

Abstract
The boronium-carbonium continuum was extended to include hypercoordinated protonated methanes and their boron analogs. The 11B NMR chemical shifts of the hypercoordinated hydriodo boron compounds and the 13C NMR chemical shifts of the corresponding isoelectronic and isostructural carbocations were calculated by using the GIAO-MP2 method. The data show good linear correlation between 11B and 13C NMR chemical shifts, which indicates that the same factors that determine the chemical shifts of the boron nuclei also govern the chemical shifts of carbon nuclei of these hypercoordinated hydriodo compounds.
A trivalent carbocation is isoelectronic with the corresponding neutral trivalent boron compounds. Olah et al. (1) in 1971 discussed the relationship between (CH3)3C+ and B(CH3)3 based on infrared and Raman spectroscopy. These studies provided unambiguous evidence that the two species possessed analogous structures and bondings. The close relationship between the 13C NMR chemical shifts of the carbons in carbocations and the corresponding 11B NMR chemical shifts of the boron atoms in isoelectronic boron compounds was first shown by Spielvogel and Purser (2, 3) and by Nöth and Wrackmeyer (4). The general correlation equation for trigonal species is shown below.‡
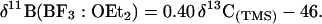 |
1 |
In Eq. 1, the δ13C is the chemical shift of the cationic carbon of the carbocation with respect to tetramethylsilane and the δ11B is the chemical shift of the corresponding boron atoms with respect to the BF3:OEt2. The empirically derived Eq. 1, however, is in good agreement with most of the available data that span some 600 ppm on the 13C NMR chemical shift scale. Recently, Prakash et al. (5) reported an extension of the relationship to cage compounds.
Williams et al. (6) derived a similar empirical Eq. 2 for the hypercoordinate (7) carbocations (carbonium ions) and their corresponding hypercoordinate boron compounds.
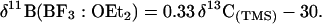 |
2 |
These relationships show that the same factors that determine the chemical shifts of the boron nuclei also govern the chemical shifts of carbon nuclei. We now extend this boronium-carbonium continuum to hypercoordinated hydriodo boron compounds and their isoelectronic and isostructural carbocations, hitherto not yet observed under long-lived superacidic stable ion conditions, based on GIAO-MP2 calculations.
RESULTS AND DISCUSSION
The structures of the hypercoordinate protonated methanes and their boron analogs have been optimized at the MP2/6–31G** level by using the gaussian-94 (8) package of programs. 13C and 11B NMR chemical shifts were calculated by the correlated GIAO-MP2 method (9, 10) (tzp/dz basis set; refs. 9 and 10) using the aces ii programs (11). 13C NMR chemical shifts are referenced to tetramethylsilane, and 11B NMR chemical shifts are referenced to BF3:OEt2.
CH5+ and BH5.
CH5+ (protonated methane) is considered the parent of nonclassical carbocations containing a five-coordinate carbon atom. The Cs symmetrical structure 1a with a three-center, two-electron (3c-2e) bond is the preferred ground-state structure for the CH5+ (12–16). The isoelectronic boron analog of CH5+ is neutral BH5 and is also Cs symmetrical 1b based on high-level ab initio calculations (17, 18). ![]()
The 13C and 11B NMR chemical shifts of 1a and 1b were calculated by using the GIAO-MP2 method (9, 10) (Table 1). Calculation of the 13C and 11B NMR chemical shifts by the correlated GIAO-MP2 method was shown to reproduce well experimental values for carbocations and boron compounds, respectively (19, 20). The calculated δ13C value of 1a is −11.5, 7.1 ppm more shielded than that of CH4 (δ13C = −4.4) at the same level of calculations. The experimental δ13C of methane is −2.3. The shielding effect in 1a, however, is expected for such a hypercoordinate (nonclassical) carbocation (6, 7). Evidence for CH5+ in the condensed phase came from hydrogen-deuterium exchange on methane in deuteriated superacids (21, 22) and ESCA studies (G.A.O., unpublished results). Direct NMR spectroscopic observation of the ion was not yet achieved and is difficult because of its expected low concentration and fast exchange in superacids.
Table 1.
GIAO-MP2/tzp/dz calculated and estimated NMR chemical shifts*
| No. | a, δ13C (GIAO-MP2) | b, δ11B (GIAO-MP2) | b, δ11B (estimated)† |
|---|---|---|---|
| 1 | −11.5 | −28.8 | −33.8 |
| 2 | −25.7 | −32.4 | −38.5 |
| 3 | −13.0 | −35.6 | −34.3 |
| 4 | 332.9 | 62.4 | 79.8 |
Calculated 13C and 11B NMR chemical shifts were referenced to tetramethylsilane and BF3:OEt2, respectively.
Estimated by using Eq. 2.
The calculated δ11B of 1b is −28.8. We also estimated the δ11B values in 1b by applying Eq. 2 and by using the δ13C value of 1a. This yielded a δ11B value of −33.8 for the neutral boron compound 1b. The boron chemical shift of −33.8 is reasonably close to the GIAO-MP2 calculated value of −28.8 and thus reconfirms the close relationship between the 11B and 13C chemical shifts of isoelectronic carbon and boron analogs 1a and 1b, respectively.
CH62+ and BH6+.
The calculated parent six-coordinate carbocation, diprotonated methane (CH62+), has two two-electron, three-center (2e-3c) bonding interactions in its minimum-energy structure 2a (C2v) (23, 24). The calculated δ13C value of the dication 2a is −25.7, 14.2 ppm more shielded than that of five-coordinate monocation CH5+ 1a. ![]()
The structure and energetics of the parent hexacoordinate boronium ion BH6+ 2b, isoelectronic with CH62+ 2a, recently have been studied (25). The C2v symmetric form 2b with two three-center, two-electron (3c-2e) bonds was found to be a stable minimum for BH6+. Structure 2b is isostructural with CH62+ 2a. Recently, DePuy et al. (26) were able to generate the BH6+ in the gas phase. We calculated δ11B of 2b as −32.4, 3.6 ppm more shielded than that of five-coordinate neutral BH5 1b. The estimated δ11B of 2b obtained from applying Eq. 2 and using a δ13C value of −25.7 for 2a is −38.5 ppm, close to that of the GIAO-MP2 calculated value of −32.4 ppm.
CH73+ and BH72+.
The structures of parent heptacoordinate carbocation (27), CH73+, and its isoelectronic boron analog (26), BH72+, recently have been reported. The C3v symmetric forms 3a and 3b were found to be the stable minima for CH73+ and BH72+, respectively. The 13C and 11B NMR chemical shifts of 3a and 3b were calculated. The calculated δ13C of 3a is −13.0, and the δ11B value of 3b is −35.6. δ11B for 3b, estimated by the application of Eq. 2 and by using the δ13C value in 3a was assessed to be −34.3 ppm, which shielded by only 1.3 ppm more than the GIAO-MP2 calculated value of −35.6 ppm. ![]()
CH42+ and BH4+.
The planar C2v symmetrical structures containing a three-center, two-electron (3c-2e) bond are preferred for both CH42+ 4a (28) and BH4+ 4b (25). The calculated δ13C value of 4a is 332.9. The estimated δ11B value in 4b obtained by applying Eq. 2 is 79.8, differing by 17.4 ppm from the GIAO-MP2 values of 62.4. ![]()
Correlation of 13C and 11B NMR Chemical Shifts.
The calculated 13C NMR chemical shifts of studied methonium ions, in general, correlate well with the calculated 11B NMR chemical shifts of the corresponding isoelectronic boron analogs (Fig. 1). This correlation indicates that these small hypercoordinate methonium ions and their boron analogs also follow Eq. 2, which was derived for their nonclassical systems (6). The correlation line derived from Fig. 1 corresponds closely to the correlation line of Eq. 2.
Figure 1.

Calculated 13C NMR chemical shifts vs. calculated 11B NMR chemical shifts of structures 1–4.
In conclusion, we have extended the 11B–13C NMR chemical shift relationship of boronium-carbonium ions to include the parent hypercoordinated methonium ions and their boron analogs. By using the correlated GIAO-MP2 method the 11B NMR chemical shifts of the hydriodo boron compounds and the 13C NMR chemical shifts of their corresponding isoelectronic and isostructural carbonium ions were calculated. The data showed good linear correlation between 11B and 13C NMR chemical shifts. This finding indicates that these small parent carbonium and boronium ions also follow Eq. 2, derived for the nonclassical systems (6). It seems that the same factors that determine the chemical shifts of the boron nuclei also govern the chemical shifts of the carbon nuclei within the hypercoordinated hydriodo compounds.
Acknowledgments
Support of our work by the National Science Foundation is gratefully acknowledged.
Footnotes
This is paper 40 in the series “Chemistry in Superacids.” Paper no. 39 is ref. 29.
References
- 1.Olah G A, Demember J R, Commeyras A, Bribes J L. J Am Chem Soc. 1971;93:459–463. [Google Scholar]
- 2.Spielvogel B F, Purser J M. J Am Chem Soc. 1971;93:4418–4426. [Google Scholar]
- 3.Spielvogel B F, Nutt W R, Izydore R A. J Am Chem Soc. 1975;97:1609–1610. [Google Scholar]
- 4.Nöth H, Wrackmeyer B. Chem Ber. 1974;107:3089–3092. [Google Scholar]
- 5.Prakash G K S, Rasul G, Yudin A K, Williams R E. In: Borane, Carborane, Carbocation Continuum. Casanova J, editor. New York: Wiley Interscience; 1998. , Ch. 8, pp. 147–167. [Google Scholar]
- 6.Williams R E, Prakash G K S, Field L D, Olah G A. In: Advances in Boron and Boranes. Liebman J F, Greenberg A, Williams R E, editors. New York: VCH; 1988. pp. 191–224. [Google Scholar]
- 7.Olah G A, Prakash G K S, Williams R E, Field L D, Wade K. Hypercarbon Chemistry. New York: Wiley; 1987. pp. 191–213. [Google Scholar]
- 8.Frisch M J, Trucks G W, Schlegel H B, Gill P M W, Johnson B G, Robb M A, Cheeseman J R, Keith T A, Peterson G A, Montgomery J A, et al. gaussian 94. Pittsburgh, PA: Gaussian; 1995. , Revision A.1. [Google Scholar]
- 9.Gauss J. J Chem Phys Lett. 1992;191:614–620. [Google Scholar]
- 10.Gauss J. J Chem Phys. 1993;99:3629–3643. [Google Scholar]
- 11.Stanton J F, Gauss J, Watts J D, Lauderdale W, Bartlett R J. aces II: An Ab Initio Program System. Gainesville: Univ. of Florida; 1991. [Google Scholar]
- 12.Olah G A, Rasul G. Acc Chem Res. 1997;30:245–250. [Google Scholar]
- 13.Marx D, Parrinello M. Nature (London) 1995;375:216–218. [Google Scholar]
- 14.Schreiner P R, Kim S-J, Schaefer H F, Schleyer P v R. J Chem Phys. 1993;99:3716–3720. [Google Scholar]
- 15.Scuseria G E. Nature (London) 1993;366:512–513. [Google Scholar]
- 16.Müller H, Kützelnigg W. J Chem Phys. 1997;106:1863–1869. [Google Scholar]
- 17.Schreiner P R, Schaefer H F, Schleyer P v R. J Chem Phys. 1994;101:7625–7632. [Google Scholar]
- 18.Watts J D, Bartlett R J. J Am Chem Soc. 1995;117:825–826. [Google Scholar]
- 19.Schleyer P v R, Maerker C, Buzek P, Sieber S. In: Stable Carbocation Chemistry. Prakash G K S, Schleyer P v R, editors. New York: Wiley; 1997. pp. 19–74. and references therein. [Google Scholar]
- 20.Bühl M, Gauss J, Hoffmann M, Schleyer P v R. J Am Chem Soc. 1993;115:12385–12390. [Google Scholar]
- 21.Olah G A, Klopman G, Schlosberg R H. J Am Chem Soc. 1969;91:3261–3268. [Google Scholar]
- 22.Olah G A, Schlosberg R H. J Am Chem Soc. 1968;90:2726–2727. [Google Scholar]
- 23.Lammertsma K, Olah G A, Barzaghi M, Simonetta M. J Am Chem Soc. 1982;104:6851–6852. [Google Scholar]
- 24.Lammertsma K, Barzaghi M, Olah G A, Pople J A, Schleyer P v R, Simonetta M. J Am Chem Soc. 1983;105:5258–5263. [Google Scholar]
- 25.Rasul G A, Olah G A. Inorg Chem. 1997;36:1278–1281. doi: 10.1021/ic960843k. [DOI] [PubMed] [Google Scholar]
- 26.DePuy C H, Gareyev R, Hankin J, Davico G E. J Am Chem Soc. 1997;119:427–428. [Google Scholar]
- 27.Olah G A, Rasul G. J Am Chem Soc. 1996;118:8503–8504. [Google Scholar]
- 28.Wong M W, Radom L. J Am Chem Soc. 1989;111:1155–1156. [Google Scholar]
- 29.Olah, G. A., Orlinkov, A., Oxyzoglou, A. B. & Prakash, G. K. S. (1998) Russ J. Org. Chem., in press.