

Abstract
It is frequently stated that UV light would cause massive destruction of prebiotic organic compounds because of the absence of an ozone layer. The elevated UV flux of the early sun compounds this problem. This applies to organic compounds of both terrestrial and extraterrestrial origin. Attempts to deal with this problem generally involve atmospheric absorbers. We show here that prebiotic organic polymers as well as several inorganic compounds are sufficient to protect oceanic organic molecules from UV degradation. This aqueous protection is in addition to any atmospheric UV absorbers and should be a ubiquitous planetary phenomenon serving to increase the size of planetary habitable zones.
It is widely held that there was no oxygen in the primitive atmosphere and therefore no UV protective ozone layer, although there are dissenting opinions (1–3). This is considered to be a serious problem for the accumulation of prebiotic organic compounds on the earth (4–6) and on Mars (7). This problem would have been worsened by the elevated UV production of the early sun (8–10). Protection from UV radiation is one of the motivations for proposing an origin of life in the submarine vents (11), benthic regions (6, 12), and in deep subsurface environments (13). Most attempts to deal with this problem have involved atmospheric absorbers such as H2S (6), SO2 (14), S8 (14), and organic hazes (6, 15). We show here that even in the absence of atmospheric shielding there would have been sufficient UV absorbers in the ocean to allow for the accumulation of organic material. These include organic polymers from electric discharges and HCN (hydrogen cyanide) polymerizations, solubilized elemental sulfur, and inorganics such as Cl−, Br−, Mg2+, and SH−, or Fe2+. There would have been complete UV protection if there were a frozen ocean (16), an oil slick (17), or large amounts of organic foams. Oceanic UV protectors increase the size of planetary habitable zones and thereby increase the number of planets on which life may have arisen.
MATERIALS AND METHODS
Spectrophotometry.
Absorbance measurements were made by a Hewlett Packard HP 89532A UV-Visible Spectrophotometer. All measurements were made by using 1-cm or 1-mm path length quartz cuvettes. Samples were diluted to obtain 1-cm absorbance measurements between 0.1 and 1.
Reagents.
Reagents were purchased from Fisher. Sea water was collected from the Scripps Institute of Oceanography pier (La Jolla, CA) and filtered through a 0.22-μm filter.
Organic Polymer Synthesis.
HCN polymer was obtained from the polymerization of 0.1 M KCN brought to pH 8 at room temperature. Spark discharge polymer was obtained from the action of a Tesla coil on a gas mixture of 200 mm CH4, 200 mm NH3, and 500 ml H2O in a 3-liter flask for 6 days (18).
Solubilization of Sulfur by Tholin Polymers.
Various concentrations of polymer were stirred with elemental sulfur overnight followed by filtration through a 0.22-μm filter.
UV Destruction of Organic Polymers (Tholins).
Solutions of spark discharge polymer or HCN polymer were dissolved in water in 2-cm diameter Vycor tubes and degassed by three consecutive freeze-thaw cycles. These were placed in a Rayonet Photochemical Reactor (New England Ultraviolet, Branford, CT, 16 × 2.2 W, 2,537 Å Hg lamps) at room temperature along with a control tube containing an identical solution in Pyrex. Samples were removed for absorbance measurements at appropriate time intervals. UV flux was estimated at 1 × 10−7E⋅cm−2⋅s−1 (where E = einstein) by potassium ferrioxalate actinometry (19).
RESULTS AND DISCUSSION
In the absence of greenhouse gases the surface temperature of the early Earth would have been −40°C, and there would have been a frozen ocean 300–1,000 m thick (16). This would have offered complete shielding of dissolved organic compounds from damaging UV radiation by scattering of the UV light. The ice layer on Europa offers similar protection.
It has been suggested that the primitive ocean could have been covered with an oil slick some 1–10 m thick (17) if the C/O ratio of early atmospheric constituents (CH4, CO, CO2) exceeded unity (20, 21). This oil slick could have been produced from the photopolymerization of methane or from meteorites containing hydrocarbons similar to those found in the Murchison meteorite (22). Such oil slicks would have provided strong protection because of absorbance and scattering of UV light.
A layer of foam over the entire ocean also would protect the organic compounds. It is not clear whether there was sufficient prebiotic synthesis of surfactants. However, the action of UV light on pyrene and hexadecane readily forms surface active compounds (22). These would be very effective UV protectors because of scattering effects.
It is sometimes stated that UV destruction of dissolved organic material would result from the UV transparency of pure water (23). Ocean water, however, is opaque to UV in the 200- to 220-nm region (24) (absorbance = 0.22 cm−1 at 218 nm), mainly because of dissolved species such as NaCl, NaBr, and Mn2+ and dissolved organic material. It should be noted that although bromide ion is present in 1/450 the concentration of chloride ion, its molar absorbtivity at 218 nm is 7,800 × higher. Together these compounds would offer essentially complete protection for aliphatic amino acids (25).
Sea water, however, does not afford protection to the nucleobases (adenine, uracil, guanine, and cytosine) of RNA, central to an RNA world, which absorb near 260 nm. The following discussion will focus mainly on protection at 260 nm, the region in which nucleic acid bases and aromatic amino acids absorb (25).
In the modern ocean the concentration of UV-absorbing organic matter is small (24, 26), with a 1-cm absorbance of 0.01 at 218 nm. These organics are of biological or industrial origins, and their low steady-state concentrations are governed by biodegradation and photooxidation processes (26). The prebiotic ocean would not have had these loss channels, and the concentration of organics would have been able to build up.
Prebiotic syntheses usually produce large amounts of organic polymers (18) called “tholins” (27) that often are ignored because they are difficult to analyze. They do, however, absorb strongly in the UV region. The yield of polymer varies with the details of the experiment but usually is between 5% and 60% of the input carbon. We will include polycyclic aromatic hydrocarbons (22) with these tholins for the purposes of this discussion. Prebiotic cyanide polymerizations produce a very insoluble and intractable polymer in large yield (60–90% of the input carbon) along with soluble polymers (28). Polymers of this type as well as more carbon-rich polymers occur in carbonaceous chondrites and would be expected in interstellar dust and cometary material, as well as on other planets (27, 29). We have examined the UV absorbance of the product of a spark discharge experiment and of an HCN polymerization. These results are shown in Fig. 1 and expressed as the absorbance of a solution containing 330 mg⋅liter−1 of reactant carbon. This represents an amount equivalent to 1% of the Earth’s crustal carbon (100 g⋅cm−2) dissolved in an ocean of the present size (300 liter⋅cm−2). The choice of this concentration is discussed below. It can be seen that both processes produce strong UV absorbers.
Figure 1.

The absorbance spectra of aqueous prebiotic organic polymers produced by the action of a spark discharge on a mixture of methane, water, and ammonia for 6 days and a 0.1 M KCN (brought to pH 8 with HCl) polymerization, each dissolved as 330 mg of carbon per liter solutions.
We measured the loss of absorbance of HCN polymer and the spark discharge polymer by UV light. The cyanide polymer is rapidly photolyzed, but the spark discharge polymer is more stable to photolysis. Approximately 10% of the spark discharge polymer’s absorbance at 260 nm is resistant to photodestruction under anaerobic conditions.
It is difficult to estimate the percentage of terrestrial carbon that was dissolved as polymer in the primitive ocean. If it is assumed that the majority of the organic material produced in the atmosphere had a relatively short residence time and ultimately became dissolved in the ocean, then it is possible to derive the absorbance of the primitive ocean based on the polymer yield over time. Estimates of the carbon outgassing rates from hydrothermal vents 3 billion years ago (15) range between 3.3 × 1012 mol C per yr to 5 × 1013 mol C per year. Because the oceans pass through the midocean ridge hydrothermal vents every 10 million years, where the intense heat would result in the destruction of organic compounds, we will take this as the upper limit for organic accumulation (30). If this outgassed carbon was released in the form of methane, rather than the present form of CO2 because of a more reduced mantle (21), and was photoreacted every year, and the concentration was allowed to build up to a steady state over 10 million years then carbon concentrations of 0.2–3.0 g⋅liter−1 are obtained. Assuming the polymer is similar to the product of a spark discharge, this gives 1-cm absorbances of 3.1–46.5 at 260 nm. It follows that given efficient atmospheric syntheses, a steady state will arise quickly on a geologic time scale, which will result in a self-shielding buildup of dissolved organic compounds.
The UV-absorbing organic material found in the Murchison meteorite (31) supports this calculation. A kilogram of meteorite contains 18 g of carbon, which has an absorbance of 1.3 cm−1 at 260 nm when dissolved in 1 liter (31). If the 10,000 g⋅cm−2 of carbon in the Earth’s crust contained soluble UV absorbers in the same proportions as in the Murchison meteorite and were dissolved in the oceans (300 liter⋅cm−2) the absorbance at 260 nm would be 2.4 cm−1. This is close to the absorbance obtained from cyanide polymer (A260 cm−1 = 6.72 for 330 mg⋅liter−1) and spark polymer (A260 cm−1 = 3.93 for 330 mg⋅liter−1), assuming 1% of the Earth’s crustal carbon was dissolved in these forms in the ocean.
It is clear that percentages this high or higher will be very protective of the dissolved organic material. Much lower percentages give inadequate protective coverage, and the inorganic species would assume greater importance. This may pose a problem for the survival of the small amounts of organic material delivered by extraterrestrial infall if this were the only source of prebiotic organic compounds (32). In other words, the UV protection of organic compounds requires efficient prebiotic syntheses, unless there is efficient inorganic protection.
The most important inorganic UV absorbers appear to be Fe2+ and H2S. There would either be an excess of Fe2+ or of SH− because of the insolubility of FeS (33). We consider first the case in which H2S is in excess. For an ocean of the present pH (≈8.2) most of the dissolved H2S would be present as SH− as the pK1 of H2S is 6.9. The uncertainty is in the concentration of SH−. Archaen concentrations of H2S have been estimated (33, 34) between 0 and 5 mM.
Any reasonable model with SH− in excess would have substantial absorbance in the region of λ < 270 nm (λmax ≈ 235 nm, ɛ 260 nm ≈ 231). For 1 mM SH− concentrations a 1-cm absorbance of 0.23 at 260 nm is obtained, thereby protecting the nucleic acid bases and other organic compounds. Although submarine vents and other sources of H2S could have established millimolar concentrations of SH−, the escape of H2S into the atmosphere would be a major sink because of UV photolysis,
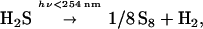 |
although the photochemistry is much more complex than indicated in the equation (35, 36). The quantum yield (37) for the above reaction is approximately 1. The elemental sulfur (S8) produced in this reaction can react with SH− to give polysulfides. These are formed easily under dilute oceanic conditions (38), are fairly stable in aqueous solution at moderate temperatures (39), and also absorb strongly in the 260-nm region.
The elemental sulfur produced in the above reaction is itself a strong UV absorber (ɛ260 approximately 5,500), but is limited by its low solubility in water (1.8 × 10−7 M). However, elemental sulfur is solubilized by anionic and cationic organic micelles (40), increasing solubility by factors of up to 4,000. We measured an increase by a factor of 15 in S8 solubility in a 100 mg⋅liter−1 solution of electric discharge polymer. The cyanide polymer exhibited no measurable ability to solubilize sulfur. A significant fraction of the organic material of the Murchison meteorite is composed of amphiphilic micelle-forming species (31), which are likely to solubilize S8. A prebiotic oil slick also would dissolve large amounts of S8. Using mineral oil, a mixture of high molecular weight hydrocarbons, as a model we found absorbtivities at 260 nm of 0.7 cm−1 for mineral oil itself and 5.5 cm−1 for S8-saturated mineral oil.
If Fe2+ is in excess then its absorbance replaces that of HS− in importance (33, 41, 42). Again, there is uncertainty in the concentration of Fe2+ in the primitive ocean assuming Fe2+ ≫ H2S. Archaen oceanic concentrations between 0 and 50 mM have been estimated (33, 34, 42–44) for Fe2+. The Archaen banded iron formations (45) provide some evidence for significant concentrations of Fe2+. Assuming a rate of weathering of 1 km3 of rock per yr, a mean rock density of 3 g⋅cm−3, and a 10% average iron content, 3 × 1014 g of Fe2+ per yr (5.4 × 1012 mol⋅yr−1) would have been added to the ocean. This is equal to adding 1.1 × 10−6 mol⋅cm−2⋅yr−1. Disregarding loss channels this would lead to millimolar oceanic concentrations in as little as 2.5 × 105 yr. The molar absorbtivity of iron(II) at 260 nm is approximately 1,630, giving a 1-cm absorbance at 260 nm of 1.63 for 1 mM concentrations.
It may be difficult to achieve significant concentrations of Fe2+ in surface waters because of photooxidation (45). Depending on the rate of mixing of surface waters, the rate of upwelling, and the pH of the early ocean, this could have been a significant sink for oceanic iron(II). A half-life of surface Fe2+ from 2 days to 1 week at pH 8.5 and 25°C has been estimated (44). Estimated values for H2S-rich and Fe2+-rich models are shown in Figs. 2 and 3.
Figure 2.

The absorbance spectrum of a model Archaen marine environment composed of 330 mg of carbon per liter HCN polymer, 1 mM SH−, 0.5 M NaCl, 1 mM NaBr, and 50 mM MgCl2. The contribution from NaCl, NaBr, and MgCl2 is approximately equivalent to the absorbance of modern seawater.
Figure 3.

The absorbance spectrum of a model Archaen marine environment composed of 330 mg of carbon per liter spark polymer, 1 mM Fe2+, 0.5 M NaCl, 1 mM NaBr, and 50 mM MgCl2.
CONCLUSIONS
This analysis shows that there would have been a wide variety of both organic and inorganic UV absorbers in the primitive ocean that would have protected the UV-sensitive organic compounds such as the purines and pyrimidines, with amino acids being protected effectively by aqueous salts. Our models suggest that it is entirely possible that incident UV flux could have been attenuated to minimal intensities after passage through as little as 2 mm of ocean water (<1% T at 218 nm and 260 nm).
We recognize that any one of these concentration estimates may be in considerable error. However, entirely removing any one of the absorbers still leaves a variety of alternative absorbers that still would offer adequate protection.
It is likely that some of these oceanic UV absorbers would have persisted well after the origin of life. When this UV shield disappeared, organisms would have needed to develop mechanisms to protect themselves from UV radiation, such as nucleic acid repair enzymes, pigmentation, or by living at sufficient depth to avoid UV damage.
These results have considerable bearing on discussions of extrasolar planetary habitable zones (46, 47). Concern has been expressed that the origin and evolution of life may not be possible around some stars because of the intense UV environment (48–50), especially in the late T-Tauri phase of stellar evolution (8–10). It is clear that dissolved organics and inorganics, which are likely to be present in an aqueous environment, can offer sufficient protection against this radiation. This is in addition to any protection offered by atmospheric absorbers. It therefore is probable that planets providing suitable havens for the accumulation of prebiotic compounds, and presumably for the origin of life, are more common than previously thought.
Acknowledgments
We thank Dr. James Lyons for helpful comments and the National Aeronautics and Space Administration Specialized Center for Research and Training (NSCORT) in Exobiology for grant support.
References
- 1.Towe K M. Nature (London) 1978;274:657–661. [Google Scholar]
- 2.Levine J S. J Mol Evol. 1982;18:161–172. doi: 10.1007/BF01733042. [DOI] [PubMed] [Google Scholar]
- 3.Carver J H. Nature (London) 1981;292:136–138. [Google Scholar]
- 4.Hull D E. Nature (London) 1960;186:693–694. [Google Scholar]
- 5.Hulett H R. J Theor Biol. 1969;24:56–72. doi: 10.1016/s0022-5193(69)80006-8. [DOI] [PubMed] [Google Scholar]
- 6.Sagan C. J Theor Biol. 1973;39:195–200. doi: 10.1016/0022-5193(73)90216-6. [DOI] [PubMed] [Google Scholar]
- 7.Stoker C R, Bullock M A. J Geophys Res. 1997;102:10881–10888. doi: 10.1029/97je00667. [DOI] [PubMed] [Google Scholar]
- 8.Canuto V M, Levine J S, Augustsson T R, Imhoff C L. Nature (London) 1982;296:816–820. [Google Scholar]
- 9.Canuto V M, Levine J S, Augustsson T R, Imhoff C L, Giampapa M S. Nature (London) 1983;305:281–286. [Google Scholar]
- 10.Gaustad J E, Vogel S N. Origins Life Evol Biosphere. 1982;12:3–8. doi: 10.1007/BF00926907. [DOI] [PubMed] [Google Scholar]
- 11.Baross J A, Hoffman S E. Origins Life Evol Biosphere. 1985;15:327–345. [Google Scholar]
- 12.Margulis L, Walker J C G, Rambler M. Nature (London) 1976;264:620–624. [Google Scholar]
- 13.Gold T. Proc Natl Acad Sci USA. 1992;89:6045–6049. doi: 10.1073/pnas.89.13.6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasting J F, Zahnle K J, Pinto J P, Young A T. Origins Life Evol Biosphere. 1989;19:252–253. doi: 10.1007/BF01808144. [DOI] [PubMed] [Google Scholar]
- 15.Sagan C, Chyba C. Science. 1997;276:1217–1221. doi: 10.1126/science.276.5316.1217. [DOI] [PubMed] [Google Scholar]
- 16.Bada J L, Bigham C, Miller S L. Proc Natl Acad Sci USA. 1994;91:1248–1250. doi: 10.1073/pnas.91.4.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasaga A C, Holland H D, Dwyer M J. Science. 1971;174:53–55. doi: 10.1126/science.174.4004.53. [DOI] [PubMed] [Google Scholar]
- 18.Miller S L. Science. 1953;117:528–529. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 19.Hatchard C G, Parker C A. Proc R Soc London Ser A. 1956;235:518–536. [Google Scholar]
- 20.Khare B N, Sagan C, Zumberge J E, Sklarew D S, Nagy B. Icarus. 1981;48:290–297. [Google Scholar]
- 21.Kasting J F, Eggler D H, Raeburn S P. J Geol. 1993;101:245–257. doi: 10.1086/648219. [DOI] [PubMed] [Google Scholar]
- 22.Deamer D W, Harang E A, Seleznev S A. Origins Life Evol Biosphere. 1989;19:291–292. [Google Scholar]
- 23.Berkner L V, Marshall L C. J Atmos Sci. 1965;22:225–261. [Google Scholar]
- 24.Ogura N, Hanya T. Nature (London) 1966;212:758. [Google Scholar]
- 25.McLaren A D, Shugar D. Photochemistry of Proteins and Nucleic Acids. New York: Pergamon; 1964. [Google Scholar]
- 26.Zafirou O C, Jousset-Dubien J, Zepp R G, Zika R G. Environ Sci Technol. 1984;18:358A–371A. [Google Scholar]
- 27.Sagan C, Khare B N. Nature (London) 1979;277:102–107. [Google Scholar]
- 28.Ferris J P, Joshi P C, Edelson E H, Lawless J G. J Mol Evol. 1978;11:293–311. doi: 10.1007/BF01733839. [DOI] [PubMed] [Google Scholar]
- 29.Matthews C N. In: Circumstellar Habitable Zones: Proceedings of the First International Conference. Doyle L R, Sagan C, editors. Menlo Park, CA: Travis House Publications; 1996. pp. 317–329. [Google Scholar]
- 30.Stribling R, Miller S L. Origins Life Evol Biosphere. 1987;17:261–273. doi: 10.1007/BF02386466. [DOI] [PubMed] [Google Scholar]
- 31.Deamer D W, Pashley R M. Origins Life Evol Biosphere. 1989;19:21–38. doi: 10.1007/BF01808285. [DOI] [PubMed] [Google Scholar]
- 32.Chyba C, Sagan C. Nature (London) 1992;355:125–132. doi: 10.1038/355125a0. [DOI] [PubMed] [Google Scholar]
- 33.Walker J C G, Brimblecombe P. Precambrian Res. 1985;28:205–222. doi: 10.1016/0301-9268(85)90031-2. [DOI] [PubMed] [Google Scholar]
- 34.Stumm W, Morgan J J. Aquatic Chemistry. 2nd Ed. New York: Wiley; 1981. [Google Scholar]
- 35.Kasting J F, Zahnle K J, Pinto J P, Young A T. Origins Life Evol Biosphere. 1989;19:95–108. doi: 10.1007/BF01808144. [DOI] [PubMed] [Google Scholar]
- 36.Kasting J F. Origins Life Evol Biosphere. 1990;20:199–231. doi: 10.1007/BF01808105. [DOI] [PubMed] [Google Scholar]
- 37.Wojciechowski K, Baj H, Forys M. J Chem Soc Faraday Trans 2. 1979;75:1085–1094. [Google Scholar]
- 38.Boulègue J, Michard G. C R Acad Sci. 1973;277:2613–2616. [Google Scholar]
- 39.Giggenbach W F. Inorg Chem. 1974;13:1724–1728. [Google Scholar]
- 40.Steudel R, Holdt G. Angew Chem Int Ed Engl. 1988;27:1358–1359. [Google Scholar]
- 41.Walker J C G. Nature (London) 1983;302:518–520. [Google Scholar]
- 42.Osterberg R. Origins Life Evol Biosphere. 1997;27:481–484. doi: 10.1023/a:1006532427925. [DOI] [PubMed] [Google Scholar]
- 43.Holland H D. Chemical Evolution of the Atmosphere and Oceans. Princeton: Princeton Univ. Press; 1984. pp. 379–388. [Google Scholar]
- 44.Mauzerall D C. In: Encyclopedia of Earth System Science. Nierenberg W A, editor. Vol. 3. San Diego: Academic; 1992. pp. 445–453. [Google Scholar]
- 45.Braterman P S, Cairns-Smith A G, Sloper R W. Nature (London) 1983;303:163–164. [Google Scholar]
- 46.Schwartzman D W, Volk T. In: Bioastronomy: The Search for Extraterrestrial Life. Heidmann J, Klein M J, editors. Berlin: Springer; 1991. pp. 155–162. [Google Scholar]
- 47.Doyle L R, Sagan C, editors. Circumstellar Habitable Zones: Proceedings of the First International Conference. Menlo Park, CA: Travis House Publications; 1996. [Google Scholar]
- 48.Hart M H. Icarus. 1978;33:23–39. [Google Scholar]
- 49.Hart M H. Icarus. 1979;37:351–357. [Google Scholar]
- 50.Kasting J F, Whittet D C B, Sheldon W A. Origins Life Evol Biosphere. 1997;27:413–420. doi: 10.1023/a:1006596806012. [DOI] [PubMed] [Google Scholar]