

Abstract
Many bacterial toxins target small Rho GTPases in order to manipulate the actin cytoskeleton. The depolymerization of the actin cytoskeleton by the Vibrio cholerae RTX toxin was previously identified to be due to the unique mechanism of covalent actin cross-linking. However, identification and subsequent deletion of the actin cross-linking domain within the RTX toxin revealed that this toxin has an additional cell rounding activity. In this study, we identified that the multifunctional RTX toxin also disrupts the actin cytoskeleton by causing the inactivation of small Rho GTPases, Rho, Rac and Cdc42. Inactivation of Rho by RTX was reversible in the presence of cycloheximide and by treatment of cells with CNF1 to constitutively activate Rho. These data suggest that RTX targets Rho GTPase regulation rather than affecting Rho GTPase directly. A novel 548 amino acid region of RTX was identified to be responsible for the toxin-induced inactivation of the Rho GTPases. This domain did not carry GAP or phosphatase activities. Overall, these data show that the RTX toxin reversibly inactivates Rho GTPases by a mechanism distinct from other Rho modifying bacterial toxins.
Keywords: Rho GTPase, Vibrio cholerae RTX toxin, actin, Lethal Factor, Cytotoxic Necrotizing Factor
Introduction
Vibrio cholerae is a Gram-negative bacterium that causes the diarrheal disease cholera. The bacterium colonizes the intestinal epithelium after the consumption of contaminated food or water. After colonization, V. cholerae elicits disease through the secretion of enterotoxins. The major virulence factor, the ADP-ribosylating cholera toxin (CT), is responsible for the profuse watery diarrhea associated with the disease (Kaper 1995). However, both clinical isolates and genetically modified live attenuated vaccine strains of V. cholerae that lack the ctx genes have been shown to cause mild diarrheal disease (Satchell, 2003). Other accessory toxins secreted by V. cholerae - the hemagglutinin protease, hemolysin, and the RTX (repeats-in-toxin) toxin - are hypothesized to be coordinately contributing to this milder form of cholera disease (Fullner et al., 2002; Haines et al., 2005)
The V. cholerae RTX toxin is a predicted 484,000 Da bacterial protein that is expressed by El Tor O1 and O139 V. cholerae strains responsible for the current cholera pandemic (Lin et al., 1999; Chow et al., 2001). This toxin is also expressed by non-O1/non-O139 isolates, and is believed to contribute to the emergence of these strains as pathogens (Dalsgaard et al., 2001; Faruque et al., 2004). Although its role in pathogenesis remains unclear, the RTX toxin is associated with the stimulation of the proinflammatory immune response during acute infection, and therefore may contribute to the reactogenicity of live attenuated vaccine strains (Fullner et al., 2002; Satchell, 2003; Haines et al., 2005).
Characterization of the RTX toxin revealed that it is distinct from other members of the RTX family of toxins. First, the V. cholerae RTX toxin is secreted from the bacterium by an atypical Type 1 secretion system that requires two distinct transport ATPases for export of the toxin (Boardman and Satchell, 2004). In addition, approximately one-quarter of this large toxin is comprised of 18-20 amino acid glycine rich repeats that are localized at the extreme N- and C-terminus of RTX as opposed to the C-terminal nonapeptide repeats of most RTX toxins (Lin et al., 1999). These repeat regions are hypothesized to be important for toxin binding and translocation into cells, while the internal portion of the toxin is thought to carry cytopathic activities (Sheahan et al., 2004). Also unlike other RTX toxins, the V. cholerae RTX toxin is not a pore-forming toxin, but rather causes rounding of a broad range of cells grown in culture due to the depolymerization of actin stress fibers (Lin et al., 1999; Fullner and Mekalanos, 2000). This depolymerization of actin occurs by a mechanism unique to this toxin, resulting in the formation of covalently cross-linked actin dimers, trimers and higher order multimers (Fullner and Mekalanos, 2000). The actin cross-linking activity has been mapped to a 47.8 kDa domain within the RTX toxin located at amino acids 1963-2419. This actin cross-linking domain (ACD) induced both cell rounding and the cross-linking of actin when expressed as a transgene in eukaryotic cells (Sheahan et al., 2004). Subsequent studies demonstrated that purified ACD was sufficient to induce cell rounding and cross-linking of purified G-actin when delivered to the cell cytoplasm and directly catalyzed the cross-linking of purified actin in vitro in the absence of other host cell factors (Cordero et al., in press)
An in-frame deletion of the coding region for the ACD within the rtxA gene (RTXΔACD) on the V. cholerae chromosome eliminated actin cross-linking activity, further confirming that the ACD carries the cross-linking activity. Unexpectedly, cells treated with V. cholerae expressing RTXΔACD still rounded. However, this RTXΔACD-induced rounding was slower, occurring after 3 hours of incubation as compared to the rounding typically observed after 75-90 minutes in the presence of actin cross-linking. These rounded cells were also phenotypically distinct from cells incubated with wild-type toxin in that the cells were not as spherical and were less refractive to light. These results revealed that RTX is a multi-functional toxin that causes cell rounding by at least two distinct mechanisms (Sheahan et al., 2004).
In the current study, we further investigated the mechanism of cell rounding in the absence of actin cross-linking. We found that, both in the presence and absence of actin cross-linking, the RTX toxin was able to induce the depolymerization of the actin cytoskeleton through the inactivation of Rho, Rac, and Cdc42. These three proteins are extensively characterized members of the Rho GTPase family that are involved in the formation of stress fibers, lamellipodia and filopodia, respectively. These GTPases cycle between an active membrane-localized GTP-bound state and an inactive GDP-bound state. The activation state is regulated by guanine nucleotide exchange factors (GEFs) that mediate exchange of GDP for GTP and GTPase activating proteins (GAPs) that stimulate the GTPase activity of the Rho family proteins. The inactive GDP-bound form can also be sequestered in the cytosol in complex with guanine nucleotide dissociation inhibitors (GDI), adding an additional level of regulation for GTPase activation (Hall and Nobes, 2000).
Due to the critical role of the Rho family GTPases in many cellular processes, numerous bacterial toxins have adapted different mechanisms to target these signaling proteins. There are four general mechanisms utilized by bacterial toxins for inactivation of Rho family GTPases: 1) covalent modification by attachment of sugar moieties that block the ability of Rho, Rac, and Cdc42 to be activated by GEFs (Chardin et al., 1989; Just et al., 1995; Sehr et al., 1998); 2) cleavage of C-terminal isoprenyl groups that facilitate interaction of Rho, Rac, and Cdc42 with the membrane (Shao et al., 2002); 3) mimicry of GAPs to stimulate the inherent GTPase activity of Rho, Rac, and Cdc42 (Goehring et al., 1999; Black and Bliska, 2000; Krall et al., 2000); and 4) dephosphorylation of focal adhesion proteins disrupting upstream signaling that could indirectly downregulating Rho GTPase activity (Persson et al., 1997; Schoenwaelder and Burridge, 1999). Our studies, specifically focused on Rho, demonstrate that the RTX toxin does not use a mechanism similar to other bacterial toxins to inactivate Rho, suggesting it employs a novel mechanism for Rho GTPase inactivation.
Results
RTX depolymerizes actin in the absence of actin cross-linking activity
Previous studies showed that the V. cholerae RTX toxin can induce cell rounding both in the presence and absence of actin cross-linking. The cell rounding induced by RTXΔACD, as well as by wild-type toxin, is not a result of any pore forming activity of the toxin causing cell lysis. (Sheahan et al., 2004). A mechanism by which numerous bacterial toxins round cells without directly affecting cell viability or membrane integrity is to induce depolymerization of actin stress fibers. To test if RTXΔACD affects the state of actin polymerization, human laryngeal epithelial cells (HEp-2) incubated with V. cholerae for 4 hours were fixed and stained with TRITC-labeled phalloidin to visualize F-actin by fluorescence microscopy. As shown in Figure 1, control cells incubated with a V. cholerae strain with a null mutation in rtxA (RTX-) or mock treated with phosphate buffered saline (PBS) exhibited a normal cuboidal shape with stress fibers visibly crossing the cell, filopodia extending from the cells, and stress fibers and lamellipodia present at the edges. By contrast, cells incubated with V. cholerae secreting wild-type toxin (RTX+) were rounded with little or no staining of actin observed, which is consistent with 100% cross-linking of actin in these cells by 3 hours (Fullner and Mekalanos, 2000). Actin in cells treated with the RTXΔACD-expressing V. cholerae strain appeared clumped and condensed with no distinct actin structures, demonstrating that cell rounding in response to RTXΔACD was due to actin depolymerization.
Figure 1.

RTX causes depolymerization of actin in the absence of actin cross-linking. HEp-2 cells were incubated with either PBS or V. cholerae strains with a null mutation in the rtxA gene (RTX-), a wild type rtxA gene (RTX+), or an rtxA gene with an in-frame deletion of the ACD (RTXΔACD) for 4 hours at 37°C at a multiplicity of infection (M.O.I) of 200. Cells were then fixed with 4% parafomaldehyde, permeabilized with 0.1% Triton X-100, and stained with TRITC-phalloidin. Scale bar represents 10 μm.
Constitutive activation of the Rho GTPases by CNF1 prevents RTXΔACD- induced cell rounding
Bacterial toxins that cause actin depolymerization either target actin assembly directly through modifications of actin or indirectly by targeting the small GTPase regulatory pathways (Aktories and Barbieri, 2005). Cytotoxic necrotizing factor (CNF1) from E. coli modifies the GTPase proteins Rho, Rac and Cdc42 by deamidation of Gln43 (Schmidt et al., 1997; Lerm et al., 1999). This modification causes constitutive activation of these proteins by inhibiting GTPase activity, even in the presence of GAPs, locking Rho GTPases in an active state (Aktories and Barbieri, 2005). Constitutive activation of the Rho GTPases can counteract the activity of most toxins that inhibit Rho GTPases, but has no effect on toxins that target actin directly (Fiorentini et al., 1995).
To investigate whether constitutive activation of the Rho GTPases could prevent RTXΔACD induced cell rounding, HEp-2 cells were pretreated with CNF1 for 1 hour followed by incubation with either the RTX- or RTXΔACD V. cholerae strains for 4 hours. Cells were then fixed and stained with TRITC-labeled phalloidin to observe the actin stress fibers. Constitutive activation of the Rho GTPases by CNF1 caused an increase in stress fiber formation as well as membrane ruffling in control cells (Figure 2). Pretreatment of HEp-2 cells with CNF1 prevented the cell rounding and actin depolymerization associated with RTXΔACD compared to cells incubated with RTXΔACD alone (Figure 2). These data indicated that RTXΔACD functions through the Rho GTPase signaling pathways and does not directly target actin.
Figure 2.
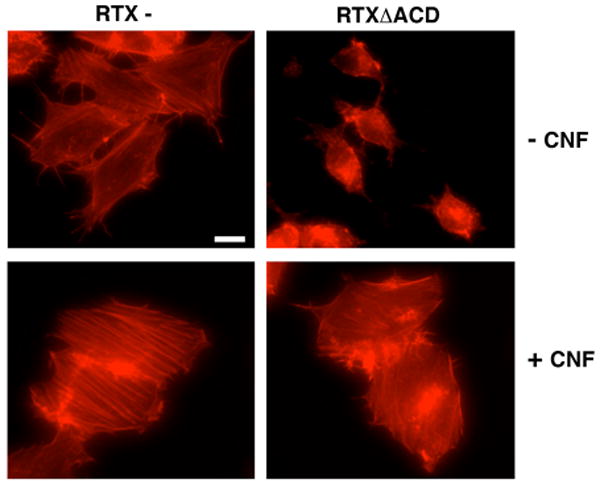
Constitutive activation of the Rho GTPases prevents RTXΔACD induced cell rounding. HEp-2 cells were pretreated with 0.9 mg/mL CNF1 for 1 hour before incubation with the V. cholerae strains with an rtxA mutant (RTX-) or an rtxA gene with an in-frame deletion of the ACD (RTXΔACD) for 4 hours at an M.O.I of 200. All cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton, and stained with TRITC-phalloidin before observation by fluorescence microscopy. Scale bar represents 15 μm.
RTX inactivates the small GTPase Rho
To determine whether RTXΔACD-induced cell rounding is due to the inactivation of the Rho GTPases, an affinity precipitation assay was performed to assess whether the RTX toxin causes a decrease in the level of active Rho-GTP in cells (Ren et al., 1999). HEp-2 cells were incubated with various V. cholerae strains as a source of RTX toxin at a multiplicity of infection (M.O.I.) of 200. Four hours following addition of bacteria, cells were collected and cell lysates were incubated with a glutathione-S-transferase fusion of the Rho binding domain (GST-RBD) from Rhotekin bound to glutathione agarose. GST-RBD specifically interacts with the active GTP-bound form of Rho in the cell lysate such that Rho-GTP, but not inactive Rho-GDP, is pelleted by centrifugation when bound to the GST-RBD agarose beads. Total Rho and Rho-GTP levels were then determined by Western blotting with an anti-RhoA antibody.
As shown in Figure 3A, active Rho-GTP was precipitated by GST-RBD from lysates of HEp-2 cells that were incubated with either PBS or an rtxA null strain (RTX-) of V. cholerae indicating that active Rho-GTP is normally present in HEp-2 cells. However, GST-RBD added to lysates from HEp-2 cells incubated with either the V. cholerae strain secreting wild-type toxin (RTX+), or RTXΔACD did not precipitate any detectable Rho-GTP. Although exposure to the RTX toxin had an effect on the activation state of RhoA (Figure 3A, upper panel), there was no effect on the amount of total RhoA within these cell lysates (Figure 3A, lower panel). Similar studies were performed using a pan-specific Rho antibody that recognizes RhoA, RhoB, and RhoC, demonstrating that all forms of Rho were inactivated in toxin-treated cells (data not shown). From this experiment, we concluded that the RTX toxin inactivates Rho, and that this activity is independent of actin cross-linking catalyzed by the ACD.
Figure 3.
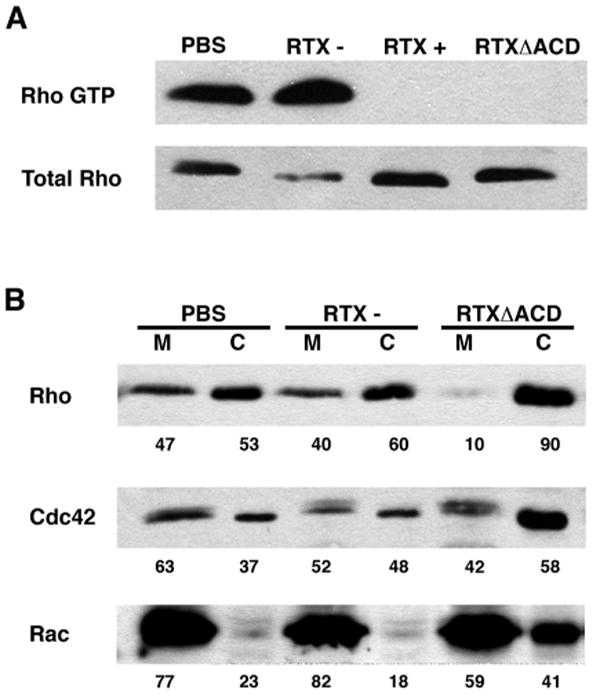
RTX inactivates small Rho GTPases. HEp-2 cells were incubated with either PBS, a V. cholerae strain with a wild type rtxA gene (RTX+), an rtxA mutant (RTX-), or an rtxA gene with an in-frame deletion in the ACD (RTXΔACD) for 4 hours at an M.O.I. of 200. (A) Cells were lysed and the active Rho was precipitated using GST-RBD coated agarose beads. Rho-GTP and total Rho levels were detected by Western blotting with an anti-RhoA antibody. (B) Cells were lysed and fractionated. Equivalent membrane (M) and cytosolic (C) fractions were subjected to SDS-PAGE followed by Western blotting with anti-RhoA, anti-Cdc42, and anti-Rac antibodies. Numbers represent the percentage of each protein in the membrane verses cytosolic fractions determined by densitometric analysis.
RTXΔACD causes the relocalization of Rho, Rac and Cdc42
Rho GTPases are localized at the membrane when in their active GTP-bound conformation, while inactive GDP-bound forms can be sequestered in the cytosol by Rho-GDI proteins (Krall et al., 2002). To further confirm that RTXΔACD inactivates Rho, subcellular fractionation experiments were performed to determine whether the toxin causes the relocalization of Rho to the cytosol. After incubation with V. cholerae, HEp-2 cells were collected and lysates were centrifuged at 100,000g to separate the membrane and cytosolic fractions. Western blotting with an anti-RhoA antibody revealed that RhoA was equally distributed between the membrane and cytosolic fractions of HEp-2 cells that were incubated with either PBS or an rtxA null strain of V. cholerae (RTX-) (Figure 3B). However, exposure of HEp-2 cells to the RTXΔACD toxin relocalized the Rho GTPase such that 90% of RhoA is present in the cytosolic fraction (Figure 3B), further demonstrating that Rho is inactivated in RTXΔACD treated cells.
Rac and Cdc42 are two other predominant members of the small Rho GTPase family that regulate actin dynamics. To determine if RTXΔACD specifically targets Rho or globally targets this family of small GTPases, the subcellular localization of Rac and Cdc42 was determined. Western blotting of membrane and cytosolic fractions with anti-Rac and anti-Cdc42 revealed that RTXΔACD redistributed approximately 20% of Rac from the membrane to the cytosol (Figure 3B). A slight, but reproducible relocalization was detected for Cdc42 (Figure 3B). From these experiments, we conclude that the RTXΔACD toxin causes the inactivation of Rho, Rac and Cdc42 with the most dramatic effect on Rho. Therefore, subsequent studies were focused on the mechanism of Rho inactivation.
RTXΔACD inactivation of Rho GTPase is reversible
Bacterial toxins have adapted many well-characterized mechanisms to target the Rho GTPases (Aktories and Barbieri, 2005). To identify the mechanism by which the RTX toxin inactivates the GTPase Rho, experiments were designed to determine whether RTX utilizes a known mechanism analogous to other bacterial toxins that target Rho. If the RTX toxin inactivates Rho in a similar manner to the Clostridial glucosyltransferase Toxins A (TcdA) and B (TcdB) or the cysteine protease YopT from Yersinia spp., constitutive activation of the Rho GTPases should not reverse the cell rounding induced by the toxin (Fiorentini et al., 1995; Sorg et al., 2001). To test this hypothesis, HEp-2 cells were incubated with the RTX- or RTXΔACD strains of V. cholerae for 4 hours followed by the removal of the toxin and recovery in media supplemented with gentamicin in the presence or absence of CNF1. After 4 hours of recovery, cells were fixed and stained with TRITC-labeled phalloidin to visualize the actin stress fibers. As shown in Figure 4, cells exposed to RTXΔACD recovered in the absence of CNF1 are still round with clumped and condensed actin staining. However, the cell morphology and actin cytoskeleton of cells incubated with RTXΔACD rapidly recovered in the presence of CNF1 resulting in more defined actin structures especially at the cell periphery (Figure 4). Control cells showed that the addition of CNF1 enhanced the actin stress fiber network (Figure 4). This experiment demonstrated that constitutive activation of the Rho GTPases with CNF1 enhanced the reversal of the cytotoxic effects by the RTXΔACD toxin. From this experiment, we concluded that the RTX toxin does not function similar to the Clostridial glucosyltransferase toxins that covalently modify Rho, since CNF1 can reverse the effects of RTXΔACD. In addition, since constitutively active Rho is equally susceptible to proteolysis as normal Rho, this experiment indicates that the RTX toxin does not proteolytically target the Rho GTPases in a mechanism similar to YopT.
Figure 4.
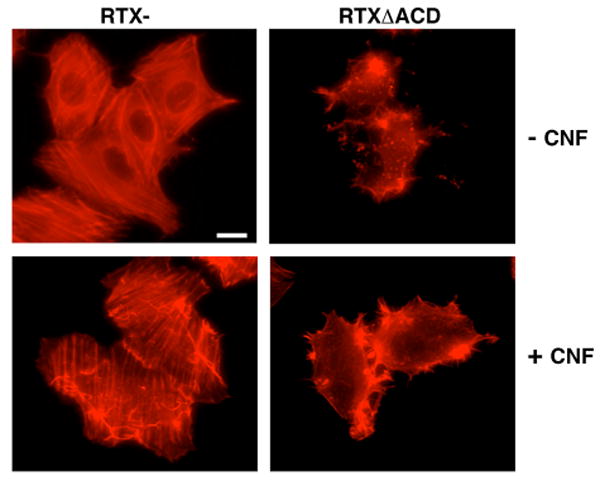
Constitutive activation of the Rho GTPases rapidly reverses RTXΔACD-induced cell rounding. HEp-2 cells were incubated for 4 hours at an M.O.I of 200 with V. cholerae strains with an rtxA mutant (RTX-) or an rtxA gene with an in-frame deletion of the ACD (RTXΔACD), washed and then incubated with gentamicin in the presence or absence of CNF1 (1.5 mg/mL) for 4 hours. All cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton and stained with TRITC-phalloidin before observation by fluorescence microscopy. Scale bar represents 15 μm.
Constitutive activation of the Rho GTPases by CNF1 rapidly reverses the destruction of the actin cytoskeleton induced by ADP-ribosylation of Rho by the C3 exoenzyme from Clostridia spp. However, this covalent modification of the GTPase Rho is irreversible and requires re-synthesis of the protein to reverse the cytotoxic effects (Barth et al., 1999). To further investigate the mechanism of RTXΔACD inactivation of Rho, the reversibility of RTXΔACD inactivation of the Rho GTPase was determined upon removal of the toxin. HEp-2 cells were incubated with the RTX- or RTXΔACD strains of V. cholerae or PBS for 4 hours. Cells were then either collected (T=0) or the culture media was removed and the cells were washed to remove the toxin-secreting bacteria. Gentamicin was added to the culture media to eliminate any remaining bacteria, and the cells were collected after 2, 4, 6, and 24 hours of recovery. Collected cells were lysed and subjected to the GST-RBD affinity precipitation assay to detect the amount of Rho-GTP. As shown in Figure 5A, active RhoA was detected within 2 hours following the removal of non-adhering bacteria. After 24 hours of recovery, the amount of Rho-GTP from cells previously incubated with RTXΔACD was equivalent to the amount of Rho-GTP detected from cells incubated with the PBS and RTX- controls. These cells exhibited a normal cell morphology, and phallodin staining showed that actin stress fibers had regenerated by 24 hours (Figure 5C). The addition of cycloheximide (CHX) to the culture medium to inhibit eukaryotic cell protein synthesis did not affect the reversal of Rho-GTP levels within the cell after exposure to RTXΔACD (Figure 5B). However, cells recovered in the presence of CHX still appeared round under the microscope and had a depolymerized actin cytoskeleton after 24 hours of recovery (Figure 5C). This phenotype was not observed in the PBS and RTX- controls recovered in CHX indicating that this cell rounding was not due to incubation with CHX (Figure 5C). Therefore, we concluded that RTX toxin inactivation of Rho is reversible, and that this reversibility does not require de novo protein synthesis. However, synthesis of other cellular components is required for cells to regain a normal morphology. The reversibility of the reaction suggests that Rho is not covalently modified by RTXΔACD; or, if Rho is targeted directly, the host cell can reverse this modification on Rho.
Figure 5.
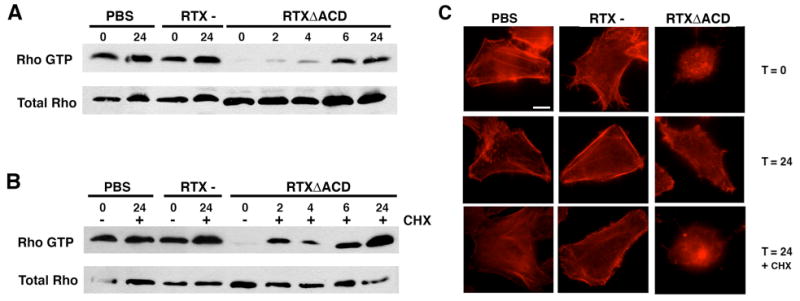
RTX inactivation of Rho is reversible. HEp-2 cells were incubated for 4 hours with V. cholerae strains with an rtxA mutant (RTX-) or a rtxA gene with an in-frame deletion of the ACD (RTXΔACD) at an M.O.I of 200. Cells were then washed to remove bacteria and allowed to recover in the presence of gentamicin (100 μg/mL) alone (A) or with CHX (20 μM)(B). Cells were then harvested at the indicated time points and the active Rho was precipitated using GST-RBD agarose beads. Western blotting with an anti-RhoA was performed to detect active Rho and total Rho in cell lysates. (C) Cells were fixed at the indicated time points with 4% paraformaldehyde, permeabilized with 0.1% Triton and stained with TRITC-phalloidin before observation by fluorescence microscopy. Scale bar represents 10 μm.
Identification of a second cell rounding domain within RTX
Further characterization of the mechanism of RTX inactivation of the Rho GTPases required the identification of the region of the toxin responsible for this activity. Previously, we identified the domain responsible for actin cross-linking through a sequence alignment analysis with the putative RTX toxin from V. vulnificus, which does not cross-link actin (Sheahan et al., 2004). Experiments revealed that the V. vulnificus RTX toxin caused transient cell rounding followed by cell lysis. The observed cell rounding correlated with Rho inactivation (our unpublished results). Therefore, a comparative sequence analysis of the V. cholerae RTX toxin with the putative RTX toxin from V. vulnificus as well as 8 other Vibrio-type RTX proteins was performed in order to identify other discrete domains of the RTX toxin. Sequence analysis identified two domains that were conserved between the V. cholerae and V. vulnificus RTX toxins (Figure 6A). These domains are located at amino acids 2552-3099 (green) and 3209-3375 (blue) within the V. cholerae RTX toxin. Each domain was cloned into the eukaryotic expression vector pEGFP-N3 and transfected into HEp-2 cells to determine whether expression of these transgenes caused cytotoxicity. Transient expression of the region encoding RTX amino acids 2552-3099 as a C-terminal EGFP fusion caused cell rounding, whereas cells transfected with EGFP alone appear similar to untransfected cells (Figure 6B). Cells transiently expressing RTX amino acids 3209-3375 looked similar to cells expressing EGFP and did not cause any cell rounding (data not shown). Since these data demonstrate that expression of RTX amino acids 2552-3099 caused cell rounding, we hypothesized that this region is responsible for the inactivation of the small Rho GTPases.
Figure 6.

Expression of a domain conserved between the V. cholerae and V. vulnificus RTX toxins causes cell rounding. (A) Schematic representation of the V. cholerae RTX (VcRTX) and V. vulnificus RTX (VvRTX) toxins depicting the conserved N- and C-terminal repeat regions (grey boxes), conserved domains (green and blue boxes) as well as the domains unique to each toxin (red and pink boxes). (B) HEp-2 cells transiently transfected with pEGFP-N3 or pRID-EGFP were observed by fluorescence microscopy at 550-575 nm to detect GFP and 440-470 nm to detect Hoechst stain. Scale bar represents 10 μm.
RTX amino acids 2552-3099 is the Rho Inactivation Domain (RID)
In order to associate this cell-rounding domain with Rho inactivation, the Rho-GTP affinity precipitation assay was performed. However, this assay is not sensitive enough to detect a change in the activation state of Rho in transiently transfected cells due to high level of Rho-GTP from untransfected cells. To address this issue, the well-characterized cellular entry mechanism of the Bacillus anthracis anthrax toxin was utilized to deliver this domain of RTX as a fusion to the N-terminal 254 amino acids of lethal factor (LFN) into the cell. Briefly, protective antigen (PA) binds to the anthrax toxin receptor, is cleaved by furin to its active form PA63 and undergoes oligomerization into a heptamer (Molloy et al., 1992; Milne et al., 1994; Bradley et al., 2001). The LFN fusion protein then binds to [PA63]7, the complex enters the cell through receptor-mediated endocytosis, and acidification of the vacuole triggers PA to form a pore and translocate the LFN fusion protein into the cytosol (Gordon et al., 1988; Koehler and Collier, 1991; Elliott et al., 2000). This method has previously been used to facilitate the entry of enzymatic domains of the bacterial toxins C. difficile Toxin B, Corynebacterium diphtheriae diptheria toxin as well as the ACD from the RTX toxin (Milne et al., 1995; Spyres et al., 2001; Cordero et al., in press). The DNA corresponding to RTX amino acids 2552-3099 was cloned into the pABII vector (Spyres et al., 2001) as an N-terminal fusion to LFN and a 6x-His tag to create the plasmid pLFNRID.
After expression in E. coli, the proteins PA, LFN, and LFNRID were purified by Ni-affinity chromatography from the soluble fraction. The eluted proteins were analyzed by SDS-PAGE to assess their purity (Figure 7A). HEp-2 cells were then incubated with LFN or LFNRID in either the presence or absence of PA at a 3:7 molar ratio, respectively (Mogridge et al., 2002). After 4 hours of incubation, cell rounding was observed with LFNRID only in the presence of PA (Figure 7B). After extensive incubation, this cell rounding was reversed suggesting that degradation of LFNRID allows the reversal of the cytotoxic effects (data not shown). This experiment indicated that this activity is similar to the rounding associated with the RTXΔACD inactivation of the Rho GTPases. Further analysis revealed that the cell rounding induced by the addition of PA and LFNRID was correlative to a decrease in the amount of Rho-GTP in these cells as determined by the affinity precipitation assay (Figure 7C). Based on these data, we concluded that amino acids 2552-3099 is the region of the RTX toxin responsible for the inactivation of the Rho GTPases, and this region was named the Rho GTPase inactivation domain (RID).
Figure 7.

Cellular delivery of purifed LFNRID causes cell rounding and inactivates the small GTPase Rho. (A) PA, LFN and LFNRID were purified by Ni2+ affinity chromatography and purity was assessed by SDS-PAGE and Coomasie Blue staining. MW represents molecular weight marker. (B) HEp-2 cells were incubated for 4 hours in the presence of PA (28 nm) with either LFN (12 nM) or LFNRID (12 nM). Phase contrast images were acquired at ×200 magnification. Scale bar represents 30 μm. (C) HEp-2 cells were incubated for 4 hours in the presence of LFNRID alone (12 nM), PA (28 nM) with LFN (12 nM) or PA (28 nM) with LFNRID (12 nM). Cells were harvested and the active Rho was pulled down in an affinity precipitation assay. Western blotting was performed to detect active Rho and total Rho in cell lysates.
RID does not function as a GAP or phosphatase
The Type III bacterial effectors ExoS and ExoT from Pseudomonas aureginosa, YopE from Yersinia spp. and SptP from Salmonella spp. reversibly inactivate the Rho GTPases by mimicking eukaryotic GAP proteins (Black and Bliska, 2000; Fu and Galan, 1999; Goehring et al., 1999; Krall et al., 2000). Since the RTX toxin does not inactivate the Rho GTPases by covalent modification or proteolysis, it may modulate the activity of Rho by functioning as a GAP. Incubation of recombinant LFNRID with RhoA loaded with [γ-32P]GTP did not increase the intrinsic hydrolysis of GTP by RhoA as measured by a filter binding assay (Figure 8A). These data revealed that recombinant LFNRID does not have GAP activity, thus eliminating that the RTX toxin functions by stimulating the intrinsic GTPase activity of the Rho GTPases.
Figure 8.

LFNRID does not function as a GAP or phosphatase. (A) 2 μM His-RhoA loaded with [γ-32P]GTP was incubated with a buffer control, 100 nM LFNRID or p20RhoGAP (positive control) for 10 minutes at 37°C and subjected to a filter binding assay to determine the %GTP bound to Rho. Data represent the average of duplicates from three independent experiments. (B) p-Nitrpohenyl phosphate (120 μg) was incubated with buffer control, 100 nM of LFN, LFNRID or calf intestinal alkaline phophatase (CIP) for 15 minutes at 37°C. Absorbance was measured at 405 nm. Data represent the average of triplicates from three independent experiments.
Upstream signaling pathways of the Rho GTPases are regulated through phosphorylation events. Cellular GEFs and GAPs can also be activated and inactivated by phosphorylation (Moon and Zheng, 2003; Rossman et al., 2005) The type III effector from Yersinia spp. YopH is a phosphatase that has been shown to target members of the focal adhesions, disrupting the actin cytoskeleton and causing reversible cell rounding (Guan and Dixon, 1990; Persson et al., 1997). However, experiments revealed that recombinant LFNRID does not have phosphatase activity in vitro since it did not stimulate the hydrolysis of the substrate p-Nitrophenyl Phosphate (Figure 8B). Taken together, the experimental evidence presented in this study suggests that the RTX toxin is utilizing a unique mechanism to target the Rho GTPases and disrupt the actin cytoskeleton.
Discussion
In this study, we demonstrated that the cell rounding activity of the RTX toxin observed in the absence of actin cross-linking is due to the inactivation of the small GTPase Rho. Additional experiments revealed that the toxin caused the dramatic relocalization of the GTPase Rho and Rac and to a lesser extent Cdc42, suggesting that the toxin affects all three GTPases. Since the RTX toxin caused the most dramatic effect on Rho, the observed relocalization of Rac and Cdc42 could be an indirect effect RTX induced inactivation of the Rho GTPase. These results indicated that the multifunctional RTX toxin targets the actin cytoskeleton by two different mechanisms: actin cross-linking and inactivation of the small Rho GTPases.
Bacterial toxins utilize four general mechanisms to disrupt the Rho GTPases leading to the depolymerization of the actin cytoskeleton. These mechanisms include covalent modification, proteolytic cleavage, mimicry of host cell GAPs and dephosphorylation of proteins regulating upstream signaling pathways. Constitutive activation of the GTPases does not prevent or reverse the cell rounding elicited by YopT from Yersinia spp., which acts by cleaving the isoprenylated C-terminal cysteine releasing the GTPases from the membrane (Sorg et al., 2001; Shao et al., 2002). The cytotoxic effects of the glucosytransferase TcdB are prevented by constitutive activation of the Rho GTPases, but not reversed (Fiorentini et al., 1995). Our experiments revealed that RTXΔACD-induced cell rounding and actin depolymerization was prevented and rapidly reversed by CNF1-induced constitutive activation of the Rho GTPases. These results suggested that the RTX toxin is not inactivating the GTPases in a manner similar to Yop T or TcdB. Therefore, these experiments established that the RTX toxin does not inactivate the Rho GTPases through either proteolytic inactivation or glucosylation.
Previous studies of the C3 exoenzyme from C. botulinum demonstrated that de novo synthesis of Rho is required to reverse the cell rounding associated with ADP-ribosylation of Rho (Barth et al., 1999). RTXΔACD inactivation of the Rho GTPases was determined to be reversible. This reversibility did not require de novo protein synthesis, since the addition of CHX did not inhibit the restoration of active Rho-GTP in these cells. These data eliminated covalent modification of the Rho GTPase as the mechanism of RTX inactivation of Rho.
Cellular delivery of recombinant LFNRID encoding amino acids 2552-3099 from RTX was able to inactivate the small GTPase Rho out of the context of the full-length RTX toxin. Identification of the domain responsible for RTX inactivation of Rho GTPases allowed further characterization of the mechanism utilizing purified protein. In vitro experiments revealed that recombinant LFNRID does not have GAP or phosphatase activity (data not shown). These experiments demonstrated that the RID of the RTX toxin is not inactivating the GTPase Rho by employing a mechanism similar to the type III effectors that either act as GAPs or phosphatases. However, we are unable to rule out that RID requires activation by a host cell factor. Based upon our experimental evidence, we concluded that the RTX toxin is utilizing a unique mechanism to target the Rho GTPases and disrupt the actin cytoskeleton.
Therefore, we hypothesize that the novel mechanism of RTX inactivation of the Rho GTPases acts (1) upon the regulation of the activation state of these GTPases, (2) through the activation of cellular GAPs or inactivation of cellular GEFs, or (3) possibly through the interference with the signaling pathways upstream of the GAPs and GEFs. Future studies to precisely determine the mechanism by which RTX inactivates the GTPases will focus on the RID region of the toxin. RID is present in three other putative Vibrio-type RTX toxins from V. vulnificus and the Xenorhabdus species pneumophila and bovinii sharing 87, 56 and 54 percent identity, respectively. The presence of this domain in other putative RTX toxins suggests that this is a common activity shared by Vibrio-type RTX toxins. However, a PSI-BLAST search of this domain did not reveal homology to any other proteins of known function (Altschul et al., 1997). In the future, mutational analysis of RID, as well as a broad screen to identify potential cellular targets will be performed to further investigate and define the mechanism by which the RID domain causes Rho GTPase inactivation.
As we reported, RTX inactivation of the Rho GTPases is independent from actin cross-linking activity. However, future experiments will focus on whether Rho GTPase inactivation is necessary for efficient RTX cross-linking of actin. In vivo and in vitro studies have shown that G-actin is the preferred substrate for actin cross-linking ((Cordero et al., in press); C. Cordero, unpublished data). It is thus possible that RTX-induced inactivation of the Rho GTPases increases the intracellular pool of G-actin to serve as the substrate for the ACD enhancing the rate of actin cross-linking. Alternatively, depletion of G-actin by actin cross-linking could stimulate feedback regulation of the Rho GTPase pathways enhancing the rate of rounding through the RID.
There is also the possibility that the RTX toxin may simply have two independent activities to disrupt the cytoskeleton and cause cell rounding reversibly and irreversibly. The RTX toxin is not the first toxin reported to have dual cell rounding activities. For example, the type III effectors ExoS and ExoT from P. aureginosa have bifunctional activity: GAP and ADP-ribosylation. These two activities act independently from each other and target different substrates (Aktories and Barbieri, 2005). Future studies will investigate whether the two cell rounding activities from the RTX toxin act together or independently from each other in the context of the full-length toxin.
Experimental Procedures
Cell Lines, Bacterial Strains, and Reagents
HEp-2 cells were cultured at 37°C with 5% CO2 in DMEM containing 50 units/ml penicillin, 50 μg/ml streptomycin, and 10% FBS (Invitrogen, Carlsbad, CA). V. cholerae strains KFV119 (RTX+), KFV92 (RTX-) and CCO5 (RTXΔACD) (Sheahan et al., 2004) were grown at 30°C in Luria broth (LB) containing 100 μg/ml streptomycin. All restriction enzymes were obtained from New England Biolabs (Beverly, MA). All chemicals were purchased from Sigma (St. Louis, MO). CNF1 was purified using the pCNF24 plasmid as previously described (Mills et al., 2000).
Cloning of the Rho Inactivation Domain (RID)
Primers RID-NheI 5′ GAGCTAGCCGCCACCATGGGTGCATGGCAATACAACGCCACA 3′ and RID-BamH1 5′ GCCGGATCCATTTTCAATCGCCACGTTTTC 3′ (IDT, Coralville, IA) were used to amplify the DNA corresponding to aa 2552-3099 of RTX with flanking NheI and BamH1 sites (underlined) from N16961 genomic DNA. This fragment was cloned into the EcoRV site of pBluescript and then subcloned into pEGFP-N3 (Clontech, Mountain View, CA) as an NheI-BamH1 fragment to create an in-frame fusion to egfp. Plasmid DNA prepared by using the Qiaprep Spin Miniprep kit was sequenced to confirm gene sequence and egfp fusions.
Recombinant LFNRID Cloning, Expression, and Purification
Primers RID-LFNF 5′ GAGGATCCCCGGGAGGTGCATGGCAATACAACGCCACA 3′ and RID-LFNR 5′ GAGGATCCCTAATTTTCAATCGCCACGTTTTC 3′ were used to amplify the RID from the V. cholerae N16961 genomic DNA with flanking BamHI sites (underlined). This fragment was cloned into pBluescript and then subcloned into pABII as an N-terminal fusion to amino acids 1-254 of B. anthracis Lethal Factor. Plasmids pABII and PA-pET15B were used for purification of the proteins PA and LFN. PA, LFN, and LFNRID were expressed in E. coli strain BL21 (λ DE3) (Spyres et al., 2001). Overnight cultures were diluted 1:500 into LB containing 100 μg/ml ampicillin and grown at 37°C to a density of OD600=0.6. Protein expression was induced with 0.2 mM IPTG (Denville Scientific, Metuchen, NJ) for 2 hours at 37°C. Bacterial pellets were then resuspended in 20 mM Tris, 500 mM NaCl and 5 mM imidazole pH 8.0 and lysed on ice for 30 minutes with 1 mg/ml lysozyme. Triton X-100 was added to a concentration of 1% and the lysates were sonicated with a Bronson Digital Sonifier 450 at 40% amplitude for 2 minutes. Insoluble debris was pelleted at 27,000g, and the 6x-His tagged proteins were purified by affinity chromatography using a HisTrap HP column on the ÄKTA purifier (GE Healthcare, Piscataway, NJ). The column was washed with 20 mM Tris, 500 mM NaCl, and 60 mM imidazole pH 8.0 and eluted with 250 mM imidazole. Proteins were then dialyzed into 20 mM Tris, 500 mM NaCl pH 7.5. Protein purity was assessed on Coomassie Blue R250 stained SDS-PAGE gel and concentration was determined by using a BCA acid protein assay kit (Pierce, Rockford, IL).
Transient Transfection
HEp-2 cells were plated at approximately 50% confluency on coverslips 24 hours before transfection. Cells were transfected using the FuGene 6 reagent (Roche Applied Sciences, Indianapolis, IN) mixed with plasmid DNA at a 3:2 reagent:DNA ratio in DMEM. This mixture was preincubated for 45 minutes prior to the addition to cultured cells followed by incubation at 37°C with 5% CO2 for 24 hours.
Microscopy
Phase contrast images were acquired at ×200 magnification of HEp-2 cells that had been incubated for 4 hours at 37°C with 28 nM PA with either 12 nM LFN or LFNRID. Transfected cells on coverslips were washed with PBS, fixed with 4% paraformaldehyde, and stained with Hoechst 33342 (0.5 μg/ml) 24 hours after transfection. Coverslips were then mounted using Anti-Fade mounting media (Molecular Probes, Eugene, OR). Transfected cells were observed for expression of EGFP at ×1000 magnification under fluorescence microscopy at 550-575 nm and 440-470 nm to visualize the nucleus. For actin staining, fixed cells were permeabilized with 0.1% Triton X-100 for 10 minutes and then incubated with 0.1 ug/ml of Tetramethylrhodamine B iso-Thiocyanate (TRITC) Phalloidin conjugate for 30 minutes at RT. Phalloidin stained cells were visualized under fluorescence microscopy at 610-675 nm at either ×400 or ×1000 magnification as indicated in figure legends. Images were captured using an inverted Leica DMIRE2 microscope with a C4742-95-12ERG digital charge-coupled device (CCD) camera (Hamamatsu Photonics, Tokyo) in conjunction with the OPENLAB software (Improvision, Coventry, UK) for image processing.
Affinity Precipitation Assay
Approximately 107 HEp-2 cells were incubated with either PBS, KFV92, KFV119, CCO5 or in the presence of PA with either LFNRID or LFN at a molar ratio of 7:3 for 4 hours at 37 °C. Cells were washed with cold PBS, collected and lysed in 1 ml of Rho assay buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Np-40, 10 mM MgCl2, 1 mM EDTA, 2% glycerol, 10 μg/mL aprotinin and leupeptin). Lysates were centrifuged at 14,000g for 5 minutes at 4°C to remove insoluble debris. Lysates (800 μl) were then incubated with 40 μg of GST-RBD agarose beads (Cytoskeleton, Denver, CO) for 1 hour at 4°C to measure Rho-GTP levels. Beads were collected, washed 3× with Rho assay buffer, resuspended in SDS-PAGE buffer, and boiled for 5 minutes. Total Rho was measured from 40 μl of lysate. Total Rho and Rho-GTP levels were detected by Western blotting.
Subcellular Localization
Approximately 107 HEp-2 cells were incubated with PBS, KFV92, KFV119 or CCO5 for 4h at 37 °C. Cells were washed with cold PBS, collected and resuspended in 1 ml of homogenization buffer (250 mM sucrose, 3 mM imidazole pH 7.4, 0.5 mM EDTA and complete EDTA-free protease inhibitor mixture (Roche Applied Sciences, Indianapolis, IN)). The cells were lysed by 3 pulses of 10 s with a Sonicel disrupter and then centrifuged at 2,000g for 5 minutes at 4°C. The post-nuclear supernatant was then fractionated by centrifugation at 100,000g for 30 minutes at 4°C. The supernatant (cytosolic fraction) was removed, and the insoluble pellet (membrane fraction) was resuspended in an equivalent volume of homogenization buffer containing 1% Triton X-100. SDS-PAGE loading buffer was added to the cytosolic and membrane fractions and then the fractions were boiled for 5 minutes. Equivalent volumes of samples were then analyzed by Western blotting.
Western Blotting
Samples were resolved by SDS-PAGE and transferred to Hybond-C nitrocellulose (GE Healthcare, Piscataway, NJ). Western blotting was then performed with anti-RhoA (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Rac, or anti-Cdc42 (Upstate Biotechnology, Lake Placid, NY) at 1:500 dilution. The anti-mouse IgG HRP secondary antibody was used at 1:5000 dilution (Sigma, St. Louis, MO) followed by chemiluminescent detection. Densitometric analysis was performed using NIH ImageJ 1.36b software (http://rsb.info.nih.gov/ij/).
GAP Assay
The GAP assay was performed as previously described (Self and Hall, 1995). Briefly, recombinant His-RhoA (Cytoskeleton, Denver, Co) was loaded with [γ-32P]GTP (6000 Ci/mmol, 10μCi/μl) (Perkin Elmer NEN, Wellesley, MA) in loading buffer (20 mM Tris-HCl pH 7.5, 0.1 mM DTT, 25 mM NaCl, and 4 mM EDTA) for 5 min at 37° C followed by the addition of MgCl2 to a final concentration of 17 mM. To test for GAP activity, 2 μM GTP-loaded RhoA was incubated alone, with LFNRID (100 nM), or with positive control p29RhoGAP (100 nM) (Cytoskeleton, Denver, CO) in hydrolysis buffer (20 mM Tris-HCl pH 7.5, 0.1 mM DTT, 1 mM GTP, 1 mg/mL BSA) for 10 minutes at 37° C followed by dilution into 1 mL of cold assay buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl and 5 mM MgCl2). Samples were analyzed by a filter binding assay and then reported as bound GTP remaining at T=10 min as a percentage of the initial bound GTP at T=0.
pNPP Hydrolysis Assay
LFN (100 nM), LFNRID (100 nM) and as a positive control calf intestinal alkaline phosphatase (CIP) (100 nM) (Invitrogen, Carlsbad, CA) were incubated with 120 μg of p-Nitrophenyl phosphate (pNPP) in pNPP hydrolysis buffer (25 mM HEPES pH 7.4, 50 mM NaCl, 5 mM DTT, 2.5 mM EDTA supplemented with 5 μg of BSA). After 15 min of incubation at 37°C, the reaction was stopped with 13% K2HPO4. Samples were then measured at absorbance of 405 nm.
Acknowledgments
We thank A. O'Brien for the plamid pCNF24 and J. Ballard for the plasmids pABII and PA-pET15b. We thank J. Barbieri for input on the Rho GTPase relocalization assay. We also thank A. Bonebrake for technical assistance and C. Cordero for purification of PA and LFN. This work was supported by a Biomedical Research Support Program Award from the Howard Hughes Medical Institute and Public Health Services Grant AI051490 from the National Institute for Allergy and Infectious Disease (to K.J.F.S.). K.-L.S. was supported by the Ruth L. Kirschstein NRSA Predoctoral Fellowship T32-AI07476.
References
- Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol. 2005;3:397–410. doi: 10.1038/nrmicro1150. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H, Olenik C, Sehr P, Schmidt G, Aktories K, Meyer DK. Neosynthesis and activation of Rho by Escherichia coli cytotoxic necrotizing factor (CNF1) reverse cytopathic effects of ADP-ribosylated Rho. J Biol Chem. 1999;274:27407–27414. doi: 10.1074/jbc.274.39.27407. [DOI] [PubMed] [Google Scholar]
- Black DS, Bliska JB. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol. 2000;37:515–527. doi: 10.1046/j.1365-2958.2000.02021.x. [DOI] [PubMed] [Google Scholar]
- Boardman BK, Satchell KJ. Vibrio cholerae strains with mutations in an atypical type I secretion system accumulate RTX toxin intracellularly. J Bacteriol. 2004;186:8137–8143. doi: 10.1128/JB.186.23.8137-8143.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- Chardin P, Boquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. Embo J. 1989;8:1087–1092. doi: 10.1002/j.1460-2075.1989.tb03477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KH, Ng TK, Yuen KY, Yam WC. Detection of RTX toxin gene in Vibrio cholerae by PCR. J Clin Microbiol. 2001;39:2594–2597. doi: 10.1128/JCM.39.7.2594-2597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero CL, Kudryashov DS, Reisler E, Satchell KJ. The Actin Cross-linking Domain of the Vibrio cholerae RTX Toxin Directly Catalyzes the Covalent Cross-linking of Actin. J Biol Chem. doi: 10.1074/jbc.M605275200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard A, Serichantalergs O, Forslund A, Lin W, Mekalanos J, Mintz E, et al. Clinical and environmental isolates of Vibrio cholerae serogroup O141 carry the CTX phage and the genes encoding the toxin-coregulated pili. J Clin Microbiol. 2001;39:4086–4092. doi: 10.1128/JCM.39.11.4086-4092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JL, Mogridge J, Collier RJ. A quantitative study of the interactions of Bacillus anthracis edema factor and lethal factor with activated protective antigen. Biochemistry. 2000;39:6706–6713. doi: 10.1021/bi000310u. [DOI] [PubMed] [Google Scholar]
- Faruque SM, Chowdhury N, Kamruzzaman M, Dziejman M, Rahman MH, Sack DA, et al. Genetic diversity and virulence potential of environmental Vibrio cholerae population in a cholera-endemic area. Proc Natl Acad Sci U S A. 2004;101:2123–2128. doi: 10.1073/pnas.0308485100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C, Donelli G, Matarrese P, Fabbri A, Paradisi S, Boquet P. Escherichia coli cytotoxic necrotizing factor 1: evidence for induction of actin assembly by constitutive activation of the p21 Rho GTPase. Infect Immun. 1995;63:3936–3944. doi: 10.1128/iai.63.10.3936-3944.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Galan JE. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature. 1999;401:293–297. doi: 10.1038/45829. [DOI] [PubMed] [Google Scholar]
- Fullner KJ, Mekalanos JJ. In vivo covalent cross-linking of cellular actin by the Vibrio cholerae RTX toxin. Embo J. 2000;19:5315–5323. doi: 10.1093/emboj/19.20.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Boucher JC, Hanes MA, Haines GK, 3rd, Meehan BM, Walchle C, et al. The contribution of accessory toxins of Vibrio cholerae O1 El Tor to the proinflammatory response in a murine pulmonary cholera model. J Exp Med. 2002;195:1455–1462. doi: 10.1084/jem.20020318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring UM, Schmidt G, Pederson KJ, Aktories K, Barbieri JT. The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J Biol Chem. 1999;274:36369–36372. doi: 10.1074/jbc.274.51.36369. [DOI] [PubMed] [Google Scholar]
- Gordon VM, Leppla SH, Hewlett EL. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect Immun. 1988;56:1066–1069. doi: 10.1128/iai.56.5.1066-1069.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 1990;249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- Haines GK, 3rd, Sayed BA, Rohrer MS, Olivier V, Satchell KJ. Role of toll-like receptor 4 in the proinflammatory response to Vibrio cholerae O1 El tor strains deficient in production of cholera toxin and accessory toxins. Infect Immun. 2005;73:6157–6164. doi: 10.1128/IAI.73.9.6157-6164.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos Trans R Soc Lond B Biol Sci. 2000;355:965–970. doi: 10.1098/rstb.2000.0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem. 1995;270:13932–13936. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- Kaper J, Morris JG, Levine MM. Cholera. Clinical Microbiology Reviews. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler TM, Collier RJ. Anthrax toxin protective antigen: low-pH-induced hydrophobicity and channel formation in liposomes. Mol Microbiol. 1991;5:1501–1506. doi: 10.1111/j.1365-2958.1991.tb00796.x. [DOI] [PubMed] [Google Scholar]
- Krall R, Schmidt G, Aktories K, Barbieri JT. Pseudomonas aeruginosa ExoT is a Rho GTPase-activating protein. Infect Immun. 2000;68:6066–6068. doi: 10.1128/iai.68.10.6066-6068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall R, Sun J, Pederson KJ, Barbieri JT. In vivo rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS. Infect Immun. 2002;70:360–367. doi: 10.1128/IAI.70.1.360-367.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerm M, Selzer J, Hoffmeyer A, Rapp UR, Aktories K, Schmidt G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: activation of c-Jun N-terminal kinase in HeLa cells. Infect Immun. 1999;67:496–503. doi: 10.1128/iai.67.2.496-503.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE, et al. Identification of a vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci U S A. 1999;96:1071–1076. doi: 10.1073/pnas.96.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills M, Meysick KC, O'Brien AD. Cytotoxic necrotizing factor type 1 of uropathogenic Escherichia coli kills cultured human uroepithelial 5637 cells by an apoptotic mechanism. Infect Immun. 2000;68:5869–5880. doi: 10.1128/iai.68.10.5869-5880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Blanke SR, Hanna PC, Collier RJ. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol Microbiol. 1995;15:661–666. doi: 10.1111/j.1365-2958.1995.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- Mogridge J, Cunningham K, Collier RJ. Stoichiometry of anthrax toxin complexes. Biochemistry. 2002;41:1079–1082. doi: 10.1021/bi015860m. [DOI] [PubMed] [Google Scholar]
- Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13–22. doi: 10.1016/s0962-8924(02)00004-1. [DOI] [PubMed] [Google Scholar]
- Persson C, Carballeira N, Wolf-Watz H, Fallman M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. Embo J. 1997;16:2307–2318. doi: 10.1093/emboj/16.9.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Satchell KJ. Activation and suppression of the proinflammatory immune response by Vibrio cholerae toxins. Microbes Infect. 2003;5:1241–1247. doi: 10.1016/j.micinf.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387:725–729. doi: 10.1038/42735. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder SM, Burridge K. Bidirectional signaling between the cytoskeleton and integrins. Curr Opin Cell Biol. 1999;11:274–286. doi: 10.1016/s0955-0674(99)80037-4. [DOI] [PubMed] [Google Scholar]
- Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37:5296–5304. doi: 10.1021/bi972592c. [DOI] [PubMed] [Google Scholar]
- Self AJ, Hall A. Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 1995;256:67–76. doi: 10.1016/0076-6879(95)56010-6. [DOI] [PubMed] [Google Scholar]
- Shao F, Merritt PM, Bao Z, Innes RW, Dixon JE. A Yersinia effector and a Pseudomonas avirulence protein define a family of cysteine proteases functioning in bacterial pathogenesis. Cell. 2002;109:575–588. doi: 10.1016/s0092-8674(02)00766-3. [DOI] [PubMed] [Google Scholar]
- Sheahan KL, Cordero CL, Satchell KJ. Identification of a domain within the multifunctional Vibrio cholerae RTX toxin that covalently cross-links actin. Proc Natl Acad Sci U S A. 2004;101:9798–9803. doi: 10.1073/pnas.0401104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg I, Goehring UM, Aktories K, Schmidt G. Recombinant Yersinia YopT leads to uncoupling of RhoA-effector interaction. Infect Immun. 2001;69:7535–7543. doi: 10.1128/IAI.69.12.7535-7543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyres LM, Qa'Dan M, Meader A, Tomasek JJ, Howard EW, Ballard JD. Cytosolic delivery and characterization of the TcdB glucosylating domain by using a heterologous protein fusion. Infect Immun. 2001;69:599–601. doi: 10.1128/IAI.69.1.599-601.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]