

Abstract
During productive infection, human cytomegalovirus (HCMV) UL44 transcription initiates at three distinct start sites that are differentially regulated. Two of the start sites, the distal and the proximal, are active at early times, whereas the middle start site is active only at late times after infection. The UL44 early viral gene product is essential for viral DNA synthesis. The UL44 gene product from the late viral promoter affects primarily viral gene expression at late times after infection rather than viral DNA synthesis (H. Isomura, M. F. Stinski, A. Kudoh, S. Nakayama, S. Iwahori, Y. Sato, and T. Tsurumi, J. Virol. 81:6197, 2007). The UL44 early viral promoters have a canonical TATA sequence, “TATAA.” In contrast, the UL44 late viral promoter has a noncanonical TATA sequence. Using recombinant viruses, we found that the noncanonical TATA sequence is required for the accumulation of late viral transcripts. The GC boxes that surround the middle TATA element did not affect the kinetics or the start site of UL44 late transcription. Replacement of the distal TATA element with a noncanonical TATA sequence did not affect the kinetics of transcription or the transcription start site, but it did induce an alternative transcript at late times after infection. The data indicate that a noncanonical TATA box is used at late times after HCMV infection.
Human cytomegalovirus (HCMV) is a member of the betaherpesvirus family. The genome of HCMV is 240,000 bp in size with at least 150 known open reading frames (ORFs) (4). A majority of the ORFs are nonessential for viral replication in cell culture. Several ORFs are beneficial but not required for viral replication. However, approximately one-quarter, or 41 ORFs, are required for viral replication (39). The virus replicates productively in terminally differentiated cells, such as fibroblasts, epithelial and endothelial cells, and monocyte-derived macrophages (7, 8, 13, 20, 30, 31, 36).
During productive infection, HCMV genes are expressed in a temporal cascade, designated immediate early (IE), delayed early, and late. The major IE genes UL123/UL122 (IE1/IE2) play a critical role in subsequent viral gene expression and the efficiency of viral replication (14, 15, 17, 22-24). The early viral genes encode proteins necessary for viral DNA replication (26). Following viral DNA replication, delayed early and late viral genes that encode structural proteins for viral production are expressed. Several early genes of HCMV have the unusual property of three promoters: two that initiate transcription early and one late (2, 21).
The UL44 protein (pUL44), which binds double-stranded DNA, is an essential protein for viral DNA replication and interacts specifically with the viral DNA polymerase encoded by UL54 (27, 29). pUL44 increases the processivity of the viral DNA polymerase along the viral DNA template (6, 37, 40). pUL44 protein accumulates to strikingly high levels at late times after infection (9, 35). The HCMV UL44 transcription unit initiates at three distinct sites, which are separated by approximately 50 nucleotides and are differentially regulated during productive infection. Two of these start sites, the distal and the proximal, are used at early times, whereas the middle start site is not used until late times (21). Expression from the late start site is dependent upon viral DNA synthesis. We have shown that mutation of the middle TATA element did not affect the level of viral DNA synthesis, but it did significantly affect the level of late viral gene expression (16). In addition, recombinant viruses with the middle TATA element mutated grew more slowly than wild-type virus at both low and high multiplicities of infection (MOI) (16). From these results, we concluded that the late promoter of the HCMV viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products independently of the level of viral DNA synthesis.
Why the middle start site is activated at late times after infection is unclear. An important parameter governing transcription initiation is the relative concentrations of the viral DNA template and the promoter sequence. As discussed previously (21), there are several differences in the sequence surrounding the TATA element upstream of the middle start site. First, the TATA box sequence is different from the TATA elements upstream of the distal and the proximal start sites. Second, there is a region of perfect dyad symmetry located immediately 3′ of the TATA element. Third, the positioning of the transcription start site (TSS) from the TATA element is longer than those of the other TATA elements. Since the UL44 late promoter affects delayed early and late viral gene expression independently of viral DNA replication (16), it is important to determine how the middle start site is activated.
In this study, we report that the noncanonical TATA element in the UL44 middle promoter is required for the late TSS and the accumulation of late viral transcripts.
MATERIALS AND METHODS
Cells and virus.
Primary human foreskin fibroblasts (HFFs) were maintained in Eagle's minimal essential medium supplemented with 10% fetal calf serum (Sigma, St. Louis, MO), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in 5% CO2 as described previously (35). The titers of wild-type (wt-R) HCMV Towne and recombinant viruses were determined by standard plaque assays on HFFs as described previously (24). The viral DNA input was determined by infecting HFFs in 35- or 60-mm plates in triplicate and harvesting the cells at 4 h postinfection (p.i.) in PCR lysis buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.001% Triton X-100, and 0.001% sodium dodecyl sulfate) containing 50 μg/ml proteinase K. After incubation at 55°C for 100 min, the proteinase K was inactivated at 95°C for 10 min. The relative amount of input viral DNA was estimated by real-time PCR using HCMV gB primers and probes as described previously (17).
Enzymes.
Restriction endonucleases were purchased from New England Biolabs Inc. (Beverly, MA). High-fidelity and expanded high-fidelity Taq DNA polymerases were purchased from Invitrogen (Carlsbad, CA) and Roche (Mannheim, Germany), respectively, and RNasin and RNase-free DNase were purchased from Promega (Madison, WI). The enzymes were used according to the manufacturers' instructions.
Mutagenesis of HCMV BAC DNA.
A rapid homologous recombination system in Escherichia coli expressing the bacteriophage lambda recombination proteins exo, beta, and gam (provided by D. Court, NIH, Bethesda, MD) was employed as described previously (5). Bacterial artificial chromosome (BAC) DNA of HCMV Towne was obtained from F. Liu (University of California, Berkeley) (4). Double-stranded DNAs for recombination contained kanamycin resistance and streptomycin sensitivity genes (RpsLneo, purchased from Gene Bridges, Dresden, Germany) and 70 bp of homologous viral DNA sequence. To generate mutations of the UL44 promoter, the following primer pairs were used: dlUL44promoterF, 5′-GCTTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATTGGCCTGGTGATGATGGCGGGATC-3′; dlUL44promoterR, 5′-GAGCGAGCGAAAGTTTTATAGAGAGCACACACGACGACCGGGAACGCTGCGAAGACGCCCGGCGTCTAATTCAGAAGAACTCGTCAAGAAGG-3′; BACUL44TATA1neo+StF, 5′-TCGGGGATGACGCCCGACGTGCTTCTGGCCAGGATGCTCAAGTGGTACCACTGGCGCTTTAAGGTCGGAGGGCCTGGTGATGATGGCGGGATC-3′; and BACUL44TATA1neo+StR, 5′-GCGAAGACGCCCGGCGTCTAATAATACAGCCGCGCCGAGCCAGCGGGCCCCCGACTAAGAGGCACAGTACTCAGAAGAACTCGTCAAGAAGG-3′.
To generate the recombinant BAC DNA with the UL44 promoter mutated, the reverse procedure was also employed, as described previously (38). Briefly, 500 ng of single-stranded DNA for recombination containing 50 bp of homologous viral DNA sequence on either end of the mutated TATA or GC boxes was introduced into HCMV BAC DNA with the RpsLneo gene in the UL44 promoter as described previously (38). Since RpsL is a streptomycin sensitivity gene, the mutated BAC DNA was selected on the basis of increased streptomycin resistance by using the Counter Selection Modification kit (Gene Bridges) as described previously (38). The following single-stranded DNAs were used for recombination: BACUL44TATAcontrololigo, 5′-GCTTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATTATTAGACGCCGGGCGTCTTCGCAGCGTTCCCGGTCGTCGTGTGTGCTCTCTATAAAACTTTCGCTCGCTC-3′; BACmutTATA2oligo, 5′-CGCTTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATataaAGACGCCGGGCGTCTTCGCAGCGTTCCCGGTCGTCGTGTGTGCTCTCTATAAAACTTTCGCTCGCTCGCG-3′; BACUL44TATA2mut2oligo, 5′-CGCTTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATaATTAGACGCCGGGCGTCTTCGCAGCGTTCCCGGTCGTCGTGTGTGCTCTCTATAAAACTTTCGCTCGCTC-3′; BACUL44TATA2mut3oligo, 5′-TTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATTATaAGACGCCGGGCGTCTTCGCAGCGTTCCCGGTCGTCGTGTGTGCTCTCTATAAAACTTTCGCTCGCTCGCG-3′; BACUL44TATA2GCboxmutoligo-1, 5′-TGGCGCTTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCttCtCGGCTGTATTATTAGAaGatGttCtTCTTCGCAGCGTTCCCGGTCGTCGTGTGTGCTCTCTATAAAACTTTCGCTCGCTCGCGCC-3′; BACUL44TATA2GCboxmutoligo-2,5′-TGGCGCTTTAAGGTCGGAGTATATAAGTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATTATTAGAaGatGttCtTCTTCGCAGCGTTCCCGGTCGTCGTGTGTGCTCTCTATAAAACTTTCGCTCGCTCGCGCC-3′; and BACTATA1mutoligo, 5′-TCGGGGATGACGCCCGACGTGCTTCTGGCCAGGATGCTCAAGTGGTACCACTGGCGCTTTAAGGTCGGAGtattattaTACTGTGCCTCTTAGTCGGGGGCCCGCTGGCTCGGCGCGGCTGTATTATTAGACGCCGGGCGTCTTCGCA-3′. The lowercase letters represent mutant bases.
Recombinant virus isolation.
HFFs were transfected with either 5 or 10 μg of each recombinant BAC in the presence of 2 μg of plasmid pSVpp71 by the calcium phosphate precipitation method of Graham and Van der Eb (10). After 10 days, viral plaques appeared. After 7 days of 100% cytopathic effect, the extracellular fluid was collected and either not diluted or diluted 1:10 for infection of HFFs. After 5 to 7 days of 100% cytopathic effect, the extracellular fluid containing virus was stored at −80°C in 50% newborn calf serum until it was used.
PCR analysis.
PCR analysis was performed using the primer pair UL44promoterF, 5′-GCGATCCAAAACGACGTGGAAATGGCG-3′, and UL44promoterR, 5′-TGAGCGCACGGATCACAGATCGC-3′, as described previously (16). The PCR cycling program was as follows: 1 cycle of denaturation at 94°C for 2 min; 30 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, and elongation at 72°C for 1 min 30 s; and 1 cycle of elongation at 72°C for 7 min. A PCR product was cloned into a TA cloning vector and sequenced to confirm the recombination and excision (Aichi Cancer Center Research Institute Central Facility).
RNase protection assay.
For construction of the antisense UL44 control probe or UL44 mutant probe, a DNA fragment including the 5′ upstream region of the TSS of the entire UL44 transcript was amplified by PCR using the primer pair UL44 F2 RNase protection assay primer (5′-CCGCTGGCTCGGCGCGGCTG-3′) and UL44 inner primer (5′-GGATAGCCGTCTTGTACGGCTTCA-3′) and using BAC wild type (see Fig. 1) or BACGCmut2 (see Fig. 4a), respectively, for templates, as described previously (16), and cloned into the TA cloning vector pCRII (Invitrogen). The resulting clone, pUL44 pro-5, was made linear with the restriction endonuclease EcoRV and used as a template for SP6 RNA polymerase. Synthesis by SP6 RNA polymerase on linear pUL44 pro-5 DNA produced a 32P-labeled antisense RNA probe in agreement with the predicted size. Cytoplasmic RNAs from mock-infected or HCMV-infected HFFs were isolated at the indicated times after infection as described previously (2, 11). DNA replication was inhibited with 200 μg/ml phosphonoacetic acid (PAA) (Sigma, St. Louis, MO) added to the medium at the time of infection and maintained throughout infection. Twenty micrograms of RNA was hybridized to 32P-labeled antisense UL44 promoter probe at 37°C overnight before digestion with RNase T1 (100 U) as described previously (14, 19). The protected RNA fragments were subjected to electrophoresis in denaturing 6% polyacrylamide gels, followed by autoradiography on Hyperfilm MP (Amersham).
FIG. 1.

Structures of recombinant HCMV BAC DNAs. To construct theses mutants, a counterselection replaced the UL44 middle promoter with a marker cassette containing the RpsL gene, conferring increased sensitivity to streptomycin, and the neomycin resistance marker to provide kanamycin resistance. Intermediate BAC clones were isolated based on resistance to kanamycin. The integrity of these clones was checked by digestion with HindIII, and insertion of the marker cassette in the correct location was confirmed by PCR using the primer pair UL44promoterF and UL44promoterR. In a second round of homologous recombination, the entire marker cassette was replaced with the TATA2 control or TATA2 mutant sequence by counterselection using single-stranded DNA as described in Materials and Methods. The lowercase letters in the sequences indicate mutant bases. A rescued BAC with the UL44 TATA2 control sequence was used for the subsequent experiments as wt-R.
FIG. 4.

Effect of the mutated GC boxes in the UL44 middle promoter. (a) Schematic representation of the recombinant viruses replaced with GC boxes surrounding the UL44 middle TATA element. To construct the mutant BACs, we reversed the recombinant virus selection procedure using the RpsL gene as described in Materials and Methods. The lowercase letters in the sequences indicate mutant bases. (b and c) RNAs were harvested at 1, 2, and 3 days after infection with an MOI of approximately 3. After an RNase protection assay, the protected RNA fragments were subjected to electrophoresis in denaturing 6% polyacrylamide gels. Arrows 1, 2, and 3 indicate the transcripts initiating at start sites 1, 2, and 3, respectively. The arrowhead indicates the alternative transcript due to the substitution for the UL44 middle TATA element. The asterisk indicates the alternative band due to the mismatch of hybridization. (b) RNase protection assay with UL44 RNA probe. Lanes: 1, wt-R; 2, mut; 3 to 6, GCmut1; 7 to 10, GCmut2, -3, and -7 1 day p.i. (d.p.i.); 1, 2, 4, 5, 8, and 9, 2 days p.i.; 6 and 10, 3 days p.i.; 5 and 9, in the presence of PAA. (c) RNase protection assay with UL44 mut probe. Lanes: 1, UL44 mut probe lacking RNase T1; 2 to 5, GCmut1; 6 to 9, GCmut2; 10, wt-R; 2 and 6, 1 day p.i.; 3 and 4, 7 and 8, and 10, 2 days p.i.; 5 and 9, 3 days p.i.; 4 and 8, in the presence of PAA.
5′-RACE analysis.
Cytoplasmic RNA was isolated from the cells infected with RUL44TATA2 mut 2 days p.i. After treatment with DNase I (Promega, Madison, WI), RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) was performed using the FirstChoice RLM-RACE kit (Ambion, Austin, TX) following the manufacturer's instructions. Twenty micrograms of RNA was used for the RLM-RACE reaction. PCR primers to amplify the cDNA fragment containing the 5′ end of the UL44 TATA2-dependent transcript were 5′ RACE outer control primer (5′-GATCACCAATCCATTGCCGACTAT-3′) and UL44TATA2Router primer (5′-CCCGGACAGCGTGCAAGTCTCGACTAA-3′). After amplification of cDNA, a second nested PCR was performed. The primer pairs were 5′ RACE inner control primer (5′-CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG-3′) and UL44TATA2Rinner primer (5′-TAAGGAGCGGGCGCGAGCGAGCGAAA-3′). The nested-PCR product was cloned into a TA cloning vector and sequenced (Aichi Cancer Center Research Institute Central Facility).
RESULTS
Effects of the TATA sequence in the UL44 middle promoter on the kinetics of transcription and start site selection.
Figure 1 is a diagram of the UL44 gene and the three spatially distinct transcriptional start sites designated distal, middle, and proximal. The distal and proximal sites are early promoters, and the middle site is a late promoter that depends on viral DNA synthesis for activity. We showed previously that mutation of the middle TATA element significantly affected the level of late viral gene expression rather than viral DNA synthesis (16). What determines the kinetics of the UL44 late transcript start site is unclear. The UL44 early promoters have a canonical TATA sequence, “TATAA.” In contrast, the viral late or middle TATA element is a noncanonical sequence, “TATTATTA” (Fig. 1). To determine the significance of the noncanonical TATA sequence in UL44 late-gene expression from the middle promoter, we constructed recombinant viruses with the UL44 middle TATA sequence, “TATTATTA,” mutated to “TATataaA” to contain a canonical TATA sequence, “TATAA”. The lowercase letters indicate the mutated bases. Using a rapid homologous-recombination system in E. coli as described in Materials and Methods, we replaced the marker cassette with the RpsL gene (Gene Bridges), conferring increased sensitivity to streptomycin. Intermediate BAC clones were isolated based on resistance to kanamycin. The integrity of these clones was checked by digestion with HindIII, and the insertion of the marker cassette in the correct location was confirmed by PCR using the primer pair UL44promoterF and UL44promoterR (data not shown). In a second round of homologous recombination, the entire marker cassette was replaced with either the wild-type sequence (wt-R) or a mutant sequence (TATAmut) by counterselection using single-stranded DNA as described in Materials and Methods. Recombinant constructs were isolated based on increased resistance to streptomycin as described previously (38), and the UL44 promoter was amplified by PCR using the primer pair UL44promoterF and UL44promoterR. DNA sequencing confirmed the correct recombination (data not shown). The integrity of the mutant BACs was checked by digestion with HindIII (data not shown). There were no differences detected in transcription from the major IE gene promoter between the wild type, wt-R, and TATAmut (16 and data not shown).
Cytoplasmic RNA was harvested 1, 2, and 3 days after infection with either wt-R or TATAmut at an MOI of approximately 3, and an RNase protection assay was performed to detect all the transcripts derived from the different start sites. The antisense UL44 RNA probe was as described previously (16). Twenty micrograms of RNA was hybridized to 32P-labeled antisense RNA probe at 37°C overnight before digestion with RNase T1 (100 U) as described in Materials and Methods. Consistent with previous reports (16, 21), three major transcripts initiating at the spatially distinct start sites were detected 2 and 3 days after infection with wt-R (Fig. 2a, lanes 4 and 8). Transcripts initiating at start site 1 or 2 consisted of a doublet start site. The proximal and distal transcripts were similar between wt-R and TATAmut at 1, 2, and 3 days p.i. (Fig. 2a). Consistent with a previous report (16, 21), the middle transcript derived from the late promoter of wt-R was not detected in the presence of an inhibitor of viral DNA synthesis (PAA) at 48 h (Fig. 2a, lane 6). In contrast, an alternative transcript initiating upstream of start site 2 was detected with TATAmut in infected cells 1, 2, and 3 days p.i. (Fig. 2a, lanes 3, 5, and 9). The alternative transcript was not detected with wt-R in the presence of PAA at 48 h, but it was detected with TATAmut (Fig. 2a, compare lanes 6 and 7). The transcript initiating at start site 2 was also detected with TATAmut, but it was at very low levels at 2 and 3 days p.i. (Fig. 2a, lanes 5 and 9). Several minor bands with wt-R and TATAmut were also detected upstream of the proximal transcript or downstream of the distal transcript. When the same aliquot of RNA from wt-R or TATAmut was analyzed with the UL44 RNA probe, detection of these minor bands was variable for each assay (compare Fig. 2a to Fig. 3b, lanes 9 and 10; Fig. 4b, lanes 1 and 2; and Fig. 5b, lanes 6 to 8), suggesting that these transcripts do not represent heterogeneous start sites. It is likely that they represent incomplete hybridization before digestion with RNase T1. When the labeled UL44 RNA probe was synthesized, parts of the transcripts may not have reached full length due to pausing or termination of the SP6 RNA polymerase. Figure 2b shows the sequence of the UL44 middle promoter region. The precise mapping of the TSS of the middle transcripts was performed previously (21). We used RLM-RACE analysis to determine the start site of the alternative transcript with TATAmut as described in Materials and Methods. The TSS is located at a distance of 22 bp 3′ of the TATA element (Fig. 2b). From these results, we concluded that the sequence of the UL44 middle TATA nucleotides affects the kinetics and the TSS selection of the UL44 late transcript.
FIG. 2.
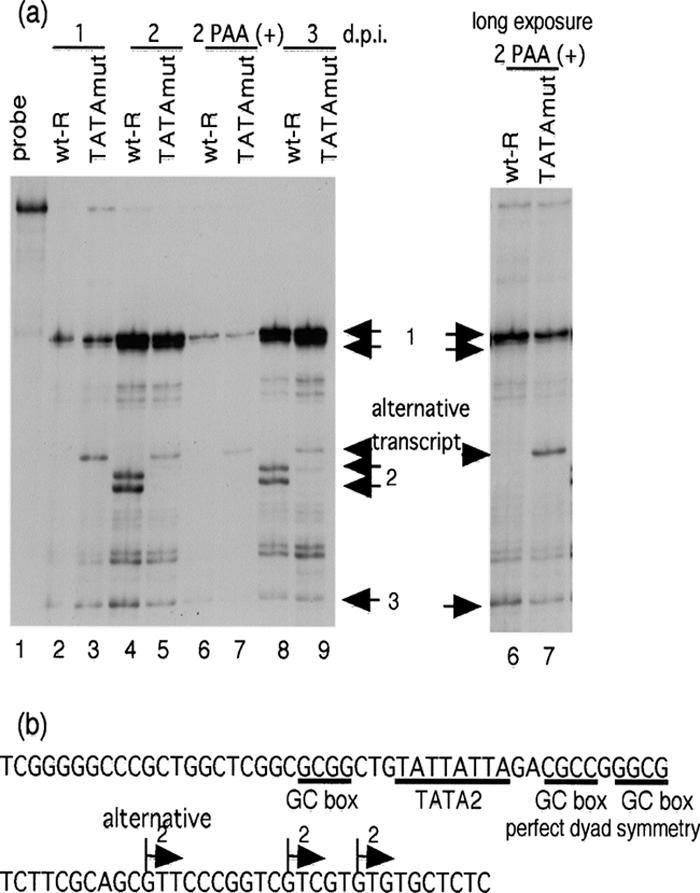
Effect of the UL44 middle TATA nucleotides on UL44 transcription in cells infected with wt-R and the recombinant virus. (a) Cytoplasmic RNAs were harvested at 1, 2, and 3 days after infection with an MOI of approximately 3. Twenty micrograms of RNA was hybridized to 32P-labeled antisense UL44 RNA probe at 37°C overnight before digestion with RNase T1. The antisense UL44 RNA probe contained sequence upstream of the TSS of all the UL44 transcripts. The protected RNA fragments were subjected to electrophoresis in denaturing 6% polyacrylamide gels. Lanes: 1, lacking RNase T1; 2, 4, 6, and 8, wt-R; 3, 5, 7, and 9, TATAmut; 2 and 3, 1 day p.i. (d.p.i.); 4 to 7, 2 days p.i.; 8 and 9, 3 days p.i.; 6 and 7, in the presence of PAA. Arrows 1, 2, and 3 indicate the transcripts initiating at start sites 1, 2, and 3, respectively. The arrowhead indicates the alternative transcript due to substitution for the UL44 middle TATA nucleotides. (b) Nucleotide sequence of the UL44 middle promoter region. The arrows above the sequence indicate the TSS of the transcripts dependent on the UL44 middle TATA element. The positions of the TATA element, GC boxes, and a region of perfect dyad symmetry are shown below the sequence.
FIG. 3.

Effect of a canonical TATA sequence in the UL44 middle promoter on the accumulation of late transcripts. (a) Schematic representation of the recombinant viruses replaced with a canonical TATA sequence. To construct the mutant BACs, we reversed the recombinant virus selection procedure using the RpsL gene (Fig. 1) as described in Materials and Methods. The lowercase letters in the sequences indicate mutant bases. (b) RNAs were harvested at 1, 2, and 3 days after infection with an MOI of approximately 3. An RNase protection assay was performed with 32P-labeled antisense UL44 RNA probe at 37°C overnight before digestion with RNase T1. The protected RNA fragments were subjected to electrophoresis in denaturing 6% polyacrylamide gels. Lanes: 1, 3, 4, and 7, mut2; 2, 5, 6, and 8, mut3; 9 and 10, wt-R; 11, mut; 12, lacking RNase T1; 1 and 2, 1 day p.i. (d.p.i.); 3 to 6 and 9 to 11, 2 days p.i.; 7 and 8, 3 days p.i.; 4, 6, and 10, in the presence of PAA. Arrows 1, 2, and 3 indicate the transcripts initiating at start sites 1, 2, and 3, respectively. The arrowhead indicates the alternative transcript due to the substitution for the UL44 middle TATA nucleotides.
FIG. 5.

Effect of the mutated TATA sequence on the UL44 distal promoter. (a) Schematic representation of the recombinant viruses replaced with a noncanonical TATA sequence in the UL44 distal promoter. To construct theses mutants, a counterselection replaced the UL44 distal promoter with a marker cassette containing the RpsL gene as described in Materials and Methods. The lowercase letters in the sequences indicate mutant bases. (b) RNAs were harvested at 1, 2, and 3 days after infection with an MOI of approximately 3. An RNase protection assay was performed with 32P-labeled antisense UL44 RNA probe at 37°C overnight before digestion with RNase T1. The protected RNA fragments were subjected to electrophoresis in denaturing 6% polyacrylamide gels. Lanes: 1, lacking RNase T1; 2 to 5, mutTATA1; 6 to 8, wt-R; 2, 1 day p.i. (d.p.i.); 3, 4, 6, and 7, 2 days p.i.; 5 and 8, 3 days p.i.; 4 and 7, in the presence of PAA. Arrows 1, 2, and 3 indicate the transcripts initiating at start sites 1, 2, and 3, respectively. The arrowhead indicates the alternative transcript due to the substitution for the UL44 distal TATA element.
The noncanonical TATA sequence in the UL44 middle promoter is required for the accumulation of late transcripts.
To confirm that the noncanonical TATA sequence is required for the accumulation of late transcripts, we made adenine and thymidine substitutions to generate the recombinant viruses TATAmut2 and TATAmut3 (Fig. 3a). To construct these recombinant viruses, we reversed the recombinant virus selection procedure (Fig. 1) as described in Materials and Methods. The correct recombination was confirmed by DNA sequencing, and the integrity of the mutant BACs was checked by digestion with HindIII (data not shown). As a result of the mutations shown in Fig. 3a, the UL44 middle TATA element in the two recombinant viruses contained a canonical TATA sequence. Cytoplasmic RNA was isolated 1, 2, and 3 days after infection with the recombinant virus at an MOI of approximately 3, and an RNase protection assay was performed with antisense UL44 RNA probe. As shown in Fig. 3b, when the TATA sequence was mutated to contain a canonical TATA sequence, “TATAA,” the accumulation of late transcripts was decreased at 2 and 3 days p.i. (compare lane 9 with lanes 1 to 8). An alternative transcript was detected with TATAmut3, as well as the middle transcripts from start site 2, at 2 and 3 days p.i. (Fig. 3b, lanes 5 and 8). These transcripts were not detected in the presence of PAA at 2 days p.i. (Fig. 3b, lanes 4 and 6). Since TATAmut contains a repeat of thymine and adenine nucleotides in front of TAA in the UL44 middle promoter (Fig. 1), the number of TA repeats in front of TAA nucleotides may determine the strength of the UL44 middle promoter with TATAmut3. While the levels of the distal transcript were similar between TATAmut2 and TATAmut3, the levels of transcripts derived from the UL44 middle promoter with mut2 were lower than those with mut3 and mut2 at 3 days p.i. (Fig. 3b, lanes 3, 5, 7, and 8). The different TATA sequences in the two recombinant viruses may affect the level of the transcript from the promoter. Several bands with wt-R and recombinant virus were also detected upstream of the proximal transcript or downstream of the distal transcript; however, there was no different transcript in these bands in wt-R, TATAmut, TATAmut2, and TATAmut3. Thus, they are due to incomplete hybridization, as described above. From these results, we concluded that the noncanonical TATA sequence in the UL44 middle promoter influences the accumulation of late transcripts.
The GC boxes surrounding the middle TATA element do not affect the kinetics and the TSS selection of the UL44 late transcript.
There is a region of perfect dyad symmetry located immediately 3′ of the UL44 middle TATA element, and the sequence contains an Sp1 binding site (Fig. 2b). It has been reported that interaction between TFIID and the TSS was dependent either on a TATA box or on Sp1 bound to upstream sites (18). To determine if this region plays a role in modulating the kinetics and the TSS selection of the middle transcript, we constructed a recombinant virus with the perfect dyad symmetry mutated as described in Materials and Methods. The correct recombination was confirmed by DNA sequencing, and the integrity of the mutant BACs was checked by digestion with HindIII (data not shown). Cytoplasmic RNA was isolated 1, 2, and 3 days after infection with the recombinant virus at an MOI of approximately 3, and an RNase protection assay was performed with antisense UL44 RNA probe. As shown in Fig. 4b, the middle transcript initiating at start site 2 was detected with the recombinant virus with the perfect dyad symmetry mutated at 2 and 3 days p.i. (Fig. 4b, lanes 8 and 10) and was not detected in the presence of PAA for 48 h (Fig. 4b, lane 9). An alternative band located between the distal and middle transcripts was also detected (Fig. 4b). The level of the distal transcript was decreased (Fig. 4b). When we constructed the antisense UL44 RNA probe, a DNA fragment including 5′ upstream of the entire TSS of the UL44 transcript was amplified by PCR using a BAC wild-type DNA as a template and the antisense UL44 RNA probe was not completely complementary to the transcribed RNA from RGCmut2. Thus, to determine if this was due to the probe design, we also performed an RNase protection assay with the antisense UL44 mut RNA probe. To construct the antisense UL44 mut RNA probe, a DNA fragment including all the 5′ upstream of the TSS was amplified using BAC GCmut2 as a template, as described in Materials and Methods. As shown in Fig. 4c, the alternative band was not detected with GCmut2 (lanes 6 to 9) but was detected with wt-R (lane 10). This indicates that our RNase protection assay partially recognized the difference of several nucleotides in the perfect dyad symmetry. From these results, we conclude that a region of perfect dyad symmetry does not modulate the kinetics and the TSS selection of the transcript from the UL44 late promoter.
There is one more GC box 5′ of the middle TATA element (Fig. 2b). To further determine if this GC box plays a role in modulating the kinetics and the TSS selection of the UL44 middle transcript, we constructed the recombinant virus with all three GC boxes surrounding the middle TATA element mutated (Fig. 4a), and an RNase protection assay was performed with the antisense UL44 RNA probe or the UL44 mut RNA probe. As shown in Fig. 4c, the middle transcript initiating at start site 2 was still detected with the recombinant virus with all three GC boxes mutated at 2 and 3 days p.i. (lanes 3 and 5) and was not detected in the presence of PAA for 48 h (lane 4). When the UL44 mut RNA probe was used for the RNase protection assay, another alternative band located between the distal and middle transcripts was detected (Fig. 4c, lanes 2 to 5). Compared to GCmut2, GCmut1 has an additional mutation in the 5′ region of the probe, and the transcribed RNA from GCmut1 is not completely complementary to the antisense UL44 mut RNA probe. Therefore, the detected alternative band located between the distal and middle transcripts (Fig. 4c, lanes 2 to 5) should be due to the mismatch of the hybridization. Minor bands were also detected with GCmut1 or GCmut2, as well as wt-R. They may have been caused by partial degradation of the isolated RNA and/or RNA probe before hybridization, as described above. However, there was no apparent alternative transcript specific for GCmut1 or GCmut2 due to the mutation of the GC boxes (Fig. 4b and c). Since the level of the UL44 transcript from GCmut1 was lower than that of wt-R or GCmut2 (Fig. 4b and c), the GC box 5′ of the TATA element may modulate the strength of the UL44 promoter. Taken together, we conclude that the GC boxes that surround the middle TATA element do not affect the kinetics and the TSS selection of the UL44 late transcript.
Replacement of the distal TATA element with a noncanonical TATA sequence.
The TATA element in the distal promoter contains a canonical sequence (Fig. 5a). To determine if the TATA sequence in the distal early promoter also has an effect on the kinetics of the transcript, we constructed a recombinant virus with a noncanonical TATA sequence using the reverse procedure described in Materials and Methods. The correct recombination was confirmed by DNA sequencing, and the integrity of the mutant BACs was checked by digestion with HindIII (data not shown). Cytoplasmic RNA was isolated 1, 2, and 3 days after infection with the recombinant virus at an MOI of approximately 3, and an RNase protection assay with an antisense UL44 RNA probe was performed. As shown in Fig. 5b, the early distal transcript was still detected with mutTATA1 at 1, 2, and 3 days p.i., and this transcript was also detected in the presence of PAA at 48 h (Fig. 5b, lane 4). The mutation did not alter the early kinetics of the distal promoter. However, an alternative transcript downstream of the distal early transcript was dependent on viral DNA replication (Fig. 5b, lanes 3 to 5). The noncanonical TATA element did not alter the TSS of the early transcript, but it did induce an alternative late transcript that was dependent on viral DNA replication.
DISCUSSION
The pUL44 protein accumulates to strikingly high levels at late times after infection (9, 35), and the UL44 gene product from the late viral transcript is required for efficient viral gene expression rather than viral DNA synthesis (16). The HCMV UL44 transcription unit initiates at three distinct sites, which are separated by approximately 50 nucleotides and are differentially regulated during productive infection. Two of these start sites, the distal and the proximal, are active at early times, whereas the middle start site is inactive until late times (21). To determine what is responsible for the activation of the middle TATA element at late times after infection, we constructed recombinant viruses with a canonical TATA sequence in the UL44 middle promoter. We found that the noncanonical TATA sequence in the UL44 middle promoter is required for accumulation of the late viral transcripts.
The transcriptional strategies of DNA viruses exhibit a number of common features. Prior to initiation of viral DNA synthesis, during the IE and early phases, infected cells are devoted to the production of viral proteins necessary for viral DNA synthesis, efficient expression of viral genes, or the other regulatory functions. Transcription of the late genes requires viral DNA synthesis. However, the molecular mechanisms of this coordinated sequential regulation are not fully understood. Previous analyses of the glycoprotein C (gC) gene in herpes simplex virus type 1 demonstrated that a specific 15-bp TATA box promoter element is required for expression of a late gene (12). The authors constructed a chimeric herpes simplex virus gene that contained the distal regulatory elements of the early thymidine kinase gene fused upstream of the 15-bp TATA sequence of the late gC gene. Synthesis of gC mRNA from the chimeric promoter showed both early and late kinetics. This interesting study showed that the cis-acting elements determine the kinetics of the early and late genes.
Originally, core promoter elements were thought to mediate basal transcription whereas gene-specific upstream regulatory elements were responsible for directing regulated gene expression. However, recent studies have demonstrated that core promoter elements can play an integral role in both environmentally induced and developmentally regulated gene expression (32). For instance, developmental-stage-specific recruitment of the TATA-binding protein (TBP) has been demonstrated for the human gamma globin gene (3). Moreover, in the case of the human osteocalcin gene, which is transcriptionally repressed by glucocorticoids, a specific binding element for the glucocorticoid receptor overlaps a noncanonical TATA box (25). Mutating this noncanonical TATA box into a canonical TATA box within the context of the osteocalcin promoter greatly decreased hormone-dependent transcriptional repression by the glucocorticoid receptor (25). TBP bound this mutated element much more strongly, which suggests a physiologically relevant role for the weak osteocalcin TATA element in the regulation of this bone-specific gene (25).
For the early and late gene transcription of DNA viruses, IE proteins recruit the general transcription factors, including TBP, to the promoter. Following the recruitment of general transcription factors, recognition of the TATA box in the core promoter by TBP constitutes the first step toward preinitiation complex formation to start early and late gene transcription. Since the difference in the TATA sequence in the UL44 late promoter presumably modulates the strength of TBP-DNA binding (33, 34), it is possible that the binding affinity of TBP to the promoter became stronger due to the replacement of the UL44 middle noncanonical TATA sequence by a canonical TATA sequence, “TATATAA” (33, 34), and this caused the shift from late kinetics to early kinetics. The molecular coupling of replication to transcription of late genes remains unclear. A part of the newly replicated DNA could serve as a template for transcription. Therefore, one hypothesis is that the increased concentration of transcriptional templates is necessary for the initiation of late UL44 transcription. The relatively weak binding affinity of TBP for the noncanonical TATA sequence at the middle promoter may explain a lack of transcription at early times after infection. However, the weak binding affinity of TBP for the middle promoter is not the only reason for the lack of early transcription, because late transcripts were not detected with the recombinant virus TATAmut2 or TATAmut3, while an alternative transcript was significantly detected with TATAmut3. Mutation to a noncanonical TATA sequence in the UL44 distal promoter with the recombinant virus mutTATA1 induced a late specific alternative transcript that was detected after viral DNA synthesis. Late specific transcription from a noncanonical TATA sequence may be simply a concentration effect after viral DNA synthesis or may reflect the presence of a viral transcription factor that specifically activates a late promoter.
The main function of the TATA box is to anchor the transcription preinitiation complex guiding RNA polymerase upstream of the TSS. Therefore, the spacing between the TATA box and the TSS is functionally important for efficient transcription (28), but the underlying mechanisms that determine the start site selection are not understood. As previously shown (1), the preferred canonical sequence for the initiation site is a pyrimidine-purine dinucleotide situated at positions −1 and +1 relative to the TSS. When the UL44 middle TATA element was replaced by a canonical sequence, the distance between the TATA box and the TSS was shortened from 32 or 37 to 22 nucleotides.
Our data indicate that the GC boxes surrounding the middle TATA sequence do not affect the kinetics and the TSS selection of the middle late transcript, while the GC box 5′ of the TATA element may modulate the strength of the UL44 promoter. The transcription factors in the core promoter, including the TATA box, must communicate with the surrounding sequence in order to either enhance or repress transcription. Therefore, further studies are required to determine the roles of the GC boxes surrounding the middle UL44 core promoter in UL44 transcription and viral replication.
In conclusion, a noncanonical TATA sequence at the middle promoter of the UL44 transcription unit is associated with the accumulation of late viral transcripts.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports, Culture and Technology of Japan (15390153, 17659138, and 16017322 to T.T. and 17590429 to H.I.); Research on Health Sciences Focusing on Drug Innovation (SH54412 to H.I.) and a Grant-in-aid for Cancer Research (13-01 to H.I.) from the Ministry of Health, Labor and Welfare; and grant AI-13562 from the National Institutes of Health (to M.F.S.).
Footnotes
Published ahead of print on 5 December 2007.
REFERENCES
- 1.Carninci, P., A. Sandelin, B. Lenhard, S. Katayama, K. Shimokawa, J. Ponjavic, C. A. Semple, M. S. Taylor, P. G. Engstrom, M. C. Frith, A. R. Forrest, W. B. Alkema, S. L. Tan, C. Plessy, R. Kodzius, T. Ravasi, T. Kasukawa, S. Fukuda, M. Kanamori-Katayama, Y. Kitazume, H. Kawaji, C. Kai, M. Nakamura, H. Konno, K. Nakano, S. Mottagui-Tabar, P. Arner, A. Chesi, S. Gustincich, F. Persichetti, H. Suzuki, S. M. Grimmond, C. A. Wells, V. Orlando, C. Wahlestedt, E. T. Liu, M. Harbers, J. Kawai, V. B. Bajic, D. A. Hume, and Y. Hayashizaki. 2006. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 38626-635. [DOI] [PubMed] [Google Scholar]
- 2.Chang, C.-P., C. L. Malone, and M. F. Stinski. 1989. A human cytomegalovirus early gene has three inducible promoters that are regulated differentially at various times after infection. J. Virol. 63281-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duan, Z. J., X. Fang, A. Rohde, H. Han, G. Stamatoyannopoulos, and Q. Li. 2002. Developmental specificity of recruitment of TBP to the TATA box of the human gamma-globin gene. Proc. Natl. Acad. Sci. USA 995509-5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 10014223-14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis, H. M., D. Yu, T. DiTizio, and D. L. Court. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 986742-6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ertl, P. R., and K. L. Powell. 1992. Physical and functional interaction of human cytomegalovirus DNA polymerase and its accessory protein (ICP36) expressed in insect cells. J. Virol. 664126-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fish, K. N., W. Britt, and J. A. Nelson. 1996. A novel mechanism for persistence of human cytomegalovirus in macrophages. J. Virol. 701855-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fish, K. N., A. S. Depto, A. V. Moses, W. Britt, and J. A. Nelson. 1995. Growth kinetics of human cytomegalovirus are altered in monocyte-derived macrophages. J. Virol. 693737-3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geballe, A. P., F. S. Leach, and E. S. Mocarski. 1986. Regulation of cytomegalovirus late gene expression: gamma genes are controlled by posttranscriptional events. J. Virol. 57864-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham, F. L., and A. J. van der Eb. 1973. A new technique for the assay of infectivity of adenovirus 5 DNA. Virology 52456-467. [DOI] [PubMed] [Google Scholar]
- 11.Hermiston, T. W., C. L. Malone, P. R. Witte, and M. F. Stinski. 1987. Identification and characterization of the human cytomegalovirus immediate-early region 2 gene that stimulates gene expression from an inducible promoter. J. Virol. 613214-3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Homa, F. L., J. C. Glorioso, and M. Levine. 1988. A specific 15-bp TATA box promoter element is required for expression of a herpes simplex virus type 1 late gene. Genes Dev. 240-53. [DOI] [PubMed] [Google Scholar]
- 13.Ibanez, C. E., R. Schrier, P. Ghazal, C. Wiley, and J. A. Nelson. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 656581-6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isomura, H., and M. F. Stinski. 2003. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J. Virol. 773602-3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isomura, H., M. F. Stinski, A. Kudoh, T. Daikoku, N. Shirata, and T. Tsurumi. 2005. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 799597-9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isomura, H., M. F. Stinski, A. Kudoh, S. Nakayama, S. Iwahori, Y. Sato, and T. Tsurumi. 2007. The late promoter of the human cytomegalovirus viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products but not on viral DNA synthesis. J. Virol. 816197-6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isomura, H., T. Tsurumi, and M. F. Stinski. 2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol. 7812788-12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaufman, J., and S. T. Smale. 1994. Direct recognition of initiator elements by a component of the transcription factor IID complex. Genes Dev. 8821-829. [DOI] [PubMed] [Google Scholar]
- 19.Lashmit, P. E., M. F. Stinski, E. A. Murphy, and G. C. Bullock. 1998. A cis-repression sequence adjacent to the transcription start site of the human cytomegalovirus US3 gene is required to down regulate gene expression at early and late times after infection. J. Virol. 729575-9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lathey, J. L., and S. A. Spector. 1991. Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J. Virol. 656371-6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach, F. S., and E. S. Mocarski. 1989. Regulation of cytomegalovirus late-gene expression: differential use of three start sites in the transcriptional activation of ICP36 gene expression. J. Virol. 631783-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meier, J. L., M. J. Keller, and J. J. McCoy. 2002. Requirement of multiple cis-acting elements in the human cytomegalovirus major immediate-early distal enhancer for viral gene expression and replication. J. Virol. 76313-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meier, J. L., and J. A. Pruessner. 2000. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J. Virol. 741602-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meier, J. L., and M. F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 711246-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer, T., J. Carlstedt-Duke, and D. B. Starr. 1997. A weak TATA box is a prerequisite for glucocorticoid-dependent repression of the osteocalcin gene. J. Biol. Chem. 27230709-30714. [DOI] [PubMed] [Google Scholar]
- 26.Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 676979-6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pari, G. S., M. A. Kacica, and D. G. Anders. 1993. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J. Virol. 672575-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponjavic, J., B. Lenhard, C. Kai, J. Kawai, P. Carninci, Y. Hayashizaki, and A. Sandelin. 2006. Transcriptional and structural impact of TATA-initiation site spacing in mammalian core promoters. Genome Biol. 7R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ripalti, A., M. C. Boccuni, F. Campanini, and M. P. Landini. 1995. Cytomegalovirus-mediated induction of antisense mRNA expression to UL44 inhibits virus replication in an astrocytoma cell line: identification of an essential gene. J. Virol. 692047-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinzger, C., A. Grefte, B. Plachter, A. S. H. Gouw, T. Hauw The, and G. Jahn. 1995. Fibroblasts, epithelial cells, endothelial cells, and smooth muscle cells are the major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76741-750. [DOI] [PubMed] [Google Scholar]
- 31.Sinzger, C., B. Plachter, A. Grefte, A. S. H. Gouw, T. H. The, and G. Jahn. 1996. Tissue macrophages are infected by human cytomegalovirus. J. Infect. Dis. 173240-245. [DOI] [PubMed] [Google Scholar]
- 32.Smale, S. T., and J. T. Kadonaga. 2003. The RNA polymerase II core promoter. Annu. Rev. Biochem. 72449-479. [DOI] [PubMed] [Google Scholar]
- 33.Stewart, J. J., J. A. Fischbeck, X. Chen, and L. A. Stargell. 2006. Non-optimal TATA elements exhibit diverse mechanistic consequences. J. Biol. Chem. 28122665-22673. [DOI] [PubMed] [Google Scholar]
- 34.Stewart, J. J., and L. A. Stargell. 2001. The stability of the TFIIA-TBP-DNA complex is dependent on the sequence of the TATAAA element. J. Biol. Chem. 27630078-30084. [DOI] [PubMed] [Google Scholar]
- 35.Stinski, M. F. 1978. Sequence of protein synthesis in cells infected by human cytomegalovirus: early and late virus-induced polypeptides. J. Virol. 26686-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 722059-2064. [DOI] [PubMed] [Google Scholar]
- 37.Weiland, K. L., N. L. Oien, F. Homa, and M. W. Wathen. 1994. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res. 34191-206. [DOI] [PubMed] [Google Scholar]
- 38.White, E. A., C. J. Del Rosario, R. L. Sanders, and D. H. Spector. 2007. The IE2 60-kilodalton and 40-kilodalton proteins are dispensable for human cytomegalovirus replication but are required for efficient delayed early and late gene expression and production of infectious virus. J. Virol. 812573-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu, D., M. C. Silva, and T. Shenk. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 10012396-12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuccola, H. J., D. J. Filman, D. M. Coen, and J. M. Hogle. 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 5267-278. [DOI] [PubMed] [Google Scholar]