

Abstract
There is concern about the rise of antifungal drug resistance, but little is known about comparative biological properties and pathogenicity of drug-resistant strains. We generated fluconazole (FLC; CO23RFLC)- or micafungin (FK; CO23RFK)-resistant strains of Candida albicans by treating a FLC- and FK-susceptible strain of this fungus (CO23S) with stepwise-increasing concentrations of either drug. Molecular analyses showed that CO23RFLC had acquired markedly increased expression of the drug-resistance efflux pump encoded by the MDR1 gene, whereas CO23RFK had a homozygous mutation in the FSK1 gene. These genetic modifications did not alter to any extent the growth capacity of the drug-resistant strains in vitro, either at 28°C or at 37°C, but markedly increased their experimental pathogenicity in a systemic mouse infection model, as assessed by the overall mortality and target organ invasion. Interestingly, no apparent increase in the vaginopathic potential of the strains was observed with an estrogen-dependent rat vaginal infection. The increased pathogenicity of drug-resistant strains for systemic infection was associated with a number of biochemical and physiological changes, including (i) marked cellular alterations associated with a different expression and content of major cell wall polysaccharides, (ii) more rapid and extensive hypha formation in both liquid and solid media, and (iii) increased adherence to plastic and a propensity for biofilm formation. Overall, our data demonstrate that experimentally induced resistance to antifungal drugs, irrespective of drug family, can substantially divert C. albicans biology, affecting in particular biological properties of potential relevance for deep-seated candidiasis.
Candida albicans, a dimorphic opportunistic human pathogen, is the most prominent cause of oropharyngeal, vaginal, and invasive candidiasis in humans (3). In particular, oropharyngeal infections are very common in human immunodeficiency virus (HIV)-infected individuals and patients with AIDS, while deep-seated infections are frequent in neutropenic patients (27, 31). The incidence of candidiasis has dramatically increased in the last 2 decades, and bloodstream infections due to Candida spp. are becoming a prime cause of morbidity and mortality for different types of immunocompromised patients (33). Host immunosuppression and the defined virulence traits possessed by C. albicans are a combination that is ideal for favoring the emergence of this fungus as an important agent of disease in humans.
The azoles, particularly fluconazole (FLC), remain among the most common antifungal drugs, but their intensive clinical use for both therapy and prophylaxis has favored the emergence of resistant strains (42). The threat of increasing resistance to azole drugs, associated with the relative scarcity of antifungal drugs, prompted the development of new drugs, such as the echinocandins (e.g., micafungin [FK]). These cytocidal drugs inhibit cell wall synthesis through the inhibition of β-1,3 glucan synthase and have rapidly become an important therapeutic option for several fungal infections (25).
Most of the biological functions related to pathogenicity and virulence of C. albicans reside in the fungal cell wall which, as the rigid external organelle of the fungus, is critical for fungal morphogenesis and host-fungus interplay (8, 12). Of the numerous factors associated with virulence in C. albicans, hyphal morphogenesis is likely to be one of the most important (22). Hypha development from yeast cells is critical for adherence, an essential first step in microbial colonization, which is in turn a key event in the initiation of the pathogenic process (5, 41). Adherence may involve both glycosylated and nonglycosylated cell wall proteins acting as adhesins (21, 41). In response to attachment to a surface, fungal cells produce biofilms, three-dimensional structures made up of cells surrounded by exopolymeric, mostly polysaccharide matrices that contribute to the infectious process and antibiotic resistance (18, 23).
Overall, antifungal drug resistance (ADR) and fungal virulence are critical issues for the host-parasite relationship in candidiasis. However, very little is known about any interrelation between drug resistance and virulence of C. albicans. Thus, we investigated whether acquisition of resistance to FLC or FK was reflected by measurable effects on pathogenicity of the fungus. For this purpose, we generated two strains resistant to FLC or FK (strain CO23RFLC or CO23RFK, respectively) from a vaginal clinical isolate of C. albicans (CO23S) which was fully susceptible to azoles and echinocandins (1). These three strains were compared for expression levels and/or mutations of drug resistance-related genes and for morphological and ultrastructural characteristics, including putative virulence traits in vitro and in vivo.
MATERIALS AND METHODS
Yeast strains and growth conditions.
The strain CO23S of C. albicans was isolated from a subject with vulvo-vaginal candidiasis and was originally susceptible to FK and FLC. It was made resistant to FK463 or to FLC by 10 growth passages in stepwise-increasing concentrations (0.01 to 8 μg/ml or 0.32 to 128 μg/ml of the respective drug) in agar-solidified yeast nitrogen base (YNB) medium, at 28°C. The resistance phenotype was stable after multiple (50) passages in culture. Strains CO23S, CO23RFLC, and CO23RFK had identical electrophoretic karyotypes, as determined by pulse-field gel electrophoresis, performed as reported in previous studies (1).
For the determination of growth curves, the three strains were incubated at 37°C or 28°C for 48 h in 200-ml flasks containing liquid YNB medium, under slight agitation, using an initial inoculum density of 0.1 (as measured spectrophotometrically at 560 nm). Growth was assessed by harvesting 1 ml of the culture at 2, 4, 6, 8, 24, and 48 h and measuring its optical density.
EM.
For scanning electron microscopy (SEM), C. albicans cells were grown in glucose-supplemented YNB medium (see above) at 28°C for 24 h. After washing twice in calcium- and magnesium-free phosphate-buffered saline (PBS), the cellular pellets resulting from centrifugation were fixed for 20 min at room temperature with 2.5% (vol/vol) glutaraldehyde in 0.01 M cacodylate buffer (pH 7.4) containing 2% (wt/vol) sucrose. After three washes in the same buffer, the cells were postfixed with 1% (wt/vol) OsO4 for 1 h, dehydrated on an ethanol gradient, critical point dried in CO2, and gold coated by sputtering. The samples were examined with a Cambridge Stereoscan 360 SEM (Cambridge Instruments, Cambridge, United Kingdom).
For a quantitative analysis of cell volume distribution on SEM images, both the major (a) and the minor (b = c) axes of 100 randomly chosen yeast cells of each of the three strains were measured. Cell volume was calculated by assuming that the yeast cells have an ellipsoid shape, by using the formula 4/3 π (a·b·c). The values shown on the histograms are means ± standard deviations (Fig. 1C).
FIG. 1.

Growth curves (A), scanning electron micrographs (EM) (B), and cell volume (C) of CO23S (wild-type strain) and CO23RFK and CO23RFLC (FK- and FLC-resistant strains, respectively) of C. albicans. O.D. 560, optical density at 560 nm. The growth curves were obtained by growing the strains at 37°C in liquid YNB medium. The cell volumes were calculated from SEM observations, as described in Materials and Methods. In panel B, parts A, B, and C indicate the strains CO23S, CO23RFK, and CO23RFLC, respectively, with corresponding cell volumes.
For transmission EM, cells were prefixed with glutaraldehyde, as described above, and then postfixed with the OsO4 solution overnight at 4°C. The cells were then dehydrated in acetone gradient and embedded in epoxy resin (Agar 100 resin; Agar Scientific Ltd., Stansted, United Kingdom) as per routine procedures. Ultrathin sections, obtained with a LKB Ultrotome Nova, were stained with uranyl acetate and lead citrate and examined with a Philips 208 transmission EM (FEI Company, Eindhoven, The Netherlands).
Determination of dry weight and polysaccharide content.
For dry weight determination, cells grown for 24 h at 30°C with shaking. Each culture (50 ml) was filtered through 0.45-μm membrane filters (Millipore). Filters were washed with 50 ml of distilled water and dried at 80°C for 12 h, and the dry weight was calculated for each strain. Each assay was performed in triplicate. For determination of alkali-acid-soluble and insoluble cell wall components, the strains CO23S, CO23RFK, and CO23RFLC were grown in 1% yeast extract-2% peptone-2% dextrose (YPD) broth at 28°C for 24 h. The cells were hydrolyzed as described by Fleet and Manners (18, 19). An aliquot of the cells were treated with 0.5 M acetic acid at 60°C for 3 h and centrifuged, and the resultant pellet was treated with 0.5 M NaOH at 90°C for 6 h. The insoluble residue was treated with 20 U/ml of a purified β-1,3 glucanase (Zymoliase 100T) overnight at 37°C. Carbohydrates were assayed by the method described by Dubois et al. (16).
MAbs.
Monoclonal antibody (MAb) AF1 was produced in mice immunized with a crude mannoprotein (MP) preparation (GMP) from yeast cells of C. albicans (6). The MAb was purified from mouse ascitic fluids by affinity chromatography on an Affiprep-protein A column equilibrated with Maps II buffer (Bio-Rad, Richmond, CA). The titer of the purified MAb preparation was assessed by indirect enzyme-linked immunosorbent assays employing GMP as the solid-phase antigen.
Immunoelectron microscopy.
For AF1 epitope localization in the postembedding procedure, thin sections obtained as described above and collected on gold grids were treated for 3 min with 0.5 mg of sodium borohydride per ml of ice-cold distilled water. After being washed in ice-cold distilled water (three times for 5 min) and in PBS containing 0.5% (wt/vol) bovine serum albumin, 0.05% Tween 20, and 5% fetal serum (three times, 5 min each time), the sections were incubated with MAb AF1 (diluted 1:10) overnight at 4°C. After being washed for 2 h at room temperature by floating the grids on drops of PBS, samples were labeled with rabbit anti-mouse immunoglobulin M (IgM) gold conjugate (diluted 1:10; Sigma) and then washed in PBS buffer for 3 h at room temperature. For the negative control, the sections were incubated with the irrelevant IgG2a monoclonal antibody or with goat anti-mouse IgG-gold alone.
Quantitative real-time RT-PCR.
Quantitative expression of the CDR1 (31), CDR2 (37), MDR1 (31), ERG11 (31), and FLU1 (4) genes was determined by real-time reverse transcription (RT)-PCR with an iCycler iQ system (Bio-Rad Laboratories, Hercules, CA), using total RNAs extracted from the exponential phase-cultured C. albicans strains as described previously (39). Primer pairs and Taqman probes for the target and reference TEF3 genes (37) were designed using Beacon Designer 3 v. 3.00 software (Premier Biosoft International, Palo Alto, CA) and synthesized by MWG Biotech (Florence, Italy) (see Table 2). RT-PCRs were carried out using reagents and conditions as reported elsewhere (39). Each reaction was run in quadruplicate. In each sample, relative mRNA expression levels of the target genes were normalized for input RNA against the level of TEF3 gene transcripts and calculated using the comparative cycle time (Ct) method (29).
TABLE 2.
Primers and fluorescent probes used for real-time RT-PCR
| Gene (accession no.) | Primer or probe | Sequencea | Gene location (5′-3′) |
|---|---|---|---|
| CDR1 (X77589) | CDR1a | AACCGTTTACGTTGAACACGATAT | 2504-2527 |
| CDR1b | ACCAACTTCACCATCTTCAATGAC | 2565-2588 | |
| CDR1pr | 6FAM-ACTCACGCCGACACCACCGTTGTT-TAMRA | 2535-2558 | |
| CDR2 (U63812) | CDR2a | TGGCTAGTGTTTATATGGCAACCT | 1725-1748 |
| CDR2b | AAGCTTCAGCAATTGACACTCTTT | 1821-1844 | |
| CDR2pr | 6FAM-TCACCACCGGAAACACCACGCACA-TAMRA | 1792-1815 | |
| MDR1 (Y14703) | MDR1a | TCTCGGGTGGATTCTTTGCTAAT | 2659-2681 |
| MDR1b | AATGGACCAAAACTAGGACCACA | 2775-2797 | |
| MDR1pr | 6FAM-ACGGCACCCAAACTCCAAGCGGC-TAMRA | 2751-2773 | |
| FLU1 (AF188621) | FLU1a | TTTGTCGTTTCTTTGCTGGGTTT | 1153-1175 |
| FLU1b | ATTAAACATATCGGCCATAACGGC | 1206-1229 | |
| FLU1pr | 6FAM-AGCCACAACCAATGGAGCAGCACC-TAMRA | 1179-1202 | |
| ERG11 (AY856351) | ERG11a | TTATTAGGGGTTCCATTTGTTTACA | 76-100 |
| ERG11b | AAATTCATAAGGTTGTTGACCATATG | 188-213 | |
| ERG11pr | 6FAM-TGCAGAACCAAACCAAGGAATCCA-TAMRA | 160-183 | |
| TEF3 (Z12822) | TEF3a | AACCGTTTACGTTGAACACGATAT | 2480-2504 |
| TEF3b | ACCAACTTCACCATCTTCAATGAC | 2565-2588 | |
| TEF3pr | Texas Red-ACTCACGCCGACACCACCGTTGTT-BHQ2 | 2535-2558 |
Abbreviations: 6FAM, 6-carboxyfluorescein; TAMRA, 6-carboxy-N,N,N′,N′-tetramethylrhodamine; Texas Red, trademarked product from Molecular Probes; BHQ2, Black Hole Quencher 2.
PCR amplification and DNA sequencing.
Purified genomic DNA was obtained from each C. albicans strain by using an EZ1 DNA tissue kit (Qiagen, Milan, Italy) and a BioRobot EZ1 workstation (Qiagen) in accordance with the manufacturer's instructions. The entire FSK1 and ERG11 open reading frames were PCR amplified using primers and reaction conditions described previously (2, 31). PCR products were purified with a MinElute PCR purification kit (Qiagen) and sequenced on both strands with a Big Dye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) using an ABI Prism 3100 genetic analyzer (Applied Biosystems) with the primers and cycling conditions already reported (2, 31).
Experimental infections. (i) Mouse systemic model.
C. albicans strains were grown to stationary phase in YNB medium at 28°C. Cells were harvested by centrifugation, washed twice in calcium- and magnesium-free PBS, and suspended to a density of 5 × 107 cells/ml as enumerated by a hemocytometer. Subsequently, group of 10 male BALB/c mice (18 to 20 g each; Charles River Laboratories, Calco, Italy) were injected intravenously with 0.2 ml (1 × 107 cells) of a suspension of each strain. Mice were observed daily for signs of morbidity and mortality for a period of 30 days. All dead animals were autopsied, and internal organ invasion by the fungus was assessed. In a separate experiment, the fungus burden in the left kidney, taken as representative target organ, was also assessed. Differences in median survival time (in days) and in the ratio of dead animals to the total number of challenged animals (D/T) were statistically assessed by the nonparametric Mann-Whitney test (for median survival time) and Fisher's exact test (for D/T). Differences in survival curves were assessed by the log-rank test.
(ii) Rat vaginal model.
A rat vaginal model was used for the experimental vaginal infection, as previously described (14). Two independent experiments with each fungal strain were conducted, and in each experiment, groups of five rats were used. Oophorectomized female Wistar rats (80 to 100 g; Charles River, Calco, Italy) were injected subcutaneously with 0.5 mg of estradiol benzoate (Estradiolo; Amsa Farmaceutici srl, Rome, Italy). Six days after the first estradiol dose, all animals were inoculated intravaginally with 107 yeast cells of each C. albicans strain tested in 0.1 ml of saline. The inoculum of each strain used for challenge was dispensed into the vaginal cavity through a syringe equipped with a multipurpose calibrated tip (Combitip; PBI, Milan, Italy). The strains had been grown previously in YPD broth at 28°C with a gyrator shaker (200 rpm), harvested by centrifugation (1,500 g), washed, counted with a hemocytometer, and suspended to the required number in saline solution. The number of cells in the vaginal fluid was counted by culturing 1-μl samples (using a calibrated plastic loop; Disponoic; PBI) taken from each animal, on S Sabouraud dextrose agar containing chloramphenicol (50 mg/liter) as previously described. The kinetics of Candida vaginal infection were monitored by the number of CFU/ml of vaginal lavage fluid. The infection was monitored for at least 21 days after the challenge, with vaginal fluid sampling usually being done at 1, 24, and 48 h and then on days 5, 7, 14, and 21. The animal experimentation referred to in this paper was approved by the ad hoc committee of the Istituto Superiore di Sanità, Rome, Italy.
Mycelial development and agar-invasive hyphal growth.
The cells were grown in YNB medium supplemented with 0.1% (wt/vol) N-acetyl-d-glucosamine (Sigma), or in RPMI 1640 medium (Sigma Chemical Co., Detroit, MI) with 10% fetal calf serum added, and incubated for 24 h at 37°C.
Hyphal growth was induced on 4% bovine calf serum containing various concentrations (0.5 to 3%) of bacteriological agar as indicated in specific experiments. Cells grown overnight at 28°C were diluted to 5 × 106 in YPD broth, and 2 μl (104 cells) was spotted on the surface of the serum agar and incubated at 37°C for 7 days. Plates were monitored daily for invasive growth.
Adherence assay and biofilm formation.
Cells were grown for 24 h at 28°C in YPD broth, washed twice with sterile PBS (10 mM phosphate buffer, 2.7 mM potassium chloride, 137 mM sodium chloride, pH 7.4), and resuspended in RPMI 1640 supplemented with morpholinepropanesulfonic acid (MOPS) at 1.5 × 103 cells/ml. After incubation for 3 h at 37°C in six-well polystyrene plates (Corning Incorporated, Corning, NY) followed by extensive washing, 1 ml of Sabouraud dextrose agar was poured into each well and allowed to solidify. After incubation at 37°C for 24 h, colonies were counted and the results were expressed as a percentage of the inoculum. The inoculum size for each cell suspension was confirmed by plating aliquots of the culture directly in Sabouraud dextrose agar plates.
For biofilm formation, cells grown as described above were seeded at a density of 1 × 106 cells/ml in presterilized, polystyrene, flat-bottom six-well microtiter plates (Corning, NY) and incubated for 48 h at 37°C (33). After biofilm formation, the medium was aspirated, and nonadherent cells were removed by thoroughly washing the biofilms three times with sterile PBS.
A semiquantitative measurement of biofilm formation was made by using an XTT [2,3-bis(2-methoxy-4-nitro-5-sulfo-phenyl)-2H-tetrazolium-5-carboxanilide] reduction assay. XTT (Sigma) was dissolved in PBS at 0.5 g/liter. The solution was sterilized by filtration through a 0.22-μm-pore-size filter. Prior to each assay, the XTT solution was thawed and supplemented with menadione (10 mM stock dissolved in acetone; final concentration, 1 μM; Sigma), an aliquot of 1 ml of the XTT-menadione solution was added per well, and the plates were incubated in the solution at 37°C for 2 h. A sample (500 μl) was then transferred from each well into a fresh 12-well plate (to eliminate interference of cells with colorimetric readings), and the colorimetric change resulting from XTT reduction was measured at 490 nm (34) biofilm cultures were grown in triplicate, and each assay was performed six times. For photographs, the biofilms were stained with crystal violet (35).
RESULTS
Growth, ultrastructure, and biochemical characteristics of CO23RFLC and CO23RFK strains.
As the first step in this investigation, the two drug-resistant strains, CO23RFLC and CO23RFK (CO23RFLC MIC, >64 μg/ml; CO23RFK MIC, >4 μg/ml), were compared to their parental strain, CO23S (FLC MIC, 0.25 μg/ml; FK463 MIC, 0.025 μg/ml), for a number of basic biochemical and physiological properties. Concerning their growth capacity at 37°C, the FLC-resistant strain had a prolonged lag phase compared to both the parental and the FK-resistant strain; nonetheless, the total growth yield after 24 to 48 h of growth was substantially the same as that of the other two strains (Fig. 1A). At 28°C, the three strains had almost overlapping growth curves in YNB medium (data not shown). At variance with growth, the three strains were quite dissimilar in growth morphology (Fig. 1B) and, particularly, cell volume (Fig. 1C). Particularly, SEM showed that sensitive C. albicans cells had their typical shape, dimensions, and surface morphology, while CO23RFK and CO23RFLC cells demonstrated overall increased cell dimensions and an increased propensity to cluster, suggesting an altered budding mechanism. Quantitative analysis of cell dimensions (Fig. 1C) revealed that the mean volume of drug-resistant cells was roughly twice that of the parental strain (9.3 ± 3.0, 17.0 ± 5.9, and 18.6 ± 5.6 μm3 for CO23S, CO23RFK, and CO23RFLU strains, respectively).
The fungal cell wall is a plastic and dynamic structure that constantly changes in response to environmental signals, with a number of highly efficient, compensatory mechanisms (28). Since this organelle could be the target of the above changes, we examined the cells of the three strains for any variation in cell extracts representative of major cell wall components. As shown in Table 1, all strains had roughly similar total hexose amounts per cell, but the two drug-resistant ones had a much higher percentage of cell wall material per cell than that of the parent strain, indicating a marked enrichment in the carbohydrate-protein complexes (mostly mannan, β-glucan, and their complexes with proteins), which was also in line with apparent abnormal cell wall deposition seen in these strains by SEM (Fig. 1 and data not shown). Although no specific measurements of these components was attempted, the isolated, purified cell wall of each strain was grossly separated into its alkali-soluble and alkali-acid-insoluble fractions, which are known to contain essentially more plastic, MP, and soluble β-glucan constituents in the former fraction and more insoluble, rigid β-glucan complexes with proteins and chitin in the latter fraction, respectively (18, 19). By this analysis, a marked relative decrease of the cell wall alkali-acid-insoluble constituent was detected for both the drug-resistant strains compared to the parental strain. Particularly, the content of the alkali-acid-insoluble material per equal cell wall amount of the CO23RFK strain was less than half that of the parental, drug-susceptible strain (Table 1). Overall, this analysis suggested an unbalance of the respective proportions of MPs, β-glucan, and other constituents in the drug-resistant strains, which appear to be relatively enriched in soluble, plastic cell wall materials and impoverished in insoluble, rigid cell wall materials.
TABLE 1.
Cell wall composition of drug-susceptible and -resistant C. albicans strains
| Strain | % Glucosea | % Cell wallb | % Alkali-acid insolublec |
|---|---|---|---|
| CO23S | 58.16 | 50.0 | 27.77 |
| CO23RFK | 52.51 | 86.4 | 12.97 |
| CO23RFLC | 49.53 | 74.8 | 18.40 |
Expressed as a percentage of cell dry weight and measured by the method of Dubois et al.
Expressed as a percentage of cell dry weight.
Expressed as a percentage of alkali-acid-insoluble cell wall material/milligram of cell wall dry weight.
We also analyzed the presence and distribution of a mannoside epitope common to major MP components, as a potential indicator of cell wall changes, by means of immunogold labeling with the specific monoclonal antibody AF1 (6). As shown in Fig. 2, the cells of the parent strain revealed the expected intense and uniform labeling of the entire cell wall profile, with numerous gold particles randomly spanning cell wall layers. In contrast, the gold particles were less numerous and irregularly distributed throughout the cell walls of both resistant strains, particularly in the FK-resistant one. All of this suggests that the deposition of the MP component and its organization within wall layers were changed in the drug-resistant strains. Figure 2 also shows the reduced thickness of the cell walls of drug-resistant strains compared to the cell wall of the drug-susceptible strain, in line with the reduced proportion of the alkali-acid-insoluble material but in an apparent contrast with the data on total cell wall amounts of these strains.
FIG. 2.
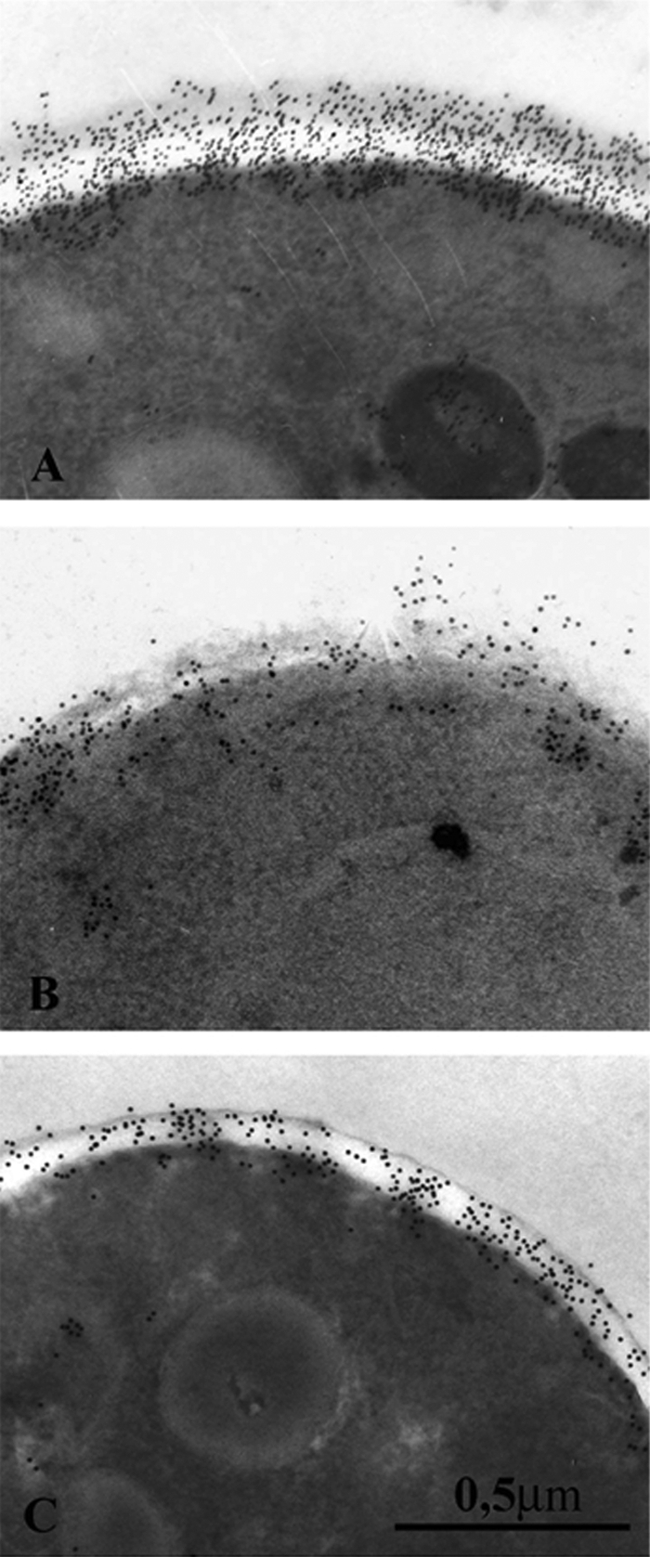
EM localization of MP constituents in freeze-substituted yeast cells of C. albicans strains CO23S (A), CO23RFK (B), and CO23RFLC (C) following postembedding labeling with the MAb AF1 followed by gold-labeled secondary antibody. For technical details, see the text.
Molecular analyses of drug resistance-related genes in CO23RFLC and CO23RFK strains.
To investigate the molecular mechanisms of the acquisition of resistance to FLC and FK by the C. albicans strains CO23RFLC and CO23RFK, the expression of the CDR1, CDR2, MDR1, and FLU1 genes, encoding efflux pumps known to be associated to azole resistance (4, 31, 37), and of the ERG11 (31) (Table 2), and mutations in the FSK1 (2) genes was analyzed. Table 3 shows the mRNA expression levels of the target genes normalized against the TEF3 gene as in-house transcript. As expected, the expression of efflux pump genes was affected only in the FLC-resistant CO23RFLC, compared to the wild-type susceptible strain CO23S. In particular, CO23RFLC exhibited strongly increased expression levels only of MDR1 (33-fold), whereas slight increases were observed for CDR1, CDR2, and FLU1 (3.7-, 3.0-, and 3.9-fold, respectively). Notably, no variation that led to an amino acid substitution was found in CO23RFLC and CO23RFK ERG11 genes, which were expressed at levels similar to those of CO23S (Table 3). In contrast, the FK-resistant strain CO23RFK displayed a S645Y substitution, which was expressed as a homozygous mutation in the FKS1 gene.
TABLE 3.
Expression levels of the CDR1, CDR2, MDR1, FLU1, and ERG11 genes for the C. albicans strains studied
| Isolate | Gene expressiona
|
||||
|---|---|---|---|---|---|
| CDR1 | CDR2 | MDR1 | FLU1 | ERG11 | |
| CO23S | 9.0 ± 0.3 | 95.0 ± 13.0 | 1.0 ± 0.09 | 38.0 ± 6.0 | 25.0 ± 3.0 |
| CO23RFLC | 34.0 ± 4.0 | 285.0 ± 25.0 | 33.0 ± 1.9 | 150.0 ± 9.0 | 29.0 ± 2.0 |
| CO23RFK | 11.0 ± 4.0 | 85 ± 3.0 | 1.5 ± 0.1 | 31.0 ± 2.0 | 31.0 ± 2.0 |
Quantification was performed by real-time RT-PCR. Values are averages from four independent experiments and represent TEF3-normalized levels of expression of the target genes (see Materials and Methods).
Virulence of C. albicans as related to the susceptible/resistant phenotype.
To study whether acquisition of FK and FLC resistance of C. albicans had any consequence on pathogenicity of this fungus, we used a murine systemic infection model having as endpoints mouse mortality and organ invasion following intravenous infection, as well as an estrogen-dependent rat vaginal infection to assess mucosal virulence. In the systemic infection model, both CO23RFK and CO23RFLC were compared to the drug-susceptible parental strain CO23S in three repeated, independent experiments. Because of the absolute comparability of mortality data from the three independent experiments with no statistically significant interexperimental variability, the data were cumulated for a Kaplan-Meyer survival curve (Fig. 3A).
FIG. 3.

Experimental pathogenicity of C. albicans strains in systemic (A) and mucosal (B) infection models. (A) Male BALB/c mice were infected intravenously with the indicated C. albicans strains (inoculum size, 107 cells). Survival was determined over the time indicated. There was a statistically significant difference (P < 0.01) between the parental and each of the two drug-resistant strains. For further details, see Materials and Methods. (B) Oophorectomized, estrogen-treated rats were infected intravaginally with the indicated C. albicans strains (inoculum size, 107 cells). At the indicated time intervals, the intravaginal burden of fungal cells was measured as described in Materials and Methods. No statistically significant differences were found among the strains.
The results consistently demonstrated that CO23S caused little overall mortality in the three cumulated experiments. In contrast, 57 or 70% lethality was measured on systemic infection by mice injected with CO23RFK or CO23RFLC, respectively, with a median survival time of less than 1 week, compared with only 10% lethality of infection with the parental, drug-susceptible strain (P < 0.01, Fisher's exact test). In all three experiments, autopsies of all dead mice demonstrated deep organ invasion by the fungus, with extensive mycelial growth in target organs (kidney and heart; data not shown). In another independent experiment, where a sublethal inoculum size (105 cells) was used and mice were all sacrificed on day 2 postchallenge, all mice (three per group) infected by the two drug-resistant strains had >104 fungal cells/kidney, whereas <102 cells were recovered from the kidney of the animals administered with the parental strain.
The virulence of the three strains in a mucosal model of candidiasis (rat vaginal model) was also assessed. In two independent experiments, the three strains showed almost overlapping kinetics of vaginal infection, with comparable times of spontaneous healing of the infection. Thus, no virulence increase was shown by the two drug-resistant strains in the above model (Fig. 3B).
In vitro mycelial morphology.
The results above demonstrated that acquisition of resistance to both FK463 and FLC was associated with a selective increase in the experimental pathogenicity for systemic infection of an originally poorly virulent strain of C. albicans in the above setting.
Since (i) the “in vivo” data showed massive hypha formation in the organs (kidney and heart) of mice injected with the drug-resistant strains, and (ii) hypha formation is a major virulence trait of the fungus, additional experiments were carried out to assess the capacity of each strain to produce hyphae in vitro. As shown in Fig. 4A, each strain was capable of producing hyphal filaments when incubated in liquid synthetic (N-acetyl-glucosamine-based) or in rich serum-based medium. However, the hyphae produced in both media by the drug-resistant cells after 24 h incubation were clearly more numerous and longer than those generated by the drug-susceptible parent strain. The three strains were also tested for agar penetration capacity, a property related to hypha formation, by spotting them onto the surface of plates containing agar concentrations ranging from 0.8 to 3% to provide various levels of resistance to fungal penetration. Upon observation at day 7 of culture, agar invasion by the drug-susceptible strain was completely absent beyond a slight border of the original colony, while the drug-resistant strains showed long branching filaments radiating into the agar from the cells on the surface. These resistant strains displayed significantly longer and more tangled hyphal growths on the colony periphery (Fig. 4B).
FIG. 4.

Hyphae and invasive hyphal growth of C. albicans strains CO23S, CO23RFK, and CO23RFLC. (A) Cells were induced to form hyphae by 24-h incubation at 37°C in YNB medium containing 0.1% (wt/vol) N-acetyl-d-glucosamine (line 1) or RPMI 1640 medium containing 10% fetal calf medium (line 2). Photomicrographs were taken with a phase contrast microscope using a 40× objective and are representative of 50% random fields observed. (B) Cells (104) of CO23S, CO23RFK, and CO23RFLC strains were suspended in 1 μl of YPD broth and spotted onto the surface of 4% serum plates containing 0.8 to 3% agar, and the plates were incubated at 37°C. Line 1, top view of spot colonies at 7 days of incubation on 0.8% agar using a 10× objective. Line 2, images of spot colony edges at 7 days of incubation on 3% agar using a 40× objective. The photographs are representative of the whole-mounted microscopic plates.
Adherence and biofilm formation.
It is well known that hyphae of C. albicans are more adhesive and tissue invasive than their yeast cell counterpart (41). Thus, from the data above, variations in adherence by the drug-resistant strains could be anticipated. We previously examined the adherence properties of the three strains to plastic surfaces, as this is an important step in the development of biofilms and may contribute to the virulence properties of the organism. As shown in Fig. 5A, there was a significant difference in the adherence to plastic among the strains, with almost twice as many adherent cells being found in both drug-resistant strains than in the control. We then examined biofilm formation by the adherent cells following incubation in fresh medium for up to 24 h. Biofilms were formed by both the mutant and the wild type, but the biofilm produced by the drug-resistant cells was more abundant than that produced by the susceptible strain, as consistently shown by microscopic observations of growth in agar plates (Fig. 5B) and by the capacity to reduce tetrazolium salt (Fig. 5C).
FIG. 5.

Adherence, biofilm, and XTT assay of C. albicans strains CO23S, CO23RFK, and CO23RFLC. (A) Percentage of plastic adherent cells of the strains CO23S, CO23RFK, and CO23RFLC. The cells were allowed to adhere to the polystyrene surface, and then 1 ml of Sabouraud dextrose agar, which was allowed to solidify, was poured onto the cells, and the mixture was incubated at 37°C for 24 h. (B) Production of biofilm on polystyrene surfaces. Biofilms were stained with crystal violet and photographed at 10× and 40× magnifications using an inverted microscope. (C) Equal numbers of cells from CO23S, CO23RFK, and CO23RFLC were suspended in 1 ml of RPMI medium and incubated in 12-well plates for the times indicated. Nonadherent cells were then removed by washing, and adherent cells were XTT assayed. Experiments were repeated three times with similar results.
DISCUSSION
In this study, we addressed the poorly investigated relationship between ADR and virulence of the human opportunistic pathogen C. albicans. ADR, in particular azole resistance, of this organism has become a clinically relevant issue (38), and most studies on the topic have been devoted to investigating its mechanisms (32). A wealth of studies have also addressed virulence trait expression in this fungus. There is substantial agreement that adherence properties and the capacity to switch the growth morphology from yeast to the mycelium, which are possibly interrelated, play a substantial role for host tissue invasion (12). Moreover, the propensity for biofilm formation has recently been shown to have an impact on both pathogenicity and drug resistance (30). While numerous reports have elucidated mechanisms of drug resistance and its evolution in Candida and, particularly, its relevance for fungus fitness in vitro (9, 11), very little is known about the potential consequences of ADR acquisition on virulence expression in vitro and in vivo.
In the absence of consistent reports, an implicit assumption circulates that, in analogy to most cases of antibiotic resistance in bacteria, ADR would not influence, or rather would decrease, the fungus virulence, and is possibly associated with changes in the fitness cost of resistance. Our expectation was similar, since pathogens showing a higher MIC of a drug are assumed to be less fit in vivo in the absence of the drug, as under the conditions of our experiments (10). In this line, Kurtz et al. (24) reported that the virulence of spontaneous or induced echinocandin-resistant mutants of C. albicans was either unimpaired or greatly reduced compared to the parental strain. In addition, the echinocandin-resistant mutant of C. albicans (CA2) has a very low pathogenicity in a systemic infection (13). Interestingly, the spontaneous resistant mutant with unimpaired virulence retained full ability to make hyphae in vitro, while the CA2 mutant was agerminative (13, 24). In this case, the mutants with reduced virulence were derived by random chemical mutagenesis, and not, as in the present investigation, by direct exposure to the antimycotics, the latter situation more closely mimicking the in vivo one. Chemical mutagenesis is a procedure that may greatly alter many gene functions and is very unlikely to occur in vivo, during antimycotic therapy.
Contrary to the above expectations, we have observed here a marked increase in the virulence of C. albicans for systemic infection when the fungus acquired ADR by exposure to the drug in vitro. More puzzling, the virulence increased to a rather equal extent when the originally susceptible strain was made resistant to either FLC or FK, two drugs belonging to distinct families, with completely different biochemical mechanisms of action and, as also shown here, different mechanisms of resistance. Finally, the increase in virulence for deep-seated infection in the mouse was not paralleled by a difference in the virulence level for vaginal infection in rats, which was a self-healing superficial infection. The data suggest that (i) virulence acquisition has nothing to do with any specific drug action or resistance mechanisms (see also below), and (ii) only the factors associated with the fungal capacity to invade host tissues have been altered in the drug-resistant strains.
We have not investigated all possible mechanisms reported in the literature to be relevant for deep-seated infection by C. albicans in the normal mouse; thus, a detailed picture of the virulence increase described here must await additional investigations. However, we have shown here that among the many changes in the biological properties shown by the drug-resistant strains, there are some which are commonly considered to be virulence traits of C. albicans. They include the capacity to generate long hyphal threads, a pronounced adherence to plastic and the ability to invade agar medium, and a greater propensity to make a biofilm; these traits are possibly interrelated.
The ability to switch from yeast to hyphal growth is likely central to all the observed phenomena and probably explains them. It is a formally established virulence trait of C. albicans, since all mutants unable to form hyphae are nonpathogenic in systemic mouse models of infection (26). Hyphae are much more adhesive and much less phagocytable than yeast cells. In addition, they have evolved a number of mechanisms to evade host innate and adaptive immune responses, including the demonstrated capacity to generate nonprotective cytokine patterns (36).
While the above properties are relevant for the virulence of C. albicans, it is quite clear from our observations that the drug-resistant strains have undergone multiple changes in the cell wall, some of which might more generally affect fungal fitness in vivo (9-11). Concerning fitness in vitro, it is of interest that the growth curves of the three strains in simple, YNB-based medium, at 37 or 28°C, do not support any marked modification of fitness by the drug-resistant strains; nonetheless, the increased filamentation in vitro would suggest that both drug-resistant strains had increased their fitness in vitro, a feature that, as discussed above, is related to a more aggressive behavior in vivo. The relationship between fitness and virulence attributes clearly warrants future investigations. Cell wall changes include (i) a marked alteration in the distribution of a major cell wall component, i.e., the MP, which contains critical adhesins (37), particularly in the FK-resistant strain; and (ii) a decreased amount of alkali-acid-insoluble material, to which MP is closely linked. This material substantially corresponds to the β-glucan interwoven fibrillar constituent of the cell wall. It is bound to chitin and confers rigidity and resistance to the cell wall itself (20). The total cellular content of hexose (essentially, glucose plus mannose) is roughly similar in all strains, suggesting that some form of compensation might have occurred in the cell wall structure to account for efficient growth on simple media and aggressive behavior in vivo by the drug-resistant strains. The specific decrease of this material relative to the other cell wall constituents might account for the marked increase in the cell volume and deformation observed for the resistant strains, as if the cell had become more extensible. Interestingly, defective cell wall β-glucan incorporation is expected, and has indeed been reported, for echinocandin drug-resistant mutants (15), whereas it is not an obvious expectation for FLC-resistant mutants, again suggesting that the changes observed with the drug-resistant strains have nothing to do with specific antimycotic actions and targets.
On the other hand, the increased secretion of β-glucan and other cell wall, plastic constituents probably represents a counterpart to the lower incorporation of this critical polysaccharide into the cell wall, and this could contribute to explaining the increased biofilm formation. C. albicans biofilm has been recently shown to consist mostly of β-glucan, which reduced the susceptibility of C. albicans cells to FLC by binding this drug extracellularly (30). It is of some speculative interest that drug resistance confers to the fungi an increased propensity to biofilm formation, as shown here, clearly suggesting that biofilm formation may also be induced in vivo by drug treatment.
How general can the phenomenon described here, i.e., increased virulence of ADR strains, be? In this context, three facts warrant careful consideration. (i) Virulence increase was detected only in the systemic infection model, not in a mucosal one. (ii) The susceptible strain was initially isolated from an HIV-positive subject with recurrent vaginitis. In a comparison with the usual virulence of C albicans on intravenous infection of mice (50% lethal dose, around 106 cells), it is clear that this vaginophatic strain is of relatively low virulence for systemic infection (50% lethal dose, around 107 cells). The virulence gained by the resistant strains makes it roughly equivalent to the “standard” level of virulence possessed by other strains of the fungus in systemic infection. This may also hold true for the increased expression of many virulence traits by a FLC-resistant strain of C. albicans and Cryptococcus neoformans, confirming what has recently been described (17, 40). (iii) The drug resistance has been acquired by in vitro treatments, consisting of repeated passages in media with increasing drug concentrations. It is not clear to what extent this may be significant in clinical situations. Azole-resistant strains have originated from refractory, chronic mucosal C. albicans infections in HIV-positive subjects (32), and it is possible that the mechanism of their ADR is not very dissimilar from the one described here, as such subjects are usually treated with repeated cycles of antifungals, often with increased concentrations.
Despite the above caveats, it remains of concern that intrinsically low-virulence strains of C. albicans may become virulent as a consequence of ADR acquisition.
Conversely, it could be speculated that the original isolate had lost in vivo, by some unknown mechanisms, the typical virulence asset of C. albicans strains and that the in vitro passages in the presence of antimycotics brought about a recovery of the lost virulence traits. This is a distinct possibility against which, however, three observations seem to argue. First, the original isolate had been of low systemic pathogenicity since its initial isolation, ruling out the possibility that long or unusual maintenance conditions in vitro before the initiation of this investigation caused loss of virulence. Second, resistant strains were as vaginopathic in the rat model infection as the original susceptible isolate, demonstrating that the strain had retained the specific virulence traits for mucosal infection. Third, the CO23S strain was isolated from a subject with recurrent candidal vaginitis. Although no detailed clinical history of the patient is available, the continuous use of vaginal medications with cycles of antimycotics is rather common for such patients. The theory of “restoration” would assume that the strain with a “normal” pathogenicity potential suffered a reduction of this potential during in vivo therapy so as to become of low pathogenicity upon isolation. All of this is in contrast to what we describe here, i.e., increased systemic virulence with drug treatment.
While a selective loss of virulence in vivo cannot be ruled out, other adaptive and/or fitness factors related to the isolation source and the different host niche are to be considered. In this context, noteworthy is the remarkable difference detected between strains of C. parapsilosis isolated from vaginitis patients and the isolates of the same fungus from patients with candidemia: the former were more pathogenic in the rat vaginal infection model, while the latter were more pathogenic in the systemic mouse infection model (7).
Finally, also noteworthy is the observation that both FK463 and FLC-resistant strains gained equal virulence upon resistance acquisition, demonstrating that the increase in virulence is independent of the mechanism of drug action and resistance itself. This is well in line with the changes in glucan-associated proteins of the cell wall which, as shown elsewhere (1), are also independent of the specific drug action. Overall, it appears that the stress of resistance acquisition upon prolonged, dosage-increasing treatments with antifungals may induce biological compensatory changes which, by unknown mechanisms, could select for acquisition by C. albicans some advantages in terms of fitness and capacity to attack the host systemically. Whatever the explanation, our data invite further studies of other well-characterized drug-resistant strains of C. albicans to resolve this important clinical issue.
Footnotes
Published ahead of print on 7 January 2008.
REFERENCES
- 1.Angiolella, L., B. Maras, A. R. Stringaro, G. Arancia, F. Mondello, A. Girolamo, A. T. Palamara, and A. Cassone. 2005. Glucan-associated protein modulations and ultrastructural changes of the cell wall in Candida albicans treated with micafungin, a water-soluble, lipopeptide antimycotic. J. Chemother. 17:409-416. [DOI] [PubMed] [Google Scholar]
- 2.Balashov, S. V., S. Park, and D. S. Perlin. 2006. Assessing resistance to the echinocandin antifungal drug caspofungin in Candida albicans by profiling mutations in FSK1. Antimicrob. Agents Chemother. 50:2058-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bastert, J., M. Schaller, H. C. Korting, and E. G. V. Evans. 2001. Current and future approaches to antimycotic treatment in the era of resistant fungi and immunocompromised hosts. Int. J. Antimicrob. Agents 17:81-89. [DOI] [PubMed] [Google Scholar]
- 4.Calabrese, D., J. Bille, and D. Sanglard. 2000. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology 146:2743-2754. [DOI] [PubMed] [Google Scholar]
- 5.Calderone, R. A., and P. C. Braun. 1991. Adherence and receptor relationships of Candida albicans. Microbiol. Res. 55:1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cassone, A., A. Torosantucci, M. Boccanera, G. Pellegrini, C. Palma, and F. Malavasi. 1988. Production and characterization of a monoclonal antibody to a cell surface, glucomannoprotein constituents of Candida albicans and other pathogenic Candida species. J. Med. Microbiol. 27:233-238. [DOI] [PubMed] [Google Scholar]
- 7.Cassone, A., F. De Bernardis, E. Pontieri, G. Carruba, C. Girmenia, P. Martino, M. Fernandez-Rodriguez, G. Quindos, and J. Ponton. 1995. Biotype diversity of Candida parapsilosis and its relationship to the clinical source and experimental pathogenicity. J. Infect. Dis. 171:967-975. [DOI] [PubMed] [Google Scholar]
- 8.Chaffin, W. L., J. L. Lopez-Ribot, M. Casanova, D. Gozalbo, and J. P. Martinez. 1998. Cell wall and secreted proteins of Candida albicans. Identification, functions and expression. Microbiol. Mol. Biol. Rev. 62:130-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowen, L. E., L. M. Kohn, and J. B. Anderson. 2001. Divergence in fitness and evolution of drug resistance in experimental populations of Candida albicans. J. Bacteriol. 183:2971-2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cowen, L. E., J. B. Anderson, and L. M. Kohn. 2002. Evolution of drug resistance in Candida albicans. Annu. Rev. Microbiol. 56:139-165. [DOI] [PubMed] [Google Scholar]
- 11.Cowen, L. E., A. E. Carpenter, O. Matangkasombut, G. R. Fink, and S. Lindquist. 2006. Genetic architecture of Hsp90-dependent drug resistance. Eukaryot. Cell 5:2184-2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cutler, J. E. 1991. Putative virulence factor of Candida albicans. Annu. Rev. Microbiol. 45:187-218. [DOI] [PubMed] [Google Scholar]
- 13.De Bernardis, F., D. Adriani, R. Lorenzini, E. Pontieri, G. Carruba, and A. Cassone. 1993. Filamentous growth and elevated vaginopathic potential of a nongerminative variant of Candida albicans expressing low virulence in systemic infection. Infect. Immun. 61:1500-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Bernardis, F., R. Lorenzini, and A. Cassone. 1999. Rat model of Candida vaginal infection, p. 735-740. In Handbook of animal models of infection. Academic Press, New York, NY.
- 15.Dijkgraaf, G. J., M. Abe, Y. Ohyab, and H. Bussey. 2002. Mutations in FSK1p affect the cell wall content of beta-1,3- and beta-1,6-glucan in Saccharomyces cerevisiae. Yeast 15:671-690. [DOI] [PubMed] [Google Scholar]
- 16.Dubois, M., K. A. Gilles, J. K. Hamilton, P. A. Rebers, and F. Smith. 1956. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28:350-356. [Google Scholar]
- 17.Fekete-Forgacs, K., L. Gyure, and B. Lenkey. 2000. Changes of virulence factors accompanying the phenomenon of induced fluconazole resistance in Candida albicans. Mycoses 43:273-279. [DOI] [PubMed] [Google Scholar]
- 18.Fleet, G. H., and D. J. Manners. 1976. Isolation and composition of an alkali-soluble glucan from the cell wall of Saccharomyces cerevisiae. J. Gen. Microbiol. 94:80-92. [DOI] [PubMed] [Google Scholar]
- 19.Fleet, G. H., and D. J. Manners. 1977. The enzymatic degradation of an alkali-soluble glucan from the cell walls of Saccharomyces cerevisiae. J. Gen. Microbiol. 98:315-345. [DOI] [PubMed] [Google Scholar]
- 20.Gow, N. A., A. J. Brown, and F. C. Odds. 2002. Fungal morphogenesis and host invasion. Curr. Opin. Microbiol. 5:366-371. [DOI] [PubMed] [Google Scholar]
- 21.Jabra-Rizk, M. A., W. A. Falkler, Jr., W. G. Merz, A. A. M. A. Baqui, J. I. Kelley, and T. E. Meiller. 2001. Cell surface hydrophobicity-associated adherence of Candida dubliniensis to human buccal epithelial cells. Rev. Iberoam. Micol. 18:17-22. [PubMed] [Google Scholar]
- 22.Kelly, M. T., D. M. MacCallum, S. D. Clancy, F. C. Odds, A. J. P. Brown, and G. Butler. 2004. The Candida albicans CaACE2 gene affects morphogenesis, adherence and virulence. Mol. Microbiol. 53:969-983. [DOI] [PubMed] [Google Scholar]
- 23.Kumamoto, C. A. 2002. Candida biofilms. Curr. Opin. Microbiol. 5:608-611. [DOI] [PubMed] [Google Scholar]
- 24.Kurtz, M. B., G. Abruzzo, A. Flattery, K. Bartizal, J. A. Marrinan, W. Li, J. Milligan, K. Nollstadt, and C. M. Douglas. 1996. Characterization of echinocandin-resistant mutants of Candida albicans: genetic, biochemical, and virulence studies. Infect. Immun. 64:3244-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurtz, M. B., and C. M. Douglas. 1997. Lipopeptide inhibitors of glucan synthase. J. Med. Vet. Mycol. 35:79-86. [DOI] [PubMed] [Google Scholar]
- 26.Lo, H. J., J. R. Kohler, B. DiDomenico, D. Loebenberg, A. Cacciapuoti, and G. R. Fink. 1997. Nonfilamentous C. albicans mutants are avirulent. Cell 90:939-949. [DOI] [PubMed] [Google Scholar]
- 27.Martinez, M., J. L. López-Ribot, W. R. Kirkpatrick, B. J. Coco, S. P. Bachmann, and T. F. Pattersons. 2002. Replacement of Candida albicans and C. dubliniensis in human immunodeficiency virus-infected patients with oropharyngeal candidiasis treated with fluconazole. J. Clin. Microbiol. 40:3135-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Lopez, R., H. Park, C. L. Meyers, C. Gil, and S. G. Filler. 2006. Candida albicans Ecm33p is important for normal cell wall architecture and interactions with host cell. Eukaryot. Cell 5:140-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meijerink, J., C. Mandingers, L. van de Locht, E. Tonnissen, F. Goodsaid, and J. Raemaekers. 2001. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 3:55-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nett J., L. Lincoln, K. Marchillo, R. Massey, K. Holoyda, B. Hoff, M. VanHandel, and D. Andes. 2007. Putative role of β-1,3 glucans in Candida albicans biofilm resistance. Antimicrob. Agents Chemother. 51:510-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perea, S., J. L. López-Ribot, W. R. Kirkpatrick, R. K. McAtee, R. A. Santillán, M. Martínez, D. Calabrese, D. Sanglard, and T. F. Patterson. 2001. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 45:2676-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perea, S., and T. F. Patterson. 2002. Antifungal resistance in pathogenic fungi. Clin. Infect. Dis. 35:1073-1080. [DOI] [PubMed] [Google Scholar]
- 33.Pfaller, M. A., D. J. Diekema, R. N. Jones, H. S. Sader, A. C. Fluit, R. J. Hollis, S. A. Messer, and the SENTRY Participant Group. 2001. International surveillance of bloodstream infections due to Candida species: frequency of occurrence and in vitro susceptibilities to fluconazole, ravuconazole, and voriconazole of isolates collected from 1997 through 1999 in the SENTRY antimicrobial surveillance program. J. Clin. Microbiol. 39:3254-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramage, G., K. V. Walle, B. L. Wickes, and J. L. López-Ribot. 2001. Standardized method for in vitro antifungal susceptibility testing of Candida albicans biofilms. Antimicrob. Agents Chemother. 45:2475-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynolds, T. B., and G. R. Fink. 2001. Bakers' yeast, a model for fungal biofilm formation. Science 291:878-881. [DOI] [PubMed] [Google Scholar]
- 36.Romani, L. 2004. Immunity to fungal infections. Nat. Rev. Immunol. 4:1-23. [DOI] [PubMed] [Google Scholar]
- 37.Sandini, S., R. La Valle, F. De Bernardis, C. Macrì, and A. Cassone. 2007. The 65 kDa mannoprotein gene of Candida albicans encodes a putative beta-glucanase adhesion required for hyphal morphogenesis and experimental pathogenicity. Cell. Microbiol. 9:1223-1238. [DOI] [PubMed] [Google Scholar]
- 38.Sanglard, D., F. Ischer, M. Monod, and J. Bille. 1997. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 143:405-416. [DOI] [PubMed] [Google Scholar]
- 39.Sanguinetti, M., B. Posteraro, B. Fiori, S. Ranno, R. Torelli, and G. Fadda. 2005. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob. Agents Chemother. 49:668-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanguinetti, M., B. Posteraro, M. La Sorda, R. Torelli, R. Santangelo, G. Delogu, and G. Fadda. 2006. Role of AFR1, an ABC transporter-encoding gene, in the in vivo response to fluconazole and virulence of Cryptococcus neoformans. Infect. Immun. 74:1352-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sudstrom, P. 2002. Adhesion in Candida spp. Cell. Microbiol. 4:461-469. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan, D. J., G. P. Moran, E. Pinjon, A. Al-Mosaid, C. Stokes, C. Vaughan, and D. Coleman. 2004. Comparison of the epidemiology, drug resistance mechanisms, and virulence of Candida dubliniensis and Candida albicans. FEMS Yeast Res. 4:369-376. [DOI] [PubMed] [Google Scholar]