V, Cross TA. Histidines: heart of the H+ channel from influenza A virus. Proc Natl Acad Sci U S A. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985;4:3021–3024. doi: 10.1002/j.1460-2075.1985.tb04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hay AJ. The action of adamantamines against influenza A viruses: inhibition of the M2 ion channel protein. Semin Virol. 1992;3:21–30. [Google Scholar]
- 38.Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol. 1993;67:5585–5594. doi: 10.1128/jvi.67.9.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duff KC, Ashley RH. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology. 1992;190:485–489. doi: 10.1016/0042-6822(92)91239-q. [DOI] [PubMed] [Google Scholar]
- 40.Hu J, Asbury T, Achuthan S, Li C, Bertram R, Quine JR, Fu R, Cross TA. Backbone structure of the amantadine-blocked transmembrane domain M2 proton channel from influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buffy JJ, Traaseth NJ, Mascioni A, Gor'kov PL, Chekmenev EY, Brey WW, Veglia G. Two-dimensional solid-state NMR reveals two topologies of sarcolipin in oriented lipid bilayers. Biochemistry. 2006;45:10939–10946. doi: 10.1021/bi060728d. [DOI] [PubMed] [Google Scholar]
- 42.Luo W, Hong M. Determination of the oligomeric number and intermolecular distances of membrane protein assemblies by anisotropic 1H driven spin diffusion NMR spectroscopy. J Am Chem Soc. 2006;128:7242–7251. doi: 10.1021/ja0603406. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishimura K, Kim S, Zhang L, Cross TA. The closed state of a H+ channel helical bundle: combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 45.Bauer CM, Pinto LH, Cross TA, Lamb RA. The influenza virus M2 ion channel protein: probing the structure of the t domain in intact cells by using engineered disulfide cross-linking. Virology. 1999;254:196–209. doi: 10.1006/viro.1998.9552. [DOI] [PubMed] [Google Scholar]
- 46.Salom D, Hill BR, Lear JD, DeGrado WF. pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry. 2000;39:14160–14170. doi: 10.1021/bi001799u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kukol A, Adams PD, Rice LM, Brunger AT, Arkin IT. Experimentally based orientational refinement of membrane protein models: a structure for the influenza A M2 H+ channel. J Mol Biol. 1999;286:951–962. doi: 10.1006/jmbi.1998.2512. [DOI] [PubMed] [Google Scholar]
- 48.Forrest LR, Tieleman DP, Sansom MSP. Defining the transmembrane helix of M2 protein from influenza A by molecular dynamics simulations in a lipid bilayer. Biophys J. 1999;76:1886–1896. doi: 10.1016/s0006-3495(99)77347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bu L, Im W, Brooks CL., III Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys J. 2007;92:854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Y, Voth GA. A computational study of the closed and open states of the influenza A M2 proton channel. Biophys J. 2005;89:2402–2411. doi: 10.1529/biophysj.105.066647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kass I, Arkin IT. How pH opens a H+ channel: the gating mechanism of influenza A M2. Structure. 2005;13:1789–1798. doi: 10.1016/j.str.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 52.Ramamoorthy A, Wei Y, Lee DK. PISEMA solid-state NMR spectroscopy. Ann Rep NMR Spectrosc. 2004;52:1–52. [Google Scholar]
- 53.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park SH, Prytulla S, De Angelis AA, Brown JM, Kiefer H, Opella SJ. High resolution NMR spectroscopy of a GPCR in aligned bicelles. J Am Chem Soc. 2006;128:7402–7403. doi: 10.1021/ja0606632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li C, Gao FP, Qin H, Chase R, Gor'kov PL, Brey WW, Cross TA. Uniformly aligned full-length membrane proteins in liquid–crystalline bilayers for structural characterization. J Am Chem Soc. 2007;129:5304–5305. doi: 10.1021/ja068402f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dürr UHN, Yamamoto K, Im SC, Waskell L, Ramamoorthy A. Solid-state NMR reveals structural and dynamical properties of a membrane-anchored electron-carrier protein, cytochrome b5. J Am Chem Soc. 2007;129:6670–6671. doi: 10.1021/ja069028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu CH, Ramamoorthy A, Opella SJ. High resolution heteronuclear dipolar solid-state NMR spectroscopy. (A).J Magn Reson. 1994;109:270–272. [Google Scholar]
- 58.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane proteins structure and topology. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J, Denny JK, Tian C, Kovacs FA, Song Z, Fu R, Quine JR, Cross TA. Imaging membrane protein helical wheels. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 60.Ramamoorthy A, Kandasamy SK, Lee DK, Kidambi S, Larson RG. Structure, topology, and tilt of cell-signaling peptides containing nuclear localization sequences in membrane bilayers determined by solid-state NMR and molecular dynamics simulation studies. Biochemistry. 2007;46:965–975. doi: 10.1021/bi061895g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu R, Truong M, Saager RJ, Cotten M, Cross TA. High resolution correlation spectroscopy in solid state NMR of aligned samples. J Magn Reson. doi: 10.1016/j.jmr.2007.06.004. in press. [DOI] [PubMed] [Google Scholar]
- 62.Hu J, Qin H, Li C, Sharma M, Cross TA, Gao FP. Structural biology of transmembrane domains: efficient production and characterization of transmembrane peptides by NMR. Protein Sci. 2007;16:2153–2165. doi: 10.1110/ps.072996707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franzin CM, Gong XM, Thai K, Yu J, Marassi FM. NMR of membrane proteins in micelles and bilayers: the FXYD family proteins. Methods. 2007;41:398–408. doi: 10.1016/j.ymeth.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franzin CM, Teriete P, Marassi FM. Structural similarity of a membrane protein in micelles and membranes. J Am Chem Soc. 2007;129:8078–8079. doi: 10.1021/ja0728371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Traaseth NJ, Buffy JJ, Zamoon J, Veglia G. Structural dynamics and topology of phospholambin in oriented lipid bilayers using multidimensional solid state NMR. Biochemistry. 2006;45:13827–13834. doi: 10.1021/bi0607610. [DOI] [PubMed] [Google Scholar]
- 66.Gor'kov PL, Chekmenev EY, Fu R, Hu J, Cross TA, Cotten M, Brey WW. A large volume flat coil probe for oriented membrane proteins. J Magn Reson. 2006;181:9–20. doi: 10.1016/j.jmr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 67.Gor'kov PL, Chekmenev EY, Li C, Cotten M, Buffy JJ, Traaseth NJ, Veglia G, Brey WW. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J Magn Reson. 2007;185:77–93. doi: 10.1016/j.jmr.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 68.Kovacs F, Cross TA. Transmembrane four-helix bundle of influenza A M2 protein channel: structural implications from helix tilt and orientation. Biophys J. 1997;73:2511–2117. doi: 10.1016/S0006-3495(97)78279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kovacs FA, Denny JK, Song Z, Quine JR, Cross TA. Helix tilt of the M2 transmembrane peptide from influenza A virus: an intrinsic property. J Mol Biol. 2000;295:117–125. doi: 10.1006/jmbi.1999.3322. [DOI] [PubMed] [Google Scholar]
- 70.Fung BM, Khitrin AK, Ermolaev K. An improved broadband decoupling sequence for liquid crystals and solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 71.Kim S, Cross TA. Uniformity, ideality and hydrogen bonds in transmembrane α-helices. Biophys J. 2002;83:2084–2095. doi: 10.1016/S0006-3495(02)73969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu J, Fu R, Cross TA. The chemical and dynamical influence of the antiviral drug amantadine on the M2 proton channel transmembrane domain. Biophys J. 2007;93:276–283. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quine JR, Achuthan S, Asbury T, Bertram R, Chapman MS, Hu J, Cross TA. Intensity and mosaic spread analysis from PISEMA tensors in solid state NMR. J Magn Reson. 2006;179:190–198. doi: 10.1016/j.jmr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 74.LoGrasso PV, Moll F, III, Cross TA. The solvent history dependence of gramicidin A conformations in hydrated lipid bilayers. Biophys J. 1988;54:259–267. doi: 10.1016/S0006-3495(88)82955-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Killian JA, Prasad KU, Hains D, Urry DW. The membrane as an environment of minimal interconversion. A circular dichroism study on the solvent dependence of the conformational behavior of gramicidin in diacylphos-phatidylcholine model membranes. Biochemistry. 1988;27:4848–4855. doi: 10.1021/bi00413a040. [DOI] [PubMed] [Google Scholar]
- 76.Bano MC, Braco L, Abad C. New high performance liquid chromatography-based methodology for monitoring the conformational transitions of self-associating hydrophobic peptides, incorporated into liposomes. J Chromatogr. 1988;458:105–116. doi: 10.1016/s0021-9673(00)90557-0. [DOI] [PubMed] [Google Scholar]
- 77.Bano MC, Braco L, Abad C. HPLC study on the ‘history’ dependence of gramicidin A conformation in phospholipid model membranes. FEBS Lett. 1989;250:67–71. doi: 10.1016/0014-5793(89)80686-6. [DOI] [PubMed] [Google Scholar]
- 78.Bouchard M, Auger M. Solvent history dependence of gramicidin-lipid interactions: a Raman and infrared spectroscopy study. Biophys J. 1993;65:2484–2492. doi: 10.1016/S0006-3495(93)81300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pinto LH, Lamb RA. Understanding the mechanism of action of the anti-influenza virus drug amantadine. TRENDS Microbiol. 1995;7:271. doi: 10.1016/s0966-842x(00)88942-8. [DOI] [PubMed] [Google Scholar]
- 80.Long SR, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]