

Abstract
A very potent and specific inhibitor of mitochondrial NADH:ubiquinone oxidoreductase (complex I), a derivative of NADH (NADH-OH) has recently been discovered (Kotlyar, A. B., Karliner, J. S., and Cecchini, G. (2005) FEBS Lett. 579, 4861–4866). Here we present a quantitative analysis of the interaction of NADH-OH and other nucleotides with oxidized and reduced complex I in tightly coupled submitochondrial particles. Both the rate of the NADH-OH binding and its affinity to complex I are strongly decreased in the presence of succinate. The effect of succinate is completely reversed by rotenone, antimycin A, and uncoupler. The relative affinity of ADP-ribose, a competitive inhibitor of NADH oxidation, is also shown to be significantly affected by enzyme reduction (KD of 30 and 500 μM for oxidized and the succinate-reduced enzyme, respectively). Binding of NADH-OH is shown to abolish the succinate-supported superoxide generation by complex I. Gradual inhibition of the rotenone-sensitive uncoupled NADH oxidase and the reverse electron transfer activities by NADH-OH yield the same final titration point (~0.1 nmol/mg of protein). The titration of NADH oxidase appears as a straight line, whereas the titration of the reverse reaction appears as a convex curve. Possible models to explain the different titration patterns for the forward and reverse reactions are briefly discussed.
The mitochondrial proton-translocating NADH:ubiquinone oxidoreductase (complex I) and its prokaryotic homologues (NDH-1)1 catalyze oxidation of NADH coupled to reduction of the membrane-located quinone. In aerobic organisms the enzymes provide at least two vitally important functions. First, they reoxidize NADH produced by multiple dehydrogenases, thus maintaining and controlling general oxidative metabolism. Second, they also serve as the energy-transducing devices accumulating free energy from the redox gap between NADH/NAD+ and the quinol/quinone pair as the proton-motive force (pmf) across the coupling membranes. In view of the structural complexity of both prokaryotic and particularly eukaryotic enzymes it seems likely that a number of other physiologically important functions of NDH-1 (13–15 different subunits) (1–3) and complex I (45 subunits) (4) remain to be discovered.
There have been a number of speculative models proposed for both the mechanism and structure of complex I. These models have relied on a variety of biochemical and biophysical approaches such as studies on catalytic activity, low-temperature EPR, and use of specific inhibitors and mutagenesis. Different aspects of those studies have been extensively reviewed (5–14). Recently, a breakthrough in the field was achieved when the 3.3 Å structure of the eight-subunit hydrophilic domain (peripheral arm) of Thermus thermophilus NDH-1 was solved (15–17). All of the known redox components except for bound quinone were visualized in the structure. This structure shows an intramolecular electron-transfer chain approximately 90 Å long. This chain begins at FMN (presumably the primary electron acceptor for NADH) and proceeds via a series of seven iron–sulfur clusters termed N-3 → N-1b → N-4 → N5 → N-6a → N-6b → N-2 all located at a distance of < 14 Å, close enough for rapid electron transfer (15, 16).
Although the partial resolution of the T. thermophilus NDH-1 structure is quite informative, a number of important mechanistic questions regarding the substrate(s)/product(s) binding and the energy coupling mechanism remain to be answered. For mechanistic models of the nucleotide substrate (product) interaction with the enzyme active site(s), only limited steady-state kinetic (18–24) and recent electrochemical (25) data are available. A very potent and specific inhibitor of the mitochondrial complex I, a derivative of NADH (tentatively identified as β-2,6-dihydroxydihydronicotinamide adenine dinucleotide), has been recently discovered (26). A selective competitive high-affinity inhibitor acting on the substrate side of the flavin is potentially a very useful tool for studies on the initial steps of NADH oxidation. In this paper we report quantitative parameters for the inhibitor and other nucleotide interactions with complex I in tightly coupled submitochondrial particles. The central finding reported here is that the nucleotide binding site(s) dramatically change the affinity to the substrate/product and to their redox-inactive analogues upon enzyme oxidoreduction.
MATERIALS AND METHODS
Bovine heart submitochondrial particles (SMPs) were prepared as described (27), and their NADH oxidase was activated by aerobic incubation with NADPH (28). After activation the SMPs were precipitated by centrifugation, suspended in a mixture comprised of 0.25 M sucrose, 50 mM Tris–HCl, 0.1 mM potassium malonate, and 0.2 mM EDTA (pH 8.0), and stored in liquid nitrogen.
NADH oxidase was assayed photometrically (ε340 = 6.22 mM−1 cm−1) in a standard mixture comprised of 0.1 mM NADH, 0.25 M sucrose, 50 mM Tris–HCl, 0.2 mM EDTA, and 0.05 μg/mL gramicidin D (pH 8.0). NADH:hexaammineruthenium(III) (HAR) reductase (21) was measured in the same mixture supplemented with 5 μM rotenone and 0.5 mM HAR. The reverse electron transfer (aerobic succinate-supported NAD+ reduction) was assayed in the standard reaction mixture (gramicidin D was excluded) supplemented with 2.5 mM potassium succinate and 1 mM NAD+. Superoxide generation was measured as the rate of super-oxide dismutase-sensitive acetylated cytochrome c reduction in a mixture containing 0.25 M sucrose, 50 mM Tris–HCl, 0.1 M potassium phosphate, and 0.2 mM EDTA (pH 8.0) supplemented with either 50 μM NADH or 5 mM potassium succinate (29).
All enzymatic assays were conducted at 25 °C. The experimental details are described in the captions to the figures and footnotes to the tables.
NADH-OH was prepared essentially as described (26), and its 60 μM solution in distilled water was aliquoted into small vials and stored in liquid nitrogen until use. The concentration of NADH-OH was determined photometrically (ε335 = 25 mM−1 cm−1) at pH 12.0 (26). All fine chemicals were from Sigma.
The protein content was determined by the buret assay using bovine serum albumin as the standard.
RESULTS
Previous studies showed that NADH-OH is a tight-binding inhibitor of complex I with a Ki of about 10−8 M estimated from the steady-state kinetics (26). During the catalytic cycle, the enzyme is reduced by NADH and oxidized by ubiquinone, thus existing in reduced and oxidized states. The ratio between these states depends on the particular kinetic mechanism the enzyme undergoes (i.e., random, ordered, ping-pong) and on the rate constants for the individual steps. Under ordinary steady-state assay conditions both binding of low concentrations of a tight inhibitor and its dissociation from the active site are slow. This creates problems in determining kinetic constants as has been discussed in a general form (30) and more specifically for complex II (succinate:quinone oxidoreductase) interaction with oxalo-acetate (31). On the other hand, tight specific binding of an inhibitor provides a unique opportunity to obtain quantitative parameters for enzyme–ligand interaction using simple conventional steady-state kinetic approaches as described below. To follow the rate of the enzyme–inhibitor interaction, the following conditions should be fulfilled: (i) the concentrations of both enzyme and inhibitor need to be adjusted so that the manipulations can be followed in a reasonable time frame; (ii) the concentration of the inhibitor should be significantly higher than Ki to analyze the process as an irreversible reaction; (iii) the concentration of the inhibitor should be significantly higher than that of the enzyme, so that inhibition will proceed as the first-order time course. After a number of pilot experiments the following conditions were used to fulfill the above criteria. The enzyme concentration used was approximately 0.5 nM (based on the complex I content in SMPs), and the lowest inhibitor concentration used was 6 nM. This ratio of inhibitor to enzyme resulted in more than 90% inhibition of the oxidized enzyme at infinite time (equilibrium). The highest inhibitor concentration used was 24 nM, which provided practically complete inhibition of the oxidized enzyme and about 80% inhibition of the reduced enzyme at infinite time (see the equilibrium constants in Table 2, below). It should also be pointed out that to keep complex I in the oxidized or reduced state in the absence of turnover the following conditions were applied. The oxidized form of the enzyme was maintained by incubating the enzyme in the absence of nucleotide substrates. The reduced form of complex I was achieved by the pmf-dependent succinate-supported reduction in the absence of NAD+, a condition which also prevents turnover of the reduced enzyme.
Table 2.
Quantitative Parameters of Complex I–Nucleotide Interactions (pH 8.0, 25 °C)
|
KD, M
|
||||
|---|---|---|---|---|
| NADH-OHa | ADP-riboseb | NAD+c | NADHd | |
| Eox | 3 × 10−10 | 2.5 × 10−5 | 8 × 10−4 | |
| Eredf | 7 × 10−9 | 4 × 10−4 | 2 × 10−5 | |
Determined as depicted in Figure 4.
Determined from the data on protection of the enzyme by NADH against “irreversible” inhibition by NADH-OH as depicted in Figure 4.
SMPs (5 μg/mL) were incubated in the standard reaction mixture containing 2.5 mM potassium succinate and different concentrations of NADH (0–120 μM). NADH-OH (28 nM) was added, and after 1 min of incubation the residual NADH oxidase was measured following the rate of NADH oxidation (final concentration of 120 μM) in the presence of 2.5 mM potassium malonate and gramicidin D (0.05 μg/mL). The decrease of NADH concentration during aerobic incubation in the presence of succinate did not exceed 1 μM. The KD value was determined from the secondary plot as depicted in Figure 4.
Ered refers to complex I reduced by succinate (energy-dependent reverse electron transfer). It should be noted that although the iron–sulfur centers N-2 and N-3 have been shown to be reduced almost completely under these conditions (28, 38), the degree of FMN reduction is not known. It seems likely that complete reduction of FMN may cause even a stronger change of NADH-OH and ADP-ribose affinities to the enzyme than those reported here.
Figure 1A shows the time course of inactivation of NADH oxidase activity by NADH-OH. As expected, a 12-fold (or more) excess of NADH-OH over the enzyme concentration caused slow inactivation of complex I following the pseudo-first-order time dependence (Figure 1B) and the first-order dependence on the inhibitor concentration (Figure 1B, inset). When complex I was reduced by succinate under energized conditions (pmf-dependent reverse electron transfer), the second-order rate constant for the enzyme inactivation decreased approximately 10-fold. The significant decrease in reactivity of NADH-OH toward its binding site in the presence of succinate could result from reduction of complex I and/or from a conformational change induced by membrane energization. As depicted in Table 1, rotenone, a potent inhibitor of reverse electron transfer, antimycin A, which inhibits succinate oxidation, and an uncoupler (gramicidin D) all brought the reactivity of NADH-OH back to the level observed for oxidized complex I.
Figure 1.

Inhibition of the NADH oxidase activity by NADH-OH. (A) SMPs (5 μg of protein/mL, about 0.5 nM complex I) were incubated with 6 (■), 12 (○), and 24 (●, □) nM NADH-OH for the time indicated on the abscissa in the standard reaction mixture (0.25 M sucrose, 50 mM Tris/Cl−, 0.2 mM EDTA, pH 8.0). The residual NADH oxidase activity was initiated by the addition of 0.1 mM NADH and gramicidin D (0.05 μg/mL). Potassium succinate (2.5 mM) was present during incubation with NADH-OH in the sample marked as □ (reduced complex I), and 2.5 mM potassium malonate was present in the NADH oxidase assay. The continuous lines are the computer-generated curves for the simple bimolecular irreversible enzyme–inhibitor interaction with the second-order rate constant of 1.6 × 108 (●, ○, ■) and 1.5 × 107 (□, reduced complex I) M−1 min−1. (B) Pseudo-first-order rate logarithmic anamorphosis of the data shown in (A). The inset shows the linear dependence of the apparent first-order rate constant on the concentration of NADH-OH.
Table 1.
Effect of Succinate on the Interaction of Complex I with NADH-OH
Next, the rate constants for dissociation of the complex I–NADH-OH complex were determined. Because of extremely high affinity of NADH-OH to the enzyme (see below), it was virtually impossible to activate the inhibited oxidized complex I by dilution, and thus, a system was designed for irreversible decomposition of free inhibitor as shown in Figure 2. It has been shown previously that NADH-OH rapidly reacts with the oxidants ferricyanide and 2,6-dichlorophenolindophenol (26). Figure 2 shows that this is also true for HAR, the most efficient electron acceptor for complex I (21, 22). Brief preincubation of NADH-OH with 0.5 mM HAR (the concentration used for the assay of the NADH dehydrogenase activity of complex I) completely abolished the inhibitory effect (Figure 2, curves 1 and 2). On the other hand, no activity was seen if the enzyme–inhibitor complex was incubated in the presence of HAR or HAR plus NADH (curves 3 and 4). The results obtained show that free NADH-OH is rapidly destroyed by HAR whereas the enzyme-bound inhibitor is not sensitive to the oxidant. These findings made it possible to determine the first-order rate constant for dissociation of the oxidized enzyme–inhibitor rate complex as shown in Figure 3. The dilution of the mixture in the absence of HAR would result in almost complete dissociation of the enzyme–inhibitor complex only if its concentration is much less than the value of KD (0.3 nM for oxidized enzyme; see Table 2). The use of such a low enzyme concentration makes accurate measurement of the rate of NADH oxidation quite challenging. However, addition of HAR to the assay makes the inhibitor dissociation irreversible and enables one to measure the kinetics of the inhibitor dissociation at much higher concentration of the enzyme than that in the absence of HAR. The affinity of the reduced enzyme is significantly lower (KD = 7 nM, Table 2) than that of the oxidized form, and dilution of the complex is sufficient to completely remove the inhibitor from the reduced enzyme. Thus, the rate of dissociation was followed after simple dilution of the enzyme–inhibitor complex in the medium containing succinate (to reduce the enzyme) and NADH (to occupy the binding site after dissociation of NADH-OH). As is seen in Figure 3, the presence of succinate (reduction of complex I) affected (increased) the dissociation rate, koff, although not as dramatically as the kon value.
Figure 2.
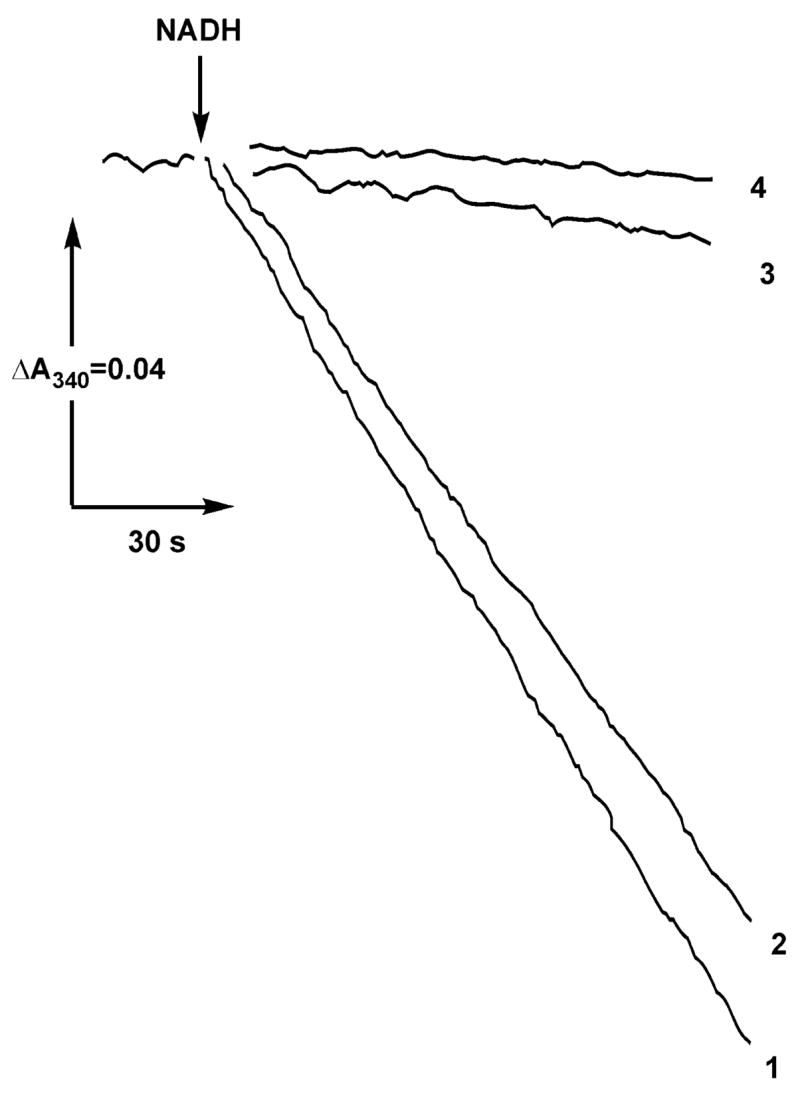
Rapid irreversible decomposition of NADH-OH by HAR and the resistance of the enzyme-bound inhibitor to the electron acceptor. Actual tracings of the NADH:HAR reductase activities after preincubation with NADH-OH and HAR are shown. (Curve 1) SMPs (25 μg/mL) were incubated for 1 min in the standard reaction mixture containing 0.5 mM HAR, and the reaction was initiated by the addition of 100 μM NADH and 5 μM rotenone as indicated. (Curve 2) NADH-OH (28 nM) was preincubated in the standard mixture containing 0.5 mM HAR for 1 min, 25 μg/mL of SMPs were then added following 1 min of incubation, and the reaction was initiated by the addition of NADH and rotenone. (Curve 3) SMPs (25 μg/mL) were incubated for 1 min with 28 nM NADH-OH, 0.5 mM HAR was added following 1 min of incubation, and the reaction was initiated by the addition of NADH and rotenone. (Curve 4) Same as curve 3. The reaction was initiated by simultaneous addition of NADH, HAR, and rotenone.
Figure 3.

Dissociation rates of the enzyme–inhibitor complex. (A) (Lines 1 and 2 (controls, ●, ■)) SMPs (25 μg/mL) were incubated in the standard reaction mixture containing either 0.5 mM HAR (■) or 5 mM succinate and 150 μM NADH (●) for the time indicated on the abscissa, and NADH oxidation was initiated by the addition of 100 μM NADH and 5 μM rotenone (■) or 5 mM potassium malonate and gramicidin D (0.05 μg/mL) (●). (Curve 3 (○)) SMPs (1 mg/mL) preincubated with 100 nM NADH-OH for 15 min at room temperature were added at zero time to the reaction mixture (final concentration of 25 μg/mL) and incubated and assayed as described for line 1 (●). (Curve 4 (□)) SMPs (1 mg/mL) preincubated with 100 nM NADH-OH for 15 min at room temperature were added at zero time to the reaction mixture (final concentration of 25 μg/mL) and incubated and assayed as described for line 2 (■). (B) Logarithmic anamorphosis of curves 3 and 4. The activities shown in (A, lines 1 and 2) were taken as v∞ to construct lines 3 and 4, corresponding to the first-order rate constants of 0.05 min−1 (oxidized enzyme) and 0.09 min−1 (succinate-reduced enzyme). Activity of 100% in (A) corresponds to the specific activities of 0.8 (μmol/min)/mg (●) and 2.4 (μmol/min)/mg (■).
The results presented above showed that the affinity of the active site-directed inhibitor is redox dependent. It was of obvious interest to find out whether the binding affinities for the natural nucleotide substrates NADH and NAD+ and the competitive inhibitor of NADH oxidation ADP-ribose (32) are also redox dependent. In contrast to NADH-OH the nucleotide substrates NAD+ and NADH and ADP-ribose have a relatively low affinity to the active site and are rapidly (in milliseconds or faster) equilibrated with the enzyme. The approach used above for estimation of NADH-OH affinity to the enzyme thus cannot be applied to the interaction of the rapidly equilibrating ligands. To estimate the affinity of these compounds, the following approach was used. Consider the system where a high-affinity inhibitor (NADH-OH (I), (KD determined as the koff/kon ratio is equal to 3 × 10−10 and 7 × 10−9 M for the oxidized (Eox) and reduced (Ered) enzyme, respectively; see Table 2) slowly and completely inhibits complex I in the pseudo-first-order reaction (see Figure 1) in the presence of a rapidly equilibrating competitive ligand (L), e.g., ADP-ribose (32):
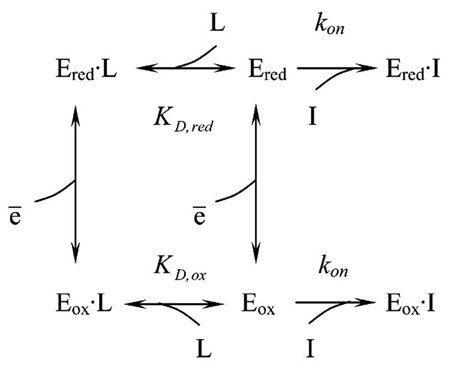
The apparent first-order rate constant (kobs) determined from the time-dependent loss of enzyme activity as shown in Figure 1 for the oxidized enzyme (in the absence of succinate, lower part of eq 1, is
| (2) |
Rearrangement of eq 2 gives
| (3) |
and KD,ox can thus be determined from the linear graph of 1/kobs versus L as the intercept on the abscissa (−KD,ox). The same dependence of 1/kobs on the ligand concentration holds for the upper part of eq 1 in the presence of reductant (Ered, in the presence of succinate).
Figure 4 depicts an example of the experimental data on the protective effect of a ligand (ADP-ribose) against inactivation of oxidized complex I by NADH-OH fitted to eqs 2 and 3 in panels A and B, respectively.
Figure 4.

Effect of ADP-ribose on irreversible inhibition of complex I by NADH-OH. (A) SMPs (25 μg/mL) were incubated for 1 min in the standard reaction mixture containing 28 nM NADH-OH and ADP-ribose (concentrations are indicated on the abscissa). The residual NADH oxidase activities (100 μM NADH and 0.05 μg/mL gramicidin D) were determined. The pseudo-first-order rate constants for NADH-OH-induced inhibition were calculated as kobs = ln(v0/v t) and plotted as a function of ADP-ribose concentration. v0 corresponds to the rate of NADH oxidation in the absence of NADH-OH, and vt is the residual activity after 1 min of incubation with NADH-OH. (B) Secondary plot of 1/kobs versus ADP-ribose concentration used to determine KD for ADP-ribose.
The KD,ox value determined by this approach (25 μM) corresponds well with Ki for competitive inhibition of NADH oxidation by ADP-ribose (26 μM) derived from conventional steady-state kinetics (32). Using this approach, the affinity of NAD+ to oxidized enzyme and that of NADH to the succinate-reduced enzyme were determined (Table 2). Obviously, the binding affinity of NAD+ to reduced complex I and NADH to the oxidized enzyme cannot be determined using this approach because of oxidation and reduction of the enzyme caused by NAD+ and NADH, respectively. The data summarized in Table 2 show that the affinity of the redox-inactive ADP-ribose is strongly affected by the reduction of some enzyme component (most likely FMN). In other words, the binding of the nucleotide per se results in a decrease of the midpoint redox potential of the enzyme-bound FMN.
Next we measured the effect of NADH-OH on the “side” reactions catalyzed by complex I, i.e., NADH- and succinate-supported superoxide generation (Figure 5). In view of the data reported above, it could be expected that a decrease of the FMN redox potential upon NADH-OH binding would increase autoxidation of the flavin, at least in the succinate-supported reaction. An opposite effect was evident: NADH-OH strongly inhibited both NADH- and succinate-supported superoxide generation.
Figure 5.
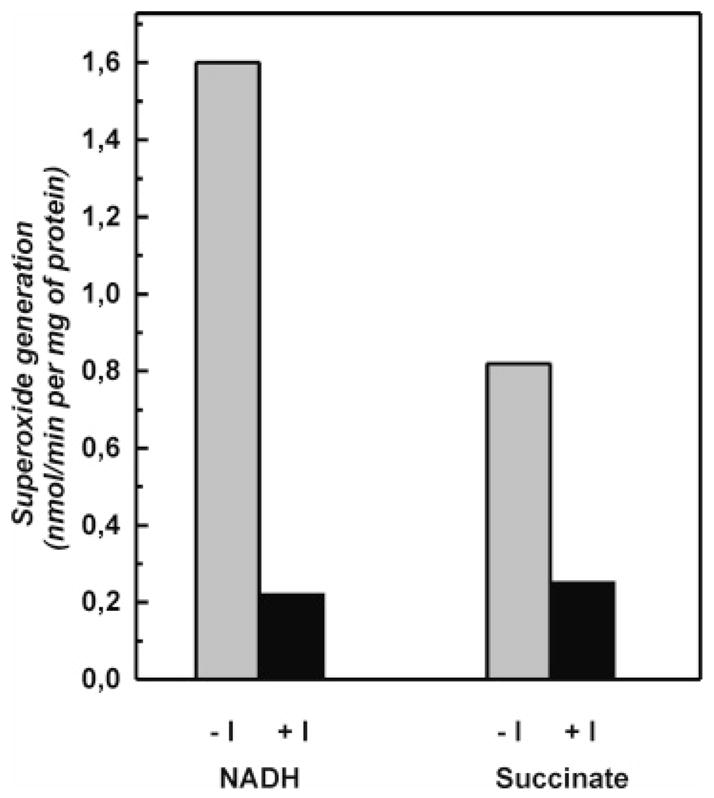
Inhibition of the complex I-mediated superoxide generation by tightly coupled submitochondrial particles. Superoxide generation (superoxide dismutase-sensitive acetylated cytochrome c reduction) was measured as described in the Materials and Methods. SMPs (1 mg/mL) were preincubated in the standard mixture for 15 min with or without 100 nM NADH-OH (I), and the rates of succinate-supported or NADH (50 μM)-supported superoxide (in the presence of 5 μM rotenone) generation were measured in the standard assay mixture containing 0.1 M potassium phosphate (pH 8.0) and 20 μM acetylated cytochrome c.
An extremely high affinity of NADH-OH to complex I (KD of 3 × 10−10 M for the oxidized enzyme) provides a unique possibility to titrate the contents of the complex I active site(s) in any particular enzyme preparation. Figure 6 shows that, under conditions where the enzyme concentration is significantly larger than KD, the titration of NADH oxidase by NADH-OH, as expected (30, 33), appeared as a straight line. Because the precise chemical structure of NADH-OH is not definitely established and the inhibitor is not very stable, only an approximate content for the nucleotide binding active site(s) of complex I (0.09 nmol/mg of protein) can be derived from the titration. This value is, however, comparable with that of the piericidin titer (0.1 nmol/mg of protein) determined with the same SMP preparation (33).
Figure 6.
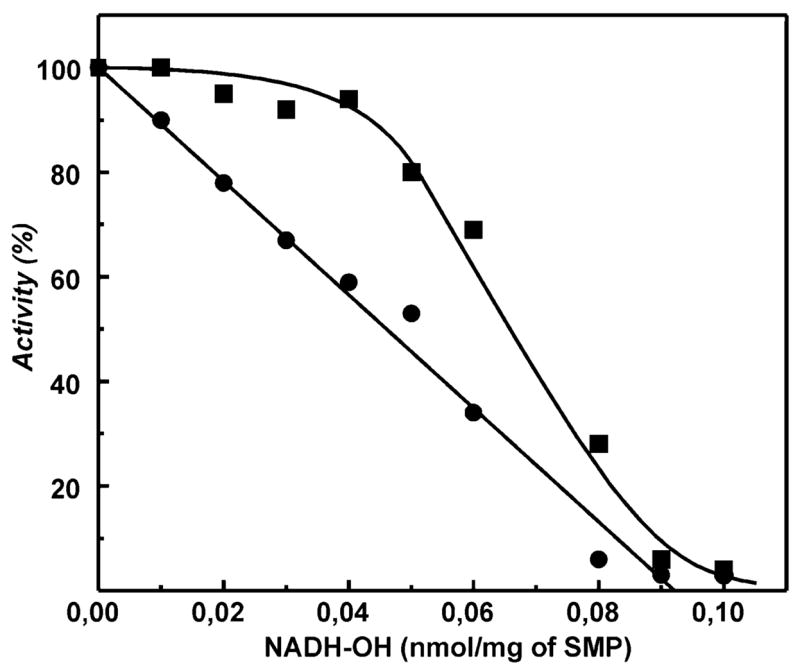
Titration curves for inhibition of uncoupled NADH oxidation and succinate-supported aerobic reverse electron transfer by NADH-OH. SMPs were preincubated in the standard reaction mixture with the indicated concentrations of NADH-OH for 15 min. The residual NADH oxidase (●, 100 μM NADH and 0.05 μg/mL gramicidin D) and reverse electron transfer (■, 1 mM NAD+ and 5 mM potassium succinate) activities were determined. The final concentration of SMPs in the assay mixture was 25 μg/mL. The SMP content in the preincubation mixture was 2 mg/mL. An activity of 100% corresponds to the specific activity (initial rate) of 0.8 and 0.1 (μmol/min)/mg of protein for NADH oxidase and reverse electron transfer, respectively. Longer preincubation of SMPs with NADH-OH did not change the titration curves.
When the initial rates of the succinate-supported reverse electron transfer activity were determined in the same sample, a convex (parabolic) titration curve was evident (Figure 6). This indicates that within some low concentration range the reverse electron transfer is much less sensitive to inhibition than the NADH oxidase activity. These results were corroborated by experiments on direct observation of the steady-state level of NADH/NAD+ reached during coupled succinate oxidation. The addition of small concentrations of NADH-OH to the system where the steady-state level of NAD+ reduction has been reached caused an increase of the NADH concentration (not shown), similar to the effects of other specific inhibitors of complex I, rotenone (34, 35) and ADP-ribose (32), previously reported.
DISCUSSION
Using a new highly specific inhibitor of complex I, we were able to demonstrate for the first time that the binding affinity of nucleotides, catalytically inactive NADH analogues, to the active site is significantly (at least 10-fold) decreased upon reduction of the enzyme (Table 2). Because FMN almost certainly serves as the primary electron acceptor for enzyme-bound NADH, it seems likely that oxidoreduction of the flavin is coupled with a conformational change of the protein. This proposal is supported by studies showing that conformational changes of complex I occur upon nucleotide binding (36) and that the three-subunit FP fragment of complex I more readily loses its flavin upon reduction of the enzyme (37). The use of ADP-ribose, a competitive inhibitor of NADH oxidation (32), was particularly beneficial for demonstration of the redox-dependent binding change since this nucleotide analogue contains no redox-active component (nicotinamide). The Nqo1 subunit of T. thermophilus is highly homologous with the mammalian complex I 51 kDa subunit. If the X-ray structure of Nqo1 is used as a model (16), the solvent-accessible positively charged area at the entrance to the FMN binding pocket is a region which may undergo this redox-dependent rearrangement. It should be noted that the actual redox dependency of the affinity during NADH oxidation turnover may be significantly larger than that experimentally observed in this study (10-fold difference). The FMN reduction level reached during pmf-dependent reverse electron transfer may be lower than that during coupled oxidation of NADH at state 4. Indeed, the rate of the succinate-supported superoxide production, which is presumably directly proportional to FMN reduction, plotted as a function of the respiratory control ratio for different preparations of SMPs, appears as a straight line and never reaches saturation (our unpublished observation). A redox-dependent change of the protein structure is of great interest for elucidation of complex I-catalyzed proton translocation. The H+/e stoichiometry determined for mammalian complex I (38–40) and the Paracoccus denitrificans enzyme (41) is equal to 4, a value which cannot be accounted for by a single proton-translocating loop mechanism (42). One coupling “subsite” is likely associated with ubiquinone reduction by the iron–sulfur center N-2 in the hydrophobic membranous phase, and different mechanisms for this coupling subsite have been proposed (43–45). Since the FMN is located almost 90 Å from the membrane plane (16), it is virtually impossible to construct any FMN-catalyzed proton-translocating loop mechanism. The alternative might be that redox-dependent conformational coupling (13, 45–48) is operating together with a direct ubiquinone reduction-associated loop mechanism which may result in the observed overall H+/e stoichiometry of 4. The redox-dependent nucleotide binding change as reported here may be a part of such a conformational coupling. A number of findings reported in the literature are in line with a significant redox-dependent conformational change of the complex I structure (17, 36, 37, 49–51). Changing of the affinity to dinucleotides upon enzyme reduction might also be due to different electronic structures of the oxidized and reduced forms of the flavin (17). It has been demonstrated that the presence of nucleotide in the active site of the enzyme affects the reduction potential of the flavin (25), consistent with the close interaction of the isoalloxazine ring and nicotinamide moiety.
We have shown previously that the substrate (NADH) and product (NAD+) nucleotides inhibit the succinate-supported complex I-mediated superoxide generation in tightly coupled SMPs (29). In the current study it is shown that NADH-OH, the nucleotide binding site directed inhibitor, strongly suppresses the succinate-supported superoxide generation (Figure 5) during reverse electron transfer. A number of possibilities for the mechanism(s) of inhibition of succinate-supported superoxide generation by NADH-OH can be considered. We have shown that NADH-OH binds much stronger to the oxidized form of the enzyme compared to the reduced one (Table 2). In other words the binding of NADH-OH results in a decrease of the midpoint redox potential of the protein-bound flavin. Data have been presented suggesting that the most probable candidate for superoxide generation from complex I is the reduced flavin moiety (29, 52). If the midpoint potential of the FMNH2/FMN couple is decreased upon NADH-OH binding in close vicinity to the flavin, this could be a reason for the suppression of succinate-supported superoxide generation in the presence of the inhibitor, which stabilizes the oxidized form. Another possibility is evident from the atomic structure of the T. thermophilus NDH1 (16, 17), which shows that accessibility of the flavin for oxygen might be provided by a solvent-accessible cavity. The inhibition of superoxide production in the presence of NADH-OH and other nucleotides might be due to steric hindrance for flavin–oxygen interaction as suggested by Sazanov (17). However, it should be pointed out that oxygen is a small, lipid-soluble molecule that does not necessarily reach the flavin from a solvent-accessible cavity. Figure 5 shows that the rate of superoxide production in the presence of succinate, when the cavity is presumably filled by water, is slightly less than that in the presence of 50 μM NADH (a concentration which is about 10 times higher than the apparent Km), i.e., under the conditions where the cavity is substantially occupied by the nucleotide. A likely explanation for the inhibitory effect of NADH-OH on the succinate-supported superoxide generation is that tight binding of the inhibitor results in a gross conformational change of a peripheral arm of complex I, which decreases the accessibility of flavin for oxygen.
The binding affinity of NADH-OH for both oxidized and reduced (by succinate) complex I is so high (3 × 10−10 and 7 × 10−9 M, respectively, Table 2) that the compound is an excellent tool for quantitative determination of the enzyme content in any particular preparation containing complex I. This is demonstrated in Figure 6, where a straight-line titration, as expected for practically irreversible inhibition (33, 35), is evident. At present we are not in a position to compare these data with a number of previously published results on titration of complex I activity by the ubiquinone junction-site-directed inhibitors, such as rotenone, piericidin, and pyridaben (33, 35, 53–55), until the exact structure of NADH-OH is established and the inhibitor is available in 100% pure form. However, the first approximation corresponds to a 1:1 ratio of NADH-OH to piericidin titers.
Strong deviation from the straight-line-type titration was evident for the reverse electron transfer activity (Figure 6). Several reasons for this type of a titration curve merit consideration. It is conceivable that the potential activity of complex I in the reverse reaction (NAD+ reduction) greatly exceeds the pmf-generating capacity of succinate oxidase. This model if considered as two homogeneous enzymes, QH2 (and pmf)-producing and QH2 (and pmf)-utilizing systems, which would result in the convex titration curve as seen in Figure 6. Another possible explanation of the observed titration behavior is that complex I in the native coupled membranes functions as a dimer. A dimeric functional model of complex I was proposed many years ago (56, 57); however, there is scant evidence that complex I functions as a dimer. The recent demonstration of the atomic structure of the hydrophilic domain of T. thermophilus NDH1 (16) clearly shows that the two-site model cannot be applied for the functioning of the bacterial enzyme. It is possible, however, that the mechanism of operation and the structure of the mammalian and bacterial enzymes are different. It has been noted that the 39 kDa subunit, not found in bacterial homologues, contains a nucleotide binding motif in addition to that found in the 51 kDa subunit (7). Further detailed studies on the structure and nucleotide binding properties of the mammalian complex I are evidently needed to sort out this complexity.
Footnotes
This study was supported by The Russian Foundation for Fundamental Research, Grants 05-04-48809 to A.D.V. and 06-04-48931 to V.G.G., NIH Research Grant R03 TW07825 funded by the Fogarty International Center (A.D.V. and G.C.), and the Department of Veterans Affairs (G.C.) and the Sandler Research Foundation (J.S.K.).
Abbreviations: pmf, proton-motive force; SMPs, submitochondrial particles; FMN, flavin mononucleotide; HAR, hexaammineruthenium-(III); NDH-1, prokaryotic homologue of the mitochondrial NADH: ubiquinone oxidoreductase (complex I).
References
- 1.Friedrich T, Steinmuller K, Weiss H. The proton-pumping respiratory complex I of bacteria and mitochondria and its homologue in chloroplasts. FEBS Lett. 1995;367:107–111. doi: 10.1016/0014-5793(95)00548-n. [DOI] [PubMed] [Google Scholar]
- 2.Dupuis A, Chevallet M, Darrouzet E, Duborjal H, Lunardi J, Issartel JP. The Complex I from Rhodobacter capsulatus. Biochim Biophys Acta. 1998;1364:147–165. doi: 10.1016/s0005-2728(98)00025-5. [DOI] [PubMed] [Google Scholar]
- 3.Yagi T, Yano T, Matsuno-Yagi A. Characteristics of the energy-transducing NADH-quinone oxidoreductase of Paracoccus denitrificans as revealed by biochemical, biophysical, and molecular biological approaches. J Bioenerg Biomembr. 1993;25:339–345. doi: 10.1007/BF00762459. [DOI] [PubMed] [Google Scholar]
- 4.Caroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 5.Ragan CI. NADH-ubiquinone oxidoreductase. Biochim Biophys Acta. 1976;456:249–290. doi: 10.1016/0304-4173(76)90001-x. [DOI] [PubMed] [Google Scholar]
- 6.Weiss H, Friedrich T, Hofhaus G, Preis D. The respiratory-chain NADH dehydrogenase (complex I) of mitochondria. Eur J Biochem. 1991;197:563–576. doi: 10.1111/j.1432-1033.1991.tb15945.x. [DOI] [PubMed] [Google Scholar]
- 7.Walker JE. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q Rev Biophys. 1992;25:253–324. doi: 10.1017/s003358350000425x. [DOI] [PubMed] [Google Scholar]
- 8.Vinogradov AD. Catalytic properties of the mitochondrial NADH-ubiquinone oxidoreductase (complex I) and the pseudo-reversible active/inactive enzyme transition. Biochim Biophys Acta. 1998;1364:169–185. doi: 10.1016/s0005-2728(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 9.Ohnishi T, Sled VD, Yano T, Yagi T, Burbaev DS, Vinogradov AD. Structure-function studies of iron-sulfur clusters and semiquinones in the NADH-Q oxidoreductase segment of the respiratory chain. Biochim Biophys Acta. 1998;1365:301–308. doi: 10.1016/s0005-2728(98)00082-6. [DOI] [PubMed] [Google Scholar]
- 10.Videira A, Duarte M. From NADH to ubiquinone in Neurospora mitochondria. Biochim Biophys Acta. 2002;1555:187–191. doi: 10.1016/s0005-2728(02)00276-1. [DOI] [PubMed] [Google Scholar]
- 11.Brandt U, Kerscher S, Dröse S, Zwicker K, Zickermann V. Proton pumping by NADH:ubiquinone oxidoreductase. A redox driven conformational change mechanism? FEBS Lett. 2003;545:9–17. doi: 10.1016/s0014-5793(03)00387-9. [DOI] [PubMed] [Google Scholar]
- 12.Hirst J. Energy transduction by respiratory complex I –an evaluation of current knowledge. Biochem Soc Trans. 2005;33:525–529. doi: 10.1042/BST0330525. [DOI] [PubMed] [Google Scholar]
- 13.Yagi T, Matsuno-Yagi A. The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry. 2003;42:2266–2274. doi: 10.1021/bi027158b. [DOI] [PubMed] [Google Scholar]
- 14.Brandt U. Energy converting NADH:quinone oxidoreductase (complex I) Annu Rev Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- 15.Hinchliffe P, Sazanov LA. Organization of iron-sulfur clusters in respiratory complex I. Science. 2005;309:771–774. doi: 10.1126/science.1113988. [DOI] [PubMed] [Google Scholar]
- 16.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 17.Sazanov LA. Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry. 2007;46:2275–2288. doi: 10.1021/bi602508x. [DOI] [PubMed] [Google Scholar]
- 18.Minakami S, Ringler RL, Singer TP. Studies on the respiratory chain-linked dihydrodiphosphopyridine nucleotide dehydrogenase. I. Assay of the enzyme in particulate and in soluble preparations. J Biol Chem. 1962;237:569–576. [PubMed] [Google Scholar]
- 19.Dooijewaard G, Slater EC. Steady-state kinetics of high molecular weight (type-I) NADH dehydrogenase. Biochim Biophys Acta. 1976;440:1–15. doi: 10.1016/0005-2728(76)90109-2. [DOI] [PubMed] [Google Scholar]
- 20.Dooijewaard G, Slater EC. Steady-state kinetics of low molecular weight (type-II) NADH dehydrogenase. Biochim Biophys Acta. 1976;440:16–35. doi: 10.1016/0005-2728(76)90110-9. [DOI] [PubMed] [Google Scholar]
- 21.Sled VD, Vinogradov AD. Kinetics of the mitochondrial NADH-ubiquinone oxidoreductase interaction with hexammineruthenium(III) Biochim Biophys Acta. 1993;1141:262–268. doi: 10.1016/0005-2728(93)90051-g. [DOI] [PubMed] [Google Scholar]
- 22.Gavrikova EV, Grivennikova VG, Sled VD, Ohnishi T, Vinogradov AD. Kinetics of the mitochondrial three-subunit NADH dehydrogenase interaction with hexammineruthe-nium(III) Biochim Biophys Acta. 1995;1230:23–30. doi: 10.1016/0005-2728(95)00015-b. [DOI] [PubMed] [Google Scholar]
- 23.Nakashima Y, Shinzawa-Itoh K, Watanabe K, Naoki K, Hano N, Yoshikawa S. Steady-state kinetics of NADH: coenzyme Q oxidoreductase isolated from bovine heart mitochondria. J Bioenerg Biomembr. 2002;34:11–19. doi: 10.1023/a:1013862502185. [DOI] [PubMed] [Google Scholar]
- 24.Fato R, Estornell E, Di Bernardo S, Pallotti F, Parenti Castelli G, Lenaz G. Steady-state kinetics of the reduction of coenzyme Q analogs by complex I (NADH:ubiquinone oxidoreductase) in bovine heart mitochondria and submitochondrial particles. Biochemistry. 1996;35:2705–2716. doi: 10.1021/bi9516034. [DOI] [PubMed] [Google Scholar]
- 25.Barker CD, Reda T, Hirst J. The flavoprotein subcomplex of complex I (NADH:ubiquinone oxidoreductase) from bovine heart mitochondria: insights into the mechanisms of NADH oxidation and NAD+ reduction from protein film voltammetry. Biochemistry. 2007;46:3454–3464. doi: 10.1021/bi061988y. [DOI] [PubMed] [Google Scholar]
- 26.Kotlyar AB, Karliner JS, Cecchini G. A novel strong competitive inhibitor of complex I. FEBS Lett. 2005;579:4861–4866. doi: 10.1016/j.febslet.2005.07.076. [DOI] [PubMed] [Google Scholar]
- 27.Kotlyar AB, Vinogradov AD. Slow active/inactive transition of the mitochondrial NADH-ubiquinone reductase. Biochim Biophys Acta. 1990;1019:151–158. doi: 10.1016/0005-2728(90)90137-s. [DOI] [PubMed] [Google Scholar]
- 28.Sh Burbaev D, Moroz IA, Kotlyar AB, Sled VD, Vinogradov AD. Ubisemiquinone in the NADH-ubiquinone reductase region of the mitochondrial respiratory chain. FEBS Lett. 1989;254:47–51. [Google Scholar]
- 29.Grivennikova VG, Vinogradov AD. Generation of superoxide by the mitochondrial Complex I. Biochim Biophys Acta. 2006;1757:553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Cha S. Tight-binding inhibitors-I. Kinetic behavior. Biochem Pharmacol. 1975;24:2177–2185. doi: 10.1016/0006-2952(75)90050-7. [DOI] [PubMed] [Google Scholar]
- 31.Kotlyar AB, Vinogradov AD. Interaction of the membrane-bound succinate dehydrogenase with substrate and competitive inhibitors. Biochim Biophys Acta. 1984;784:24–34. doi: 10.1016/0167-4838(84)90168-7. [DOI] [PubMed] [Google Scholar]
- 32.Zharova TV, Vinogradov AD. A competitive inhibition of the mitochondrial NADH-ubiquinone oxidoreductase (Complex I) by ADP-ribose. Biochim Biophys Acta. 1997;1320:256–264. doi: 10.1016/s0005-2728(97)00029-7. [DOI] [PubMed] [Google Scholar]
- 33.Grivennikova VG, Roth R, Zakharova NV, Hagerhall C, Vinogradov AD. The mitochondrial and prokaryotic proton-translocating NADH:ubiquinone oxidoreductases: similarities and dissimilarities of the quinone-junction sites. Biochim Biophys Acta. 2003;1607:79–90. doi: 10.1016/j.bbabio.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Kotlyar AB, Gutman M. The effect of ∆μH+ on the interaction of rotenone with complex I of submitochondrial particles. Biochim Biophys Acta. 1992;1140:169–174. doi: 10.1016/0005-2728(92)90006-n. [DOI] [PubMed] [Google Scholar]
- 35.Grivennikova VG, Maklashina EO, Gavrikova EV, Vinogradov AD. Interaction of the mitochondrial NADH-ubiquinone reductase with rotenone as related to the enzyme active/inactive transition. Biochim Biophys Acta. 1997;1319:223–232. doi: 10.1016/s0005-2728(96)00163-6. [DOI] [PubMed] [Google Scholar]
- 36.Mamedova AA, Holt PJ, Carrol J, Sazanov L. Substrate-induced conformational change in bacterial complex I. J Biol Chem. 2004;279:23830–23836. doi: 10.1074/jbc.M401539200. [DOI] [PubMed] [Google Scholar]
- 37.Sled VD, Vinogradov AD. Reductive inactivation of the mitochondrial three subunit NADH dehydrogenase. Biochim Biophys Acta. 1993;1143:199–203. doi: 10.1016/0005-2728(93)90143-4. [DOI] [PubMed] [Google Scholar]
- 38.Krishnamoorthy G, Hinkle PC. Studies on the electron transfer pathway, topography of iron-sulfur centers, and site of coupling in NADH-Q oxidoreductase. J Biol Chem. 1988;263:17566–17575. [PubMed] [Google Scholar]
- 39.Wikstrom M. Two protons are pumped from the mitochondrial matrix per electron transferred between NADH and ubiquinone. FEBS Lett. 1984;169:300–304. doi: 10.1016/0014-5793(84)80338-5. [DOI] [PubMed] [Google Scholar]
- 40.Galkin AS, Grivennikova VG, Vinogradov AD. H+/2e− stoichiometry in NADH-quinone reductase reactions catalyzed by bovine heart submitochondrial particles. FEBS Lett. 1999;451:157–161. doi: 10.1016/s0014-5793(99)00575-x. [DOI] [PubMed] [Google Scholar]
- 41.Grivennikova VG, Ushakova AV, Hägerhäll C, Vinogradov AD. Proton translocation catalyzed by Paracoccus denitrificans NADH:quinone oxidoreductase (NDH-1) BBA EBEC Short Rep. 2002;12:209. [Google Scholar]
- 42.Mitchell P. Chemiosmotic coupling oxidative and photosynthetic phosphorylation. Glynn Research Ltd; Bodmin, U.K.: 1966. [DOI] [PubMed] [Google Scholar]
- 43.Vinogradov AD. Kinetics, control, and mechanism of ubiquinone reduction by the mammalian respiratory chain-linked NADH-ubiquinone reductase. J Bioenerg Biomembr. 1993;25:367–375. doi: 10.1007/BF00762462. [DOI] [PubMed] [Google Scholar]
- 44.Brandt U. Proton-translocation by membrane-bound NADH:ubiquinone-oxidoreductase (complex I) through redox-gated ligand conduction. Biochim Biophys Acta. 1997;1318:79–91. doi: 10.1016/s0005-2728(96)00141-7. [DOI] [PubMed] [Google Scholar]
- 45.Ohnishi T, Salerno JC. Conformation-driven and semiquinone-gated proton-pump mechanism in the NADH-ubiquinone oxidoreductase (complex I) FEBS Lett. 2005;579:4555–4561. doi: 10.1016/j.febslet.2005.06.086. [DOI] [PubMed] [Google Scholar]
- 46.Vinogradov AD. Respiratory complex I: structure, redox components, and possible mechanisms of energy transduction. Biochemistry (Moscow) 2001;66:1086–1097. doi: 10.1023/a:1012476728710. [DOI] [PubMed] [Google Scholar]
- 47.Friedrich T. Complex I: a chimaera of a redox and conformation-driven proton pump? J Bioenerg Biomembr. 2001;33:169–177. doi: 10.1023/a:1010722717257. [DOI] [PubMed] [Google Scholar]
- 48.Zwicker K, Galkin A, Drose S, Grgic L, Kerscher S, Brandt U. The Redox-Bohr group associated with iron-sulfur cluster N2 of complex I. J Biol Chem. 2006;281:23013–23017. doi: 10.1074/jbc.M603442200. [DOI] [PubMed] [Google Scholar]
- 49.Belogrudov G, Hatefi Y. Catalytic sector of complex I (NADH:ubiquinone oxidoreductase): subunit stoichiometry and substrate-induced conformation change. Biochemistry. 1994;33:4571–4576. doi: 10.1021/bi00181a018. [DOI] [PubMed] [Google Scholar]
- 50.Yamaguchi M, Belogrudov GI, Hatefi Y. Mitochondrial NADH-ubiquinone oxidoreductase (Complex I). Effect of substrates on the fragmentation of subunits by trypsin. J Biol Chem. 1998;273:8094–8098. doi: 10.1074/jbc.273.14.8094. [DOI] [PubMed] [Google Scholar]
- 51.Marshall D, Fisher N, Grigic L, Zickermann V, Brandt U, Shannon RJ, Hirst J, Lawrence R, Rich PR. ATR-FTIR redox difference spectroscopy of Yarrowia lipolytica and bovine complex I. Biochemistry. 2006;45:5458–5467. doi: 10.1021/bi052561e. [DOI] [PubMed] [Google Scholar]
- 52.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ernster L, Dallner G, Azzone GF. Differential Effects of Rotenone and Amytal on Mitochondrial Electron and Energy Transfer. J Biol Chem. 1963;238:1124–1131. [Google Scholar]
- 54.Horgan DJ, Singer TP, Casida JE. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. 13 Binding sites of rotenone, piericidin A, and amytal in the respiratory chain. J Biol Chem. 1968;243:834–843. [PubMed] [Google Scholar]
- 55.Schuler F, Yano T, Di Bernardo S, Yagi T, Yankovskaya V, Singer TP, Casida JE. NADH-quinone oxidoreductase: PSST subunit couples electron transfer from iron-sulfur cluster N2 to quinone. Proc Natl Acad Sci USA. 1999;96:4149–4153. doi: 10.1073/pnas.96.7.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Belzen R, Albracht SP. The pathway of electron transfer in NADH:Q oxidoreductase. Biochim Biophys Acta. 1989;974:311–320. doi: 10.1016/s0005-2728(89)80249-x. [DOI] [PubMed] [Google Scholar]
- 57.van Belzen R, van Gaalen MC, Cuypers PA, Albracht SP. New evidence for the dimeric nature of NADH:Q oxidoreductase in bovine-heart submitochondrial particles. Biochim Biophys Acta. 1990;1017:152–159. doi: 10.1016/0005-2728(90)90146-u. [DOI] [PubMed] [Google Scholar]