

Abstract
The rare sugar 2,6-dideoxy-2-acetamidino-l-galactose (l-FucNAm) is found only in bacteria and is a component of cell surface glycans in a number of pathogenic species, including the O antigens of Pseudomonas aeruginosa serotype O12 and Escherichia coli O145. P. aeruginosa is an important opportunistic pathogen, and the O12 serotype is associated with multidrug-resistant epidemic outbreaks. O145 is one of the classic non-O157 serotypes associated with Shiga toxin-producing, enterohemorrhagic E. coli. The acetamidino (NAm) moiety of l-FucNAm is of interest, because at neutral pH it contributes a positive charge to the cell surface, and we aimed to characterize the biosynthesis of this functional group. The pathway is not known, but expression of NAm-modified sugars coincides with the presence of a pseA homologue in the relevant biosynthetic locus. PseA is a putative amidotransferase required for synthesis of a NAm-modified sugar in Campylobacter jejuni. In P. aeruginosa O12 and E. coli O145, the pseA homologues are lfnA and wbuX, respectively, and we hypothesized that these genes function in l-FucNAm biosynthesis. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis, Western blotting, and nuclear magnetic resonance analysis of the lfnA mutant O-antigen structure indicated that the mutant expresses 2,6-dideoxy-2-acetamido-l-galactose (l-FucNAc) in place of l-FucNAm. The mutation could be complemented by expression of either His6-tagged lfnA or wbuX in trans, confirming that these genes are functional homologues and that they are required for NAm moiety synthesis. Both proteins retained their activity when fused to a His6 tag and localized to the membrane fraction. These data will assist future biochemical investigation of this pathway.
Pseudomonas aeruginosa is a ubiquitous gram-negative microorganism and an important opportunistic pathogen which causes life-threatening infections in compromised animals and plants. It accounts for approximately 1 in 10 reported cases of common hospital-acquired infections (22) and is a principal cause of morbidity in cystic fibrosis patients (10).
One of the major virulence determinants for P. aeruginosa is a cell surface carbohydrate polymer known as B-band O antigen, which is a component of lipopolysaccharide (LPS). There are 20 major serotypes of P. aeruginosa which differ in the structure of their B-band O antigen. The chemical structure of O antigen in each of these serotypes has been elucidated by Knirel and his colleagues (36), and the gene clusters encoding B-band biosynthesis in all 20 serotypes have been cloned and sequenced (49). O antigen confers resistance to complement-mediated killing (16), and mutants deficient in O antigen are avirulent in a burnt mouse model of infection (14).
The predominant P. aeruginosa serotypes in clinical specimens, regardless of geographic location, are O1, O6, and O11 (3, 46). In contrast, the prevalence of O12 among clinical isolates varies depending upon geographical location (58) and can change over time (1); however, this serotype has made up 20% of all P. aeruginosa isolates in some clinical studies (1, 39). O12 is associated with multidrug-resistant epidemic outbreaks in European hospitals (3, 21, 30), including infection of cystic fibrosis patients (43) and burn wounds (46), and the prevalence of O12 among clinical isolates in Europe may be due to expansion of a few highly successful clones (47, 48).
Enterohemorrhagic Escherichia coli (EHEC) is a causative agent of human gastrointestinal disease with life-threatening complications in the very young or elderly. The principal characteristic feature of EHEC is expression of Shiga toxin (reviewed in reference 61). The most common E. coli O-antigen serotype associated with these pathogenic E. coli strains is O157, but one of the classic non-O157 serotypes commonly associated with Shiga toxin production is O145 (4, 5, 51). O145 strains have been identified as the etiologic agent in cases of bloody diarrhea (19), hemorrhagic colitis (8), and hemolytic-uremic syndrome (20, 28, 57).
Both the P. aeruginosa O12 and the E. coli O145 O-antigen repeats contain a 2,6-dideoxy-2-acetamidino-l-galactose (l-FucNAm) residue (12, 24, 37). This sugar is rare in nature and is found only in bacteria. Besides these etiological agents of human disease, l-FucNAm is also expressed by pathogens of animals (Salmonella enterica subsp. arizonae serotypes O21 [59] and O61 [60]) and fish (Yersinia ruckerii O1 [6] and Flavobacterium columnare ATCC 43622 [40]).
The biosynthetic pathway for expression of l-FucNAm is unknown, but synthesis of the acetamidino group on the 5- acetamido-7-acetamidino-3,5,7,9-tetradeoxy-l-glycero-α-l-manno-nonulosonic acid (Pse5NAc7Am) residue of Campylobacter jejuni flagellin glycan requires the pseA gene, and homologues of this gene exist in the P. aeruginosa O12 and the E. coli O-antigen biosynthesis loci (lfnA and wbuX, respectively). A pseA mutant glycosylates flagellin with 5,7-diacetamido-3,5,7,9-tetradeoxy-l-glycero-α-l-manno-nonulosonic acid (Pse5NAc7NAc) where Pse5NAc7Am usually is, and analysis of the sugar-nucleotide pool in this mutant indicates that PseA probably operates at a late stage in Pse5NAc7Am biosynthesis, possibly catalyzing amidotransfer to CMP-Pse5NAc7NAc (42). A pseA homologue (wbpG) is also present in the O-antigen locus of P. aeruginosa O5 which possesses the acetamidino-modified sugar 2,3-dideoxy-2-acetamido-3-acetamidino-d-mannuronic acid (d-ManNAc3NAmA) in its O antigen. The coincidence of acetamidino sugars and genes encoding PseA homologues suggests that these may be the enzymes responsible for acetamidino group generation. In each case, the sugars containing the acetamidino group are different, implying that despite a common catalytic chemistry, the enzymes may not necessarily share the same substrate specificity. These genes all encode proteins in the so-called PP-loop superfamily and are therefore predicted to bind ATP (9). A conserved domain search (41) indicates that 52% of the Pfam00733 motif (C-terminal domain of asparagine synthase) is present in the LfnA sequence (see Fig. S1 in the supplemental material). Information from the Pfam database (25) suggests that proteins with this domain use ATP to activate their substrates toward amidotransfer. Such a function would be consistent with the involvement of this protein in biosynthesis of l-FucNAm.
Here we report the genetic investigation of lfnA from P. aeruginosa O12, which was previously known as orf17 in the O-antigen biosynthesis locus. A lfnA mutant continues to produce O antigen, but with an altered structure. Results from nuclear magnetic resonance (NMR) analysis of purified polysaccharides from this mutant indicate that the O antigen lacks the acetamidino group usually present on the l-FucNAm residue, thus expressing l-FucNAc in its place. These findings indicate a role for lfnA in the formation of the acetamidino group in synthesis of l-FucNAm in P. aeruginosa O12. We also demonstrate that wbuX from E. coli O145 can cross-complement an lfnA mutation implicating wbuX in l-FucNAm biosynthesis in E. coli O145.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Bacteria were routinely propagated in Luria-Bertani (LB) broth or on LB agar containing 1.5% Bacto Agar (Difco). All strains were grown at 37°C with the following antibiotics added as appropriate: ampicillin (100 μg ml−1), carbenicillin (300 μg ml−1), kanamycin (50 μg ml−1), gentamicin (15 μg ml−1 for E. coli; 150 μg ml−1 for P. aeruginosa), and tetracycline (10 μg ml−1 for E. coli; 100 μg ml−1 for P. aeruginosa).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| JM109 | e14− (McrA−) recA1 endA1 gyrA96 thi-1 hsdR17 (rK−mK+) supE44 relA1 Δ(lac-proAB) [F′ traD36+proAB+lacIqZΔM15] | Stratagene |
| DH5α | F− φ80dlacΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) supE44 λ−thi-1 gyrA96 relA | Invitrogen |
| SM10 | thi-1 thr leu tonA lacY supE recA RP4-2-Tc::Mu Kmr | 54 |
| BL21(DE3) | F−ompT gal dcm lon hsdSB(rB− mB−) λDE3(lacI lacUV5-T7 gene 1 ind-1 sam-7 nin-5) | Novagen |
| N 00 6496 | Wild type; O145:NM | Mohamed Karmali |
| N 01 2051 | Wild type; O145:NM | Mohamed Karmali |
| N 02 5149 | Wild type; O145:NM | Mohamed Karmali |
| P. aeruginosa | ||
| PAO1 | Wild-type strain, IATSa serotype O5 (ATCC 33351) | 31 |
| O4 | Wild-type strain, IATS serotype O4 (ATCC 33351) | ATCC |
| O11 | Wild-type strain, IATS serotype O11 (ATCC 33358) | ATCC |
| O12 | Wild-type strain, IATS serotype O12 (ATCC 33359) | ATCC |
| O12 lfnA | O12 lfnA::aacC1; Gmr | This work |
| Plasmids | ||
| pEX100T | Counterselectable, mobilizable suicide vector backbone; contains oriT and sacB; Apr | 53 |
| pUCGm | Source of small, broad-host-range gentamicin cassette aacC1; Apr Gmr | 52 |
| pET28a | Protein expression vector for creating thrombin-cleavable N-terminal His6 tag gene fusions; Kmr | Novagen |
| pUCP20 | Escherichia-Pseudomonas shuttle vector derived from pUC18; Apr Cbr | 63 |
| pVLT31 | Broad-host-range vector containing the Ptac promoter for induction of downstream gene expression; Tcr | 17 |
| pKOLD2 | Suicide vector pEX100T containing the mutant lfnA allele lfnA::aacC1; Apr Gmr | This work |
| pFuc41 | pET28a-derived expression vector encoding expression of His6-Lfn; Kmr | This work |
| pUCP20-rbs-his6-lfnA | XbaI-HindIII fragment from pFuc41, containing the ribosome binding site, N-terminal His6 tag, and lfnA coding sequence cloned into pUCP20; Apr Cbr | This work |
| pVLT31-rbs-his6-lfnA | XbaI-HindIII fragment from pFuc41, containing the ribosome binding site, N-terminal His6 tag, and lfnA coding sequence cloned into pVLT31; Tcr | This work |
| pBa1ET28a-wbuX | Untagged wbuX cloned into the NcoI and BamHI sites of pET28a; Kmr | |
| pBf3ET28a-his6-wbuX | wbuX cloned into the NheI and BamHI sites of pET28a, generating an N-terminal His6 tag fusion; Kmr | This work |
| pCr1UCP20-rbs-wbuX | XbaI-HindIII fragment from pBa1ET28a-wbuX, which contains the ribosome binding site and wild-type wbuX cloned into pUCP20; Apr Cbr | This work |
| pCs2UCP20-rbs-his6-wbuX | XbaI-HindIII fragment from pBf3ET28a-his6-wbuX, which contains the ribosome binding site and His6-wbuX fusion cloned into pUCP20; Apr Cbr | This work |
IATS, International Antigenic Typing Scheme.
DNA methods.
Standard methods were used for DNA manipulations. Unless otherwise stated, enzymes and reagents were purchased from New England Biolabs or Invitrogen and used according to the manufacturer's instructions. Oligonucleotides were supplied by the University of Guelph Molecular Supercenter. PCR was performed using KOD Hot Start (Novagen) or Pwo (Roche) DNA polymerases.
Mutagenesis of lfnA in the O-antigen locus of P. aeruginosa serotype O12.
lfnA (orf17) from the O-antigen biosynthesis locus of P. aeruginosa O12 was PCR amplified using the primers 5′-AGGAAAAACATATGAGCAACCACAGCTACAAG-3′ (NdeI site underlined) and 5′-TAAGGATCCTCATCTGAATAACCTACGTTCC-3′ (BamHI site underlined) and ligated without prior restriction digestion into SmaI-cut pEX100T. The resulting plasmid was cut within the lfnA coding sequence by digestion with PpuMI, then blunted by filling in the ends with the Klenow fragment, and ligated with the SmaI-digested aacC1 (gentamicin resistance [Gmr]) cassette from pUCGm. This generated the gene knockout vector pKOLD2. This vector was transformed into P. aeruginosa O12 by conjugation between log-phase cultures of the P. aeruginosa acceptor strain and E. coli SM10 harboring pKOLD2 as the donor (54). Transconjugants were initially selected on Pseudomonas isolation agar (Difco) containing gentamicin to isolate clones in which plasmid DNA was incorporated into the chromosome. Colonies from these plates were then streaked onto LB agar plates (but without NaCl), supplemented with 10% sucrose to select for double recombinants that contained a chromosomal copy of the mutant lfnA::aacC1 allele but lacked the counterselection marker sacB, which is lethal to cells grown on sucrose (29). Allelic exchange in putative lfnA knockout strains was confirmed by PCR.
Construction of the complementation plasmid pUCP20-rbs-his6-lfnA.
The lfnA PCR product described above was digested with NdeI and BamHI and ligated into pET28a to generate plasmid pFuc41 in which lfnA is fused to the plasmid-encoded N-terminal His6 tag (His6-lfnA). This gene fusion, together with the upstream ribosome-binding site (rbs), was excised with XbaI and HindIII and ligated into the shuttle vector pUCP20 to generate the complementing vector, pUCP20-rbs-his6-lfnA.
Construction of the His6-lfnA protein expression plasmid pVLT31-rbs-his6-lfnA.
The His6-lfnA fusion, together with the upstream rbs, was excised from pFuc41 using XbaI and HindIII and cloned into these restrictions sites in the broad-host-range vector pVLT31. The resulting plasmid, pVLT31-rbs-his6-lfnA, places the His6-lfnA fusion under the control of the Ptac promoter in a broad-host-range vector.
Construction of the wbuX protein expression plasmids pBa1ET28a-wbuX and pBf3ET28a-his6-wbuX.
wbuX was PCR amplified using the primers 5′-TTTTTTTTGCTAGCTCCATGGAAAATAAAAATTATCAAATT-3′ (NheI and NcoI sites underlined) and 5′-TTTTTTTTGGATCCTCATCTGAAATACCTTTTTTC-3′ (BamHI site underlined). Cloning this fragment into pET28a using the NcoI and BamHI consensus sites generated a vector (pBa1ET28a-wbuX) for expression of wild-type, untagged WbuX in E. coli. Cloning into the NheI and BamHI sites of pET28a generated the vector pBf3ET28a-his6-wbuX for expression of His6-tagged WbuX (His6-WbuX) in E. coli.
Construction of the wbuX-based cross-complementation plasmids pCr1UCP20-rbs-wbuX and pCs2UCP20-rbs-his6-wbuX.
wbuX-containing XbaI-HindIII fragments from pBa1ET28a-wbuX and pBf3ET28a-his6-wbuX were ligated into XbaI-and-HindIII-digested pUCP20 to generate the cross-complementation vectors pCr1UCP20-rbs-wbuX and pCs2UCP20-rbs-his6-wbuX, respectively.
Electroporation protocol for P. aeruginosa.
DNA was introduced into P. aeruginosa using a novel protocol, based on the methods of Helmark and coworkers (32) and Choi and coworkers (13). An overnight culture of P. aeruginosa was used to inoculate 50 ml of growth medium (LB supplemented with 0.25 M sucrose, 27.8 mM glucose, and 0.75% [wt/vol] glycine) to give a starting optical density at 600 nm (OD600) of 0.03. The culture was shaken at 37°C until the OD600 reached approximately 0.1 when cells were harvested by centrifugation at 10,000 × g for 10 min at room temperature. The pellet was washed once in 5 ml of sterile wash solution (0.25 M sucrose, 10% [vol/vol] glycerol) at room temperature and then suspended in wash solution to give an OD600 of approximately 30. A 50-μl aliquot of this suspension was combined with 5 μg of plasmid DNA and pulsed in a 2-mm-gap electroporation cuvette (2.5 kV, 25 μF, 400 Ω) using a Gene Pulser apparatus (Bio-Rad). Immediately, 950 μl of LB was added, and the cells were recovered with shaking at 37°C for 3 h before plating on selective medium.
LPS preparation.
For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, LPS was prepared using the proteinase K digestion method of Hitchcock and Brown (34). For structural analysis, LPS was prepared using the hot aqueous phenol extraction method of Westphal and Jann (64).
SDS-PAGE.
SDS-PAGE was performed with a discontinuous gel system and 12.5% resolving gels (38). Proteins were visualized using SimplyBlue SafeStain (Invitrogen). LPS was visualized using the rapid silver staining method of Fomsgaard and coworkers (26) or by Western blotting.
Western blotting.
Western transfer of proteins and LPS was performed to BioTraceNT nitrocellulose membranes (Pall) according to standard protocols with minor modifications (11, 56). His-tagged proteins were detected using His-Probe H-3 primary antibodies (Santa Cruz Biotechnology). Rabbit polyclonal antibodies specific for P. aeruginosa O4, O11, and O12 serotypes (from a Pseudomonas typing kit prepared by Chengdu Institute of Biological Products, Ministry of Public Health, Chengdu, Sichuan, China) were used to detect the presence of O-antigen-containing LPS in these bacteria. Blots were incubated with alkaline phosphatase-conjugated secondary antibodies and then visualized by incubation with nitroblue tetrazolium (7) and 5-bromo-4-chloro-3-indolyl phosphate (BCIP) according to standard protocols.
NMR spectroscopy.
1H and 13C NMR spectra were recorded using a Varian Inova 500-MHz spectrometer for samples in D2O solutions at 25°C for the oligosaccharides with acetone standard (2.225 ppm for 1H and 31.5 ppm for 13C) using standard pulse sequences. Experiments included correlation spectroscopy (COSY), total correlation spectroscopy (mixing time 120 ms), nuclear Overhauser effect spectroscopy (mixing time 200 ms), heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond correlation (HMBC) (optimized for an 8-Hz coupling constant).
Mild acid hydrolysis of LPS.
LPS (25 mg) was hydrolyzed with 2% acetic acid (AcOH) (2 ml, 3 h, 100°C), and the precipitates of the lipid components were removed by centrifugation at top speed in a microcentrifuge (12,000 rpm). Soluble products were separated by gel chromatography on a Sephadex G50 column. Polymeric and oligosaccharide fractions were obtained. Oligosaccharide (core) fraction was further separated by anion-exchange chromatography.
Anion-exchange chromatography.
Anion-exchange chromatography was performed on a Hitrap Q column (Pharmacia). The column was washed with water for 10 min and then eluted with a linear gradient of 0 to 1 M NaCl over 60 min, with a flow rate of 3 ml min−1 and UV detection at 220 nm. Fractions were desalted by gel chromatography on a Sephadex G-15 column.
Monosaccharide analysis.
The polysaccharide, core, or LPS (0.5 mg) was hydrolyzed (0.2 ml of 3 M trifluoroacetic acid, 120°C, 2 h) and evaporated to dryness under a stream of air. The residue was dissolved in water (0.5 ml), reduced with NaBH4 (∼5 mg, 1 h), neutralized with AcOH (0.3 ml), and dried, and methanol (1 ml) was added. The mixture was dried twice by the addition of methanol, and the residue was acetylated with acetic anhydride (0.5 ml, 100°C, 30 min), dried, and analyzed by gas chromatography on a HP1 capillary column (30 m by 0.25 mm) with a flame ionization detector (Agilent 6850 chromatograph) and a temperature gradient of 170 (4 min) to 260°C at 4°C min−1.
Gel filtration chromatography.
Prior to NMR analysis, carbohydrates derived from LPS were purified on Sephadex G-50 (2.5- by 80-cm) or Sephadex G-15 (1.6- by 80-cm) columns using pyridinium acetate buffer (pH 4.5) (4 ml pyridine and 10 ml AcOH in 1 liter water) as the eluent, with monitoring of the eluate by a refractive index detector.
Protein expression and subcellular localization by differential centrifugation.
His6-LfnA and His6-WbuX expression in the P. aeruginosa lfnA mutant was done at basal expression levels from the plasmids pUCP20-rbs-his6-lfnA and pCs2UCP20-rbs-his6-wbuX, respectively. Cultures were grown overnight in LB, and isopropyl-β-d-thiogalactopyranoside (IPTG) was not added to the growth medium.
His6-Lfn expression in P. aeruginosa from the pVLT31-based expression plasmid was induced once the cultures attained an OD600 of approximately 0.3 by adding IPTG to a final concentration of 0.4 mM. The cultures were then incubated for 3 hours at 37°C.
His6-WbuX was expressed in E. coli BL21(DE3) according to protocols in the pET system manual (Novagen). Expression of His6-WbuX from the pET vector was induced, once the OD600 reached 0.4, by adding IPTG to 0.4 mM. The induced culture was then incubated overnight at 15°C, before the cells were harvested.
The cells were harvested by centrifugation (10,000 × g, 10 min) and suspended in 50 mM sodium phosphate, pH 8.0, and 300 mM NaCl. Bacterial cells were lysed on ice with 5-s ultrasound bursts generated by a model 500 sonic dismembrator (Fisher Scientific) fitted with a macrotip at 35% power for a total of 2 minutes. Cell debris was removed by centrifugation at 30,000 × g for 20 min, and membranes were isolated by ultracentrifugation at 100,000 × g for 1 to 2 h. The membrane pellets were gently washed twice with water and then suspended in SDS loading buffer (62.5 mM Bis-Tris [pH 6.8], 0.5% [wt/vol] SDS, 10% [wt/vol] glycerol, 0.005% bromophenol blue, 250 mM NaHSO3). Proteins were considered to be soluble if they remained in solution after the ultracentrifugation step.
Bioinformatics.
Protein sequences were analyzed, and sequence identities were calculated based upon alignments obtained using the position-specific iterative basic local alignment search tool (PSI-BLAST) (2).
RESULTS
Mutation of lfnA increases the relative mobility of LPS bands on SDS-PAGs.
To characterize the function of lfnA in O-antigen biosynthesis, the chromosomal copy of the gene was replaced with a mutant allele in which the coding sequence is disrupted by a gentamicin resistance cassette. LPS prepared from this lfnA knockout mutant was compared to that of wild-type bacteria using SDS-PAGE and silver staining. B-band LPS in several P. aeruginosa strains exhibits a bimodal or multimodal chain length distribution. In the P. aeruginosa serotype O5 strain, PAO1, this pattern is determined by the operation of two chain-length-regulating proteins Wzz1 and Wzz2 (15). In SDS-PAGs, this modal O-antigen length distribution is visible as clusters of intense bands in the top portion of the gel (Fig. 1). When the lfnA mutant LPS is examined using SDS-PAGE, these clusters of bands migrate faster through the gel than those of the wild-type LPS (Fig. 1A). This mutation does not interfere, however, with recognition of the O antigen by serotype O12-specific polyclonal antibodies (Fig. 1B). This suggests that the mutation has caused either a shortening of the modal O-antigen chain lengths or a change in O-antigen repeat unit structure or both. The spacing between individual bands in the SDS-PAGE profile indicates the change in electrophoretic mobility due to each additional O unit. Compared to LPS from the wild-type O12 strain, LPS from the lfnA mutant exhibited reduced spacing between bands, suggesting that this mutation caused a change in the structure of each O-antigen repeat.
FIG. 1.

SDS-PAGE analysis of LPS from wild-type P. aeruginosa serotype O12, the lfnA mutant, and the lfnA mutant containing either the complementing plasmid pUCP20-his6-lfnA, or the empty vector pUCP20. LPS was visualized by silver staining (A) and by Western blotting with polyclonal, anti-O antigen primary antibodies (B). The empty vector has no effect on LPS phenotype, whereas the complementing plasmid restores the wild-type LPS banding pattern. The positions of modal clusters of O-antigen-containing LPS in the wild-type lanes are indicated. The bands representing unsubstituted LPS core and core plus one O-antigen repeat (Core+1) are also indicated. The Core+1 is the smallest molecule to be recognized by the O-antigen-specific antibodies.
A novel electroporation protocol enabled complementation of the lfnA mutation.
We wished to complement the lfnA mutation by expressing the gene from plasmid pUCP20. This vector does not have an oriT and cannot therefore be transferred into P. aeruginosa by conjugation. Transformation of plasmid DNA into P. aeruginosa O12 strain ATCC 33359 using standard electroporation protocols usually employed for P. aeruginosa (18, 23, 55) has proven to be difficult. To alleviate this problem, we developed a novel method for preparation of electrocompetent P. aeruginosa by combining steps from two methods in the literature (13, 32). Cells were grown according to the method of Helmark and coworkers (32) but harvested at room temperature according to the protocol of Choi and coworkers (13).
Using this method, we introduced the complementing plasmid into the lfnA mutant strain, and LPS in the complemented strain showed a phenotype similar to that observed in the wild-type strain (Fig. 1).
The lfnA mutant produces LPS which contains l-FucNAc in the O-antigen repeat unit, instead of l-FucNAm.
To establish the structural basis for the closer spacing between O-antigen bands in SDS-PAGE analysis of the lfnA mutant LPS, we purified LPS from the mutant and from the complemented strain and analyzed the O-antigen repeat structure by NMR.
Oligosaccharides from the mutant and complemented strains were isolated from LPS by mild acid hydrolysis. Monosaccharide analysis confirmed the presence of fucosamine and quinovosamine in both products. In the case of the wild-type strain, mild acid hydrolysis gave a polysaccharide, although to some extent the O chain was depolymerized. The mutant LPS produced a trisaccharide and no polymer.
The polysaccharide of the wild-type strain was studied using two-dimensional NMR (COSY, total correlation spectroscopy, nuclear Overhauser effect spectroscopy, HSQC, and HMBC) (Table 2) and was confirmed to have the previously described structure of serotype O12 O antigen (12) (Fig. 2D). In particular, the amidine group was identified by its characteristic signals at 20.4 (methyl group carbon) and 169 ppm (carbonyl carbon). The amidine group was connected to N-2 of fucosamine, which was determined from HMBC correlation between the amidine CO and H-2 of the fucosamine residue from HMBC correlation. The monosaccharide sequence was determined on the basis of nuclear Overhauser effect and HMBC correlations. Thus, the polysaccharide has the following structure:
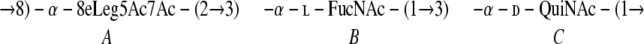 |
where 8eLeg5Ac7Ac is 5,7-diacetamido-3,5,7,9-tetradeoxy-l-glycero-d-galacto-nonulosonic acid and QuiNAc is 2-acetamido-2,6-dideoxyglucose.
TABLE 2.
NMR data for the Pseudomonas aeruginosa serotype O12 polysaccharide (40°C) and the lfnA mutant repeating unit trisaccharide (25°C)a
| Unitb | NMR spectrum | Signal (ppm) at the following positionc:
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2/3ax | 3(3eq) | 4 | 5 | 6 | 7 | 8 | 9 | ||
| PS 8eLeg5Ac7Ac (A) | 1H | 1.68 | 2.64 | 3.54 | 3.71 | 4.10 | 3.95 | 3.72 | 1.33 | |
| 13C | 173.7 | 105.0 | 42.4 | 69.2 | 53.8 | 73.6 | 54.7 | 73.2 | 14.9 | |
| PS 8eLeg5Ac7Ac, Ac (2x) | 1H | 1.98 | ||||||||
| 13C | 175.3 | 23.1 | ||||||||
| TS 8eLeg5Ac7Ac (A) | 1H | 1.87 | 2.30 | 3.94 | 3.75 | 4.12 | 3.98 | 3.75 | 1.11 | |
| 13C | 174.5 | 97.1 | 40.6 | 68.4 | 54.0 | 71.7 | 54.0 | 73.7 | 15.7 | |
| PS FucNAm (B) | 1H | 5.13 | 3.98 | 4.07 | 4.02 | 4.43 | 1.22 | |||
| 13C | 96.7 | 52.3 | 75.6 | 72.4 | 67.3 | 17.1 | ||||
| PS FucNAm, Am | 1H | 2.29 | ||||||||
| 13C | 167.5 | 20.4 | ||||||||
| TS FucNAc (B) | 1H | 4.95 | 4.08 | 3.87 | 3.79 | 4.41 | 1.18 | |||
| 13C | 98.6 | 50.8 | 69.1 | 72.4 | 67.9 | 16.7 | ||||
| TS FucNAc, Ac | 1H | 2.01 | ||||||||
| 13C | 175.7 | 23.5 | ||||||||
| PS QuiNAc (C) | 1H | 4.84 | 4.11 | 3.63 | 3.32 | 3.65 | 1.32 | |||
| 13C | 94.1 | 54.6 | 76.7 | 74.6 | 69.6 | 18.0 | ||||
| PS QuiNAc, Ac | 1H | 2.08 | ||||||||
| 13C | 175.0 | 23.3 | ||||||||
| TS QuiNAc (C) | 1H | 4.79 | 4.03 | 3.57 | 3.24 | 3.63 | 1.27 | |||
| 13C | 94.6 | 54.8 | 76.8 | 74.9 | 69.7 | 18.0 | ||||
Acetyl signals of the residues A and C in the trisaccharide (1H/13C): 1.95/23.0; 1.98/23.4; 2.03/23.4; all C-1 at 175.4 ppm.
Abbreviations: PS, wild-type polysaccharide; Ac (2x), two acetyl groups; TS, the lfnA mutant repeating unit trisaccharide; Am, acetimldoyl.
3ax, axial proton in 8eLeg5Ac7Ac residue; 3eq, equatorial proton in 8eLeg5Ac7Ac.
FIG. 2.

13C-1H correlated HSQC NMR spectra of the repeating unit trisaccharide of the lfnA mutant (A) and of the polysaccharide of the complemented lfnA mutant (B). Circles highlight the position in panel B of the characteristic methyl signal of the acetamidino group, which is not observed in the lfnA mutant. (C) Deduced structure of the lfnA mutant O polysaccharide. (D) Structure of the wild-type (or complemented lfnA mutant) O polysaccharide.
The trisaccharide obtained by mild acid hydrolysis of the lfnA mutant LPS had the nonulosonic acid at the reducing end in the anomeric configuration opposite the configuration observed in the wild-type polysaccharide, as could be concluded from the difference in position of 1H signals of its methylene group (Table 2). This trisaccharide contained four N-acetyl groups and no amidine (Table 2). The difference in the stability of the polymers with and without the amidine group, with respect to depolymerization in the conditions of mild hydrolysis, may be due to a stabilizing influence of the positively charged amidine group on the nearby anomeric center of the nonulosonic acid residue. The structure of the mild acid-released trisaccharide is therefore:
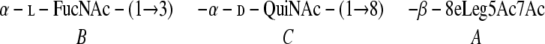 |
The trisaccharide structure represents the chemical repeating unit of the polysaccharide which is different from the biological repeating unit (12). Using these data, the structure of the lfnA mutant O polysaccharide was deduced (Fig. 2C).
The amidine signal which is absent in NMR analysis of the lfnA mutant O polysaccharide was restored in the NMR spectrum of the O polysaccharide from the complemented mutant (Fig. 2B), verifying that the O-antigen structure was restored to the wild-type polysaccharide by supplying lfnA in trans.
Expression of His6-lfnA in other P. aeruginosa serotypes has no apparent effect on the l-FucNAc sugars in their respective O-antigen repeats.
The B-band O-antigen repeats of P. aeruginosa serotypes O4 and O11 contain l-FucNAc residues (as well as other sugars). Since mutation of lfnA in P. aeruginosa O12 resulted in expression of l-FucNAc instead of l-FucNAm within the O repeat, we were interested to see whether introduction of lfnA into serotypes O4 and O11 would cause a change from l-FucNAc to l-FucNAm in the O-antigen repeats in these strains. The lfnA complementation plasmid pUCP20-his6-lfnA enabled us to answer this question: after electroporation of this plasmid into these strains, no discernible changes in the LPS were observed based upon silver-stained SDS-PAGE and Western blot analyses (data not shown).
Cross-complementation of the lfnA mutation with wbuX from enterohemorrhagic E. coli O145.
One of the closest homologues of lfnA in the DNA databases is wbuX from E. coli O145. The predicted proteins encoded by these genes share 71% sequence identity. Since E. coli O145, like P. aeruginosa O12, also expresses l-FucNAm, we hypothesized that the two genes are functionally interchangeable. wbuX was PCR amplified from three E. coli O145 strains, N 00 6496, N 01 2051, and N 02 5149. The wbuX sequence in all three strains was identical to that previously reported (NCBI protein ID AAV74532.1) (24). When wbuX was supplied on the shuttle vector pUCP20 in the lfnA mutant, it restored the wild-type LPS profile in SDS-PAGE analyses (Fig. 3). The gene was also able to complement the mutation with codons for an N-terminal His6 tag at its 5′ end (Fig. 3).
FIG. 3.

Cross-complementation of lfnA mutation by expression of wbuX in trans. LPS was analyzed by SDS-PAGE and silver staining (A) or by Western blotting using serotype O12-specific, polyclonal, primary antibodies (B). Maintenance of either one of the cross-complementation plasmids, pUCP20-wbuX or pUCP20-his6-wbuX, within the lfnA mutant restores the wild-type O-antigen banding pattern.
His6-LfnA and His6-WbuX localize to the membrane fraction.
During the process of expression and purification of His6-LfnA in E. coli, we observed that the protein localized predominantly to the membrane fraction (data not shown). Therefore, in order to establish whether a membrane location was due to intrinsic properties of the protein or just an artifact of overexpression in E. coli, we wanted to investigate the cellular localization of His6-LfnA in P. aeruginosa. We examined the subcellular localization of His6-LfnA when it was expressed from the complementing vector pUCP20-rbs-his6-lfnA in the lfnA mutant strain. The complementation experiments indicated that in these conditions, the His6-lfnA gene is able to express active protein. Cellular fractions were isolated by differential centrifugation, and His6-tagged proteins were detected by Western blotting (Fig. 4). A band of the correct apparent molecular weight and which reacted with the anti-His6 tag antibody was observed in the membrane fraction, but no His6-LfnA was detected in the soluble fraction. To examine the localization of this protein when overexpressed, His6-lfnA was subcloned into the Pseudomonas protein expression vector pVLT31, and expression of His6-LfnA was induced in P. aeruginosa strain PAO1. In these conditions also, His6-LfnA localized to the membrane fraction (data not shown).
FIG. 4.

Localization of His6-LfnA and His6-WbuX expressed in the P. aeruginosa serotype O12 lfnA mutant. Cells were grown harboring the complementation vector pUCP20-rbs-his6-lfnA (lanes 2 to 4), the cross-complementation vector pCs2UCP20-rbs-his6-wbuX (lanes 5 to 7), or the empty vector, pUCP20 (lanes 8 to 10). Cellular fractions were isolated by differential centrifugation and then analyzed by SDS-PAGE. Proteins were visualized by SimplyBlue SafeStain staining of the gel (A) or by Western blotting using anti-His6-tag primary antibodies (B). Arrows indicate the positions of His6-LfnA and His6-WbuX in the blot. Exposure of the blot to the developing solution for a longer time did not reveal any His6-tagged proteins in the soluble fraction lanes (data not shown). The positions of molecular weight markers (in thousands) are shown to the left of the gels.
With a view to future in vitro studies on these proteins, we also investigated the localization of His6-WbuX. Had His6-WbuX proven to be a soluble protein, this would have facilitated purification of this LfnA functional homologue for biochemical studies. His6-WbuX expressed from the cross-complementing plasmid pCs2UCP20-rbs-his6-wbuX in the P. aeruginosa lfnA mutant also localized to the membranes (Fig. 4), as did His6-WbuX overexpressed from the pBf3ET28a-his6-wbuX vector in E. coli BL21(DE3) (data not shown).
DISCUSSION
We have characterized the phenotype of a P. aeruginosa serotype O12 lfnA mutant by SDS-PAGE and Western blot analysis of LPS and at the molecular level by elucidating the chemical structure of the O antigen using NMR techniques. The phenotype of this mutant, which produces l-FucNAc instead of l-FucNAm in its O antigen, is similar to that of the pseA mutant in C. jejuni (42), which lacked the acetamidino group normally present on the flagellin glycan. While the pseA mutant was not complemented and therefore possible polar effects of that mutation cannot be ruled out, we have complemented the lfnA mutation by expression of His6-lfnA from a plasmid and therefore present the first unequivocal evidence that genes in this family are required for the expression of the acetamidino moieties found in bacterial glycans.
Mutation of another close homologue of lfnA, wbpG from P. aeruginosa serotype O5, results in expression of semirough LPS, i.e., the lipid A-core is substituted with only a single O-antigen unit (50). This phenotype resembles that of an O-antigen polymerase mutant, wzy, and the implication has been drawn that the acetamidino modification of the nonreducing terminal sugar in the O-antigen repeat in this serotype (2,3-dideoxy-2-acetamido-3-acetamidino-d-mannuronic acid) is required for recognition by Wzy (50). In P. aeruginosa serotype O12, the acetamidino modification is located on the middle sugar of the O repeat, and it may be that the polymerase exhibits more-relaxed substrate specificity with respect to residues which are more distant from the sites of Wzy-catalyzed glycosidic bond formation.
Successful complementation of the lfnA mutation with wbuX verifies the hypothesis that lfnA and wbuX are functional homologues in P. aeruginosa. The successful complementation with His6-tagged versions of both genes demonstrates that these fusions are functional in vivo and that these constructs will prove useful for future purification and in vitro characterization of these two proteins.
Neither LfnA nor WbuX has a predicted transmembrane helix, nor any other motifs which would indicate membrane association; therefore, neither was anticipated to localize to the membrane fraction. This subcellular localization may occur as a result of protein-protein interactions with an integral membrane protein. His6-WbuX remains entirely membrane associated, however, even when overexpressed from the pET28a vector and therefore likely to vastly outnumber potential interaction partners. This suggests that the membrane association is more likely due to intrinsic properties of the protein itself. Monotopic membrane proteins, such as prostaglandin H2 synthase, can associate with lipid bilayers through membrane binding domains which do not span the membrane but have high local concentrations of positively charged and aromatic residues (27, 45, 62). An alignment of LfnA and WbuX sequences shows several conserved stretches of sequence which are particularly enriched for these amino acid side chains. These patches are distributed throughout the sequence, and therefore, it would probably be far from trivial to engineer these proteins to make a soluble, yet still functional truncated enzyme. As a consequence, in vitro studies will require detergent solubilization of the protein to facilitate purification. While this is beyond the scope of the present work, knowledge of the LfnA and WbuX membrane localization will be useful for future studies of these proteins.
Expression of His6-lfnA in other P. aeruginosa serotypes, which produce O-antigen repeats containing l-FucNAc residues, did not affect the LPS-banding profile in SDS-PAGE analysis in these strains. Mutation of the O-antigen repeat by knocking out lfnA in P. aeruginosa O12 dramatically increased the electrophoretic mobility of O-antigen-containing LPS molecules. The most important factor causing this band shift is probably a change in the charge of the LPS molecule. The pKa of the amidine group is expected to be approximately 11 (33) so that at neutral pH, and in the SDS-PAGE running conditions, the FucNAm sugars will be positively charged, therefore retarding migration toward the anode. In this context, it is very likely that incorporation of l-FucNAm into serotype O4 and O11 LPS would result in a band shift in SDS-PAGE. Therefore, we conclude that l-FucNAm is not incorporated as a result of His6-lfnA expression in these strains. This may indicate that the lfnA substrate is not present in these serotypes. The substrate for lfnA may be UDP-l-FucNAc. This is the O-antigen precursor synthesized from UDP-d-GlcNAc by WbjB, WbjC, and WbjD in P. aeruginosa O11 (35, 44) These three genes are present in the O12 B-band locus (49), and the acetamidino group could conceivably be synthesized by direct amidotransfer to the UDP-l-FucNAc acetamido group. At present, however, there is no experimental evidence that UDP-l-FucNAc is the LfnA substrate, and amidotransfer may occur at a later stage in O-antigen biosynthesis. Alternatively, a proportion of the UDP-l-FucNAc in the sugar nucleotide pool may be converted to UDP-l-FucNAm in these experiments, but the l-FucNAm is not incorporated into LPS by downstream enzymes due to substrate specificities of the glycosyl transferase, O-antigen flippase, or O-antigen polymerase or all three. Certainly, the sugar-nucleotide pool must retain sufficient quantities of UDP-l-FucNAc in these experiments to enable expression of wild-type LPS. Another possibility is that expression of His6-lfnA alone in the other P. aeruginosa serotypes is not sufficient for amidotransfer to occur; other factors may be required.
In conclusion, we have presented genetic data to indicate the roles of lfnA and wbuX in l-FucNAm biosynthesis and substantiated these with NMR analysis of LPS prepared from the lfnA mutant. We have demonstrated that His6-tagged fusions of LfnA and WbuX retain their function and shown that these proteins localize to the membranes. This will facilitate biochemical studies of this pathway. These genes may be particularly amenable to in vitro studies, since their putative substrate UDP-l-FucNAc can by synthesized in vitro from UDP-d-GlcNAc using WbjB, WbjC, and WbjD in a well-characterized pathway (35, 44), and therefore, enzyme-substrate assays should be possible in the future.
Supplementary Material
Acknowledgments
Genomic DNA from the pathogenic E. coli O145 strains N 00 6496, N 01 2051, and N 02 5149 was a gift from Mohamed Karmali at the Public Health Agency of Canada, Laboratory for Food-borne Zoonoses, Guelph, Ontario, Canada.
This work was supported by an operating grant from the Canadian Institute of Health Research (grant MOP-14687). E.F.M. was a recipient of a studentship from the Canadian Cystic Fibrosis Foundation, and J.S.L. holds a Canada Research Chair in Cystic Fibrosis and Microbial Glycobiology.
Footnotes
Published ahead of print on 21 December 2007.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Allemeersch, D., J. Beumer, M. Devleeschouwer, S. De Maeyer, J. Dony, C. Godard, P. Osterrieth, A. Pithsy, P. Van der Auwera, and H. Van Poppel. 1988. Marked increase of Pseudomonas aeruginosa serotype O12 in Belgium since 1982. Eur. J. Clin. Microbiol. Infect. Dis. 7265-269. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bert, F., and N. Lambert-Zechovsky. 1996. Comparative distribution of resistance patterns and serotypes in Pseudomonas aeruginosa isolates from intensive care units and other wards. J. Antimicrob. Chemother. 37809-813. [DOI] [PubMed] [Google Scholar]
- 4.Beutin, L., G. Krause, S. Zimmermann, S. Kaulfuss, and K. Gleier. 2004. Characterization of Shiga toxin-producing Escherichia coli strains isolated from human patients in Germany over a 3-year period. J. Clin. Microbiol. 421099-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutin, L., S. Zimmermann, and K. Gleier. 1998. Human infections with Shiga toxin-producing Escherichia coli other than serogroup O157 in Germany. Emerg. Infect. Dis. 4635-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beynon, L. M., J. C. Richards, and M. B. Perry. 1994. The structure of the lipopolysaccharide O antigen from Yersinia ruckeri serotype 01. Carbohydr. Res. 256303-317. [DOI] [PubMed] [Google Scholar]
- 7.Blake, M. S., K. H. Johnston, G. J. Russell-Jones, and E. C. Gotschlich. 1984. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal. Biochem. 136175-179. [DOI] [PubMed] [Google Scholar]
- 8.Bopp, C. A., K. D. Greene, F. P. Downes, E. G. Sowers, J. G. Wells, and I. K. Wachsmuth. 1987. Unusual verotoxin-producing Escherichia coli associated with hemorrhagic colitis. J. Clin. Microbiol. 251486-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bork, P., and E. V. Koonin. 1994. A P-loop-like motif in a widespread ATP pyrophosphatase domain: implications for the evolution of sequence motifs and enzyme activity. Proteins 20347-355. [DOI] [PubMed] [Google Scholar]
- 10.Brauner, A., S. J. Cryz, M. Granstrom, H. S. Hanson, L. Lofstrand, B. Strandvik, and B. Wretlind. 1993. Immunoglobulin G antibodies to Pseudomonas aeruginosa lipopolysaccharides and exotoxin A in patients with cystic fibrosis or bacteremia. Eur. J. Clin. Microbiol. Infect. Dis. 12430-436. [DOI] [PubMed] [Google Scholar]
- 11.Burnette, W. N. 1981. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112195-203. [DOI] [PubMed] [Google Scholar]
- 12.Bystrova, O. V., B. Lindner, H. Moll, N. A. Kocharova, Y. A. Knirel, U. Zahringer, and G. B. Pier. 2003. Structure of the lipopolysaccharide of Pseudomonas aeruginosa O-12 with a randomly O-acetylated core region. Carbohydr. Res. 3381895-1905. [DOI] [PubMed] [Google Scholar]
- 13.Choi, K. H., A. Kumar, and H. P. Schweizer. 2006. A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64391-397. [DOI] [PubMed] [Google Scholar]
- 14.Cryz, S. J., Jr., T. L. Pitt, E. Furer, and R. Germanier. 1984. Role of lipopolysaccharide in virulence of Pseudomonas aeruginosa. Infect. Immun. 44508-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daniels, C., C. Griffiths, B. Cowles, and J. S. Lam. 2002. Pseudomonas aeruginosa O-antigen chain length is determined before ligation to lipid A core. Environ. Microbiol. 4883-897. [DOI] [PubMed] [Google Scholar]
- 16.Dasgupta, T., T. R. de Kievit, H. Masoud, E. Altman, J. C. Richards, I. Sadovskaya, D. P. Speert, and J. S. Lam. 1994. Characterization of lipopolysaccharide-deficient mutants of Pseudomonas aeruginosa derived from serotypes O3, O5, and O6. Infect. Immun. 62809-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Lorenzo, V., L. Eltis, B. Kessler, and K. N. Timmis. 1993. Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 12317-24. [DOI] [PubMed] [Google Scholar]
- 18.Diver, J. M., L. E. Bryan, and P. A. Sokol. 1990. Transformation of Pseudomonas aeruginosa by electroporation. Anal. Biochem. 18975-79. [DOI] [PubMed] [Google Scholar]
- 19.Eklund, M., K. Leino, and A. Siitonen. 2002. Clinical Escherichia coli strains carrying stx genes: stx variants and stx-positive virulence profiles. J. Clin. Microbiol. 404585-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eklund, M., F. Scheutz, and A. Siitonen. 2001. Clinical isolates of non-O157 Shiga toxin-producing Escherichia coli: serotypes, virulence characteristics, and molecular profiles of strains of the same serotype. J. Clin. Microbiol. 392829-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elaichouni, A., G. Verschraegen, G. Claeys, M. Devleeschouwer, C. Godard, and M. Vaneechoutte. 1994. Pseudomonas aeruginosa serotype O12 outbreak studied by arbitrary primer PCR. J. Clin. Microbiol. 32666-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emori, T. G., and R. P. Gaynes. 1993. An overview of nosocomial infections, including the role of the microbiology laboratory. Clin. Microbiol. Rev. 6428-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enderle, P. J., and M. A. Farwell. 1998. Electroporation of freshly plated Escherichia coli and Pseudomonas aeruginosa cells. BioTechniques 25954-958. [DOI] [PubMed] [Google Scholar]
- 24.Feng, L., S. N. Senchenkova, J. Tao, A. S. Shashkov, B. Liu, S. D. Shevelev, P. R. Reeves, J. Xu, Y. A. Knirel, and L. Wang. 2005. Structural and genetic characterization of enterohemorrhagic Escherichia coli O145 O antigen and development of an O145 serogroup-specific PCR assay. J. Bacteriol. 187758-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn, R. D., J. Mistry, B. Schuster-Bockler, S. Griffiths-Jones, V. Hollich, T. Lassmann, S. Moxon, M. Marshall, A. Khanna, R. Durbin, S. R. Eddy, E. L. Sonnhammer, and A. Bateman. 2006. Pfam: clans, web tools and services. Nucleic Acids Res. 34D247-D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fomsgaard, A., M. A. Freudenberg, and C. Galanos. 1990. Modification of the silver staining technique to detect lipopolysaccharide in polyacrylamide gels. J. Clin. Microbiol. 282627-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fowler, P. W., and P. V. Coveney. 2006. A computational protocol for the integration of the monotopic protein prostaglandin H2 synthase into a phospholipid bilayer. Biophys. J. 91401-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedrich, A. W., M. Bielaszewska, W. L. Zhang, M. Pulz, T. Kuczius, A. Ammon, and H. Karch. 2002. Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 18574-84. [DOI] [PubMed] [Google Scholar]
- 29.Gay, P., D. Le Coq, M. Steinmetz, T. Berkelman, and C. I. Kado. 1985. Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J. Bacteriol. 164918-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grattard, F., O. G. Gaudin, B. Pozzetto, A. Ros, and A. D. Mbida. 1993. Genotypic homogeneity of nosocomial Pseudomonas aeruginosa O12 strains demonstrated by analysis of protein profiles, DNA fingerprints and rRNA gene restriction patterns. Eur. J. Clin. Microbiol. Infect. Dis. 1257-61. [DOI] [PubMed] [Google Scholar]
- 31.Hancock, R. E., and A. M. Carey. 1979. Outer membrane of Pseudomonas aeruginosa: heat- and 2-mercaptoethanol-modifiable proteins. J. Bacteriol. 140902-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helmark, S., M. E. Hansen, B. Jelle, K. I. Sorensen, and P. R. Jensen. 2004. Transformation of Leuconostoc carnosum 4010 and evidence for natural competence of the organism. Appl. Environ. Microbiol. 703695-3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilal, S. H., S. W. Karickhoff, and L. A. Carreira. 1995. A rigorous test for SPARC's chemical reactivity models: estimation of more than 4300 ionization pKas. Quant. Struct.-Act. Relat. 14348-355. [Google Scholar]
- 34.Hitchcock, P. J., and T. M. Brown. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154269-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kneidinger, B., K. O'Riordan, J. Li, J. R. Brisson, J. C. Lee, and J. S. Lam. 2003. Three highly conserved proteins catalyze the conversion of UDP-N-acetyl-d-glucosamine to precursors for the biosynthesis of O antigen in Pseudomonas aeruginosa O11 and capsule in Staphylococcus aureus type 5. Implications for the UDP-N-acetyl-l-fucosamine biosynthetic pathway. J. Biol. Chem. 2783615-3627. [DOI] [PubMed] [Google Scholar]
- 36.Knirel, Y. A., O. V. Bystrova, N. A. Kocharova, U. Zahringer, and G. B. Pier. 2006. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J. Endotoxin Res. 12324-336. [DOI] [PubMed] [Google Scholar]
- 37.Knirel, Y. A., E. V. Vinogradov, A. S. Shashkov, B. A. Dmitriev, N. K. Kochetkov, E. S. Stanislavsky, and G. M. Mashilova. 1987. Somatic antigens of Pseudomonas aeruginosa. The structure of the O-specific polysaccharide chain of the lipopolysaccharide from P. aeruginosa O13 (Lanyi). Eur. J. Biochem. 163627-637. [DOI] [PubMed] [Google Scholar]
- 38.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 39.Legakis, N. J., M. Aliferopoulou, J. Papavassiliou, and M. Papapetropoulou. 1982. Serotypes of Pseudomonas aeruginosa in clinical specimens in relation to antibiotic susceptibility. J. Clin. Microbiol. 16458-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacLean, L. L., M. B. Perry, E. M. Crump, and W. W. Kay. 2003. Structural characterization of the lipopolysaccharide O-polysaccharide antigen produced by Flavobacterium columnare ATCC 43622. Eur. J. Biochem. 2703440-3446. [DOI] [PubMed] [Google Scholar]
- 41.Marchler-Bauer, A., J. B. Anderson, P. F. Cherukuri, C. DeWeese-Scott, L. Y. Geer, M. Gwadz, S. He, D. I. Hurwitz, J. D. Jackson, Z. Ke, C. J. Lanczycki, C. A. Liebert, C. Liu, F. Lu, G. H. Marchler, M. Mullokandov, B. A. Shoemaker, V. Simonyan, J. S. Song, P. A. Thiessen, R. A. Yamashita, J. J. Yin, D. Zhang, and S. H. Bryant. 2005. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 33D192-D196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNally, D. J., J. P. Hui, A. J. Aubry, K. K. Mui, P. Guerry, J. R. Brisson, S. M. Logan, and E. C. Soo. 2006. Functional characterization of the flagellar glycosylation locus in Campylobacter jejuni 81-176 using a focused metabolomics approach. J. Biol. Chem. 28118489-18498. [DOI] [PubMed] [Google Scholar]
- 43.Millesimo, M., G. de Intinis, M. G. Chirillo, T. Musso, and D. Savoia. 1996. Pseudomonas aeruginosa clinical isolates: serotypes, resistance phenotypes and plasmid profiles. Eur. J. Epidemiol. 12123-129. [DOI] [PubMed] [Google Scholar]
- 44.Mulrooney, E. F., K. K. Poon, D. J. McNally, J. R. Brisson, and J. S. Lam. 2005. Biosynthesis of UDP-N-acetyl-l-fucosamine, a precursor to the biosynthesis of lipopolysaccharide in Pseudomonas aeruginosa serotype O11. J. Biol. Chem. 28019535-19542. [DOI] [PubMed] [Google Scholar]
- 45.Nina, M., S. Berneche, and B. Roux. 2000. Anchoring of a monotopic membrane protein: the binding of prostaglandin H2 synthase-1 to the surface of a phospholipid bilayer. Eur. Biophys. J. 29439-454. [DOI] [PubMed] [Google Scholar]
- 46.Pirnay, J. P., D. De Vos, C. Cochez, F. Bilocq, J. Pirson, M. Struelens, L. Duinslaeger, P. Cornelis, M. Zizi, and A. Vanderkelen. 2003. Molecular epidemiology of Pseudomonas aeruginosa colonization in a burn unit: persistence of a multidrug-resistant clone and a silver sulfadiazine-resistant clone. J. Clin. Microbiol. 411192-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pirnay, J. P., D. De Vos, C. Cochez, F. Bilocq, A. Vanderkelen, M. Zizi, B. Ghysels, and P. Cornelis. 2002. Pseudomonas aeruginosa displays an epidemic population structure. Environ. Microbiol. 4898-911. [DOI] [PubMed] [Google Scholar]
- 48.Pitt, T. L., D. M. Livermore, D. Pitcher, A. C. Vatopoulos, and N. J. Legakis. 1989. Multiresistant serotype O 12 Pseudomonas aeruginosa: evidence for a common strain in Europe. Epidemiol. Infect. 103565-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raymond, C. K., E. H. Sims, A. Kas, D. H. Spencer, T. V. Kutyavin, R. G. Ivey, Y. Zhou, R. Kaul, J. B. Clendenning, and M. V. Olson. 2002. Genetic variation at the O-antigen biosynthetic locus in Pseudomonas aeruginosa. J. Bacteriol. 1843614-3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocchetta, H. L., L. L. Burrows, and J. S. Lam. 1999. Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 63523-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheutz, F., B. Olesen, and A. Norgaard. 2000. Two cases of human urinary tract infection complicated by hemolytic uremic syndrome caused by verotoxin-producing Escherichia coli. Clin. Infect. Dis. 31815-816. [DOI] [PubMed] [Google Scholar]
- 52.Schweizer, H. D. 1993. Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. BioTechniques 15831-834. [PubMed] [Google Scholar]
- 53.Schweizer, H. P., and T. T. Hoang. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 15815-22. [DOI] [PubMed] [Google Scholar]
- 54.Simon, R., U. Priefer, and A. Puhler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram-negative bacteria. Bio/Technology 1784-791. [Google Scholar]
- 55.Smith, A. W., and B. H. Iglewski. 1989. Transformation of Pseudomonas aeruginosa by electroporation. Nucleic Acids Res. 1710509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 764350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tozzi, A. E., A. Caprioli, F. Minelli, A. Gianviti, L. De Petris, A. Edefonti, G. Montini, A. Ferretti, T. De Palo, M. Gaido, and G. Rizzoni. 2003. Shiga toxin-producing Escherichia coli infections associated with hemolytic uremic syndrome, Italy, 1988-2000. Emerg. Infect. Dis. 9106-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vieu, J. F., G. Allos, B. Hassan-Massoud, M. O. Santos-Ferreira, and G. Tselentis. 1984. Is there a geographic epidemiology of serogroups O of Pseudomonas aeruginosa? Bull. Soc. Pathol. Exot. Filiales 77288-294. (In French.) [PubMed] [Google Scholar]
- 59.Vinogradov, E. V., Y. A. Knirel, A. S. Shashkov, N. A. Paramonov, N. K. Kochetkov, E. S. Stanislavsky, and E. V. Kholodkova. 1994. The structure of the O-specific polysaccharide of Salmonella arizonae O21 (Arizona 22) containing N-acetylneuraminic acid. Carbohydr. Res. 25959-65. [DOI] [PubMed] [Google Scholar]
- 60.Vinogradov, E. V., A. S. Shashkov, Y. A. Knirel, N. K. Kochetkov, J. Dabrowski, H. Grosskurth, E. S. Stanislavsky, and E. V. Kholodkova. 1992. The structure of the O-specific polysaccharide chain of the lipopolysaccharide of Salmonella arizonae O61. Carbohydr. Res. 2311-11. [DOI] [PubMed] [Google Scholar]
- 61.Welinder-Olsson, C., and B. Kaijser. 2005. Enterohemorrhagic Escherichia coli (EHEC). Scand. J. Infect. Dis. 37405-416. [DOI] [PubMed] [Google Scholar]
- 62.Wendt, K. U., K. Poralla, and G. E. Schulz. 1997. Structure and function of a squalene cyclase. Science 2771811-1815. [DOI] [PubMed] [Google Scholar]
- 63.West, S. E., H. P. Schweizer, C. Dall, A. K. Sample, and L. J. Runyen-Janecky. 1994. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene 14881-86. [DOI] [PubMed] [Google Scholar]
- 64.Westphal, O., and K. Jann. 1965. Bacterial lipopolysaccharides: extraction with phenol-water and further applications of this procedure. Methods Carbohydr. Chem. 583-91. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.