

Abstract
How signaling cascades influence gene regulation at the level of chromatin modification is not well understood. We studied this process using the Wingless/Wnt pathway in Drosophila. When cells sense Wingless ligand, Armadillo (the fly β-catenin) becomes stabilized and translocates to the nucleus, where it binds to the sequence-specific DNA binding protein TCF to activate transcription of target genes. Here, we show that Wingless signaling induces TCF and Armadillo recruitment to a select subset of TCF binding site clusters that act as Wingless response elements. Despite this localized TCF/Armadillo recruitment, histones are acetylated over a wide region (up to 30 kb) surrounding the Wingless response elements in response to pathway activation. This widespread histone acetylation occurs independently of transcription. In contrast to Wingless targets, other active genes not regulated by the pathway display sharp acetylation peaks centered on their core promoters. Widespread acetylation of Wingless targets is dependent upon CBP, a histone acetyltransferase known to bind to Armadillo and is correlated with activation of target gene expression. These data suggest that pathway activation induces localized recruitment of TCF/Armadillo/CBP to Wingless response elements, leading to widespread histone acetylation of target loci prior to transcriptional activation.
To package a cell's genetic material into the relatively small space provided by nuclei, linear DNA molecules are tightly wrapped around histone proteins, forming nucleosomes that are then packed together to form a higher-order structure called chromatin. This highly compacted DNA-protein assembly efficiently packages DNA but also acts as a steric barrier to transcription factors and other proteins that need to access gene-regulatory regions (45). One mechanism by which cells can regulate chromatin organization is through posttranslational modification of subunits of the histone octamers (41, 47).
One common histone modification is the acetylation of lysines in the N-terminal tails of the histone 3 (H3) and histone 4 (H4) subunits. Many studies have shown a correlation between acetylation of H3/H4 and gene activation (22, 54). Histone acetylation has been found at the transcriptional start site (TSS) of active genes as well as enhancers and conserved intergenic regions thought to be enhancers (3, 25, 55, 56).
Despite the recognized importance of histone acetylation in gene regulation, its role in many important processes has not been explored in detail. One example is Wnt/β-catenin signaling. This pathway is highly conserved and is essential for the normal development and physiology of many metazoan organisms (8, 30, 39). Wnt signaling also plays a causal role in several human cancers (19, 21, 39, 52, 53).
Activation of the pathway resulted in increased histone acetylation at Wnt response elements (WREs) in several targets (14, 29, 57), but the core promoters of these genes were not examined. In a recent study, it was reported that the regions containing WREs and TSSs contained acetylated histones, but the levels were not regulated by Wnt signaling (69). Interestingly, Wnt signaling also caused an increase in trimethylation at lysine 4 of H3 (H3K4Me3) at the c-myc WRE (57). This modification is normally observed only at the TSSs of active genes (3, 37). Clearly, a more detailed knowledge of the chromatin modifications regulated by Wnt/β-catenin signaling is needed.
While not much is known about what happens on the chromatin of Wnt targets in response to pathway activation, upstream events have been defined more extensively. In the absence of signaling, β-catenin is earmarked for proteasomal degradation by coupled phosphorylation/ubiquitination, keeping cytosolic levels low (7, 62). When Wnt binds a receptor complex at the cell surface, β-catenin turnover is inhibited (7). The stabilized protein then translocates to the nucleus, where it binds to members of the TCF family of HMG (high-mobility group) domain sequence-specific DNA binding proteins. β-Catenin binding is thought to convert TCFs from transcriptional repressors to activators (50, 65).
Once TCF recruits β-catenin to a WRE, it, in turn, recruits other coactivators required for transcription (50, 68). Among these are CREB-binding protein (CBP) and its close relative p300. Both are histone acetyl transferases (HATs) that bind to β-catenin and augment its ability to activate transcription (24, 43, 60, 61). Although CBP has been shown to be a negative regulator of the Wingless (Wg, a fly Wnt) pathway in Drosophila (66), we have recently reported that fly CBP is also required for activation of target genes by Wg. CBP binds to Armadillo (Arm, the fly β-catenin) and is recruited to WRE chromatin in a Wg- and Arm-dependent manner (35).
Although CBP and p300 can acetylate H3 and H4 tails (1, 48), it is not clear whether this is the mechanism by which they act in Wnt signaling. For example, p300 has been shown to acetylate lysines on β-catenin, which increases its affinity for TCF (32, 34, 70). There is also a report that p300 lacking HAT activity can still augment β-catenin transcriptional activation (24), raising the possibility that CBP and p300 act in a HAT-independent fashion.
Here, we report that the histone acetylation profile of activated Wg transcriptional targets in Drosophila is markedly different from that of other active genes. We find that TCF and Arm are selectively recruited to distinct WREs in target loci. In contrast to the discrete localization of TCF and Arm, Wg pathway activation induces an increase in acetylation of H3 and H4 over a wide region, tens of kilobase pairs away from the WREs. This Wg-dependent increase in histone acetylation still occurs when transcription is blocked, indicating that these two processes are not coupled. RNA interference (RNAi)-mediated knockdown of CBP does not affect TCF recruitment to WREs but does block the increase in H3/H4 acetylation and dramatically reduces Wg activation of target gene expression. The pattern of histone acetylation is roughly centered on the WREs, and unlike many active genes, the core promoter does not appear to be a primary target of this modification. These data are consistent with a model whereby localized TCF/Arm recruitment induces widespread histone acetylation that is mediated by CBP.
MATERIALS AND METHODS
Identification of TCF site clusters.
Target Explorer (http://trantor.bioc.columbia.edu/ Target_Explorer/) (59) was used to generate a weight matrix from 20 TCF sites previously identified from the regulatory regions of the following genes: sloppy paired 1 (nine sites) (33), Ultrabithorax (two sites) (72), decapentaplegic (two sites) (71), even skipped (six sites) (31), and stripe (one site) (51). A TCF site cluster was defined as at least three sites (cutoff score of 5.25) within a span of 150 bp.
Cell culture, RNAi, and DNA transfections.
Kc cells were maintained as described previously (42), except that 5% fetal bovine serum was used to supplement Schneider Drosophila medium. An S2 cell line stably transformed with a tubulin-Wg construct was a gift from R. Nusse and was used to produce Wg-conditioned medium (WCM). Control medium and WCM were collected from cultures as described previously (www.stanford.edu/∼rnusse).
For RNAi, 4 to 6 μg of double-stranded RNA (dsRNA)/106 cells was added directly to the medium. Cells were split 1:5 after 4 days and then assayed after a further 2 days of incubation. For DNA transfection, a total of 0.2 to 1.2 μg of DNA/106 cells was used. FuGENE 6 transfection reagent (Roche Applied Science, Indianapolis, IN) was used at 1 to 2 μl/μg of DNA.
Reporter assays and chromatin immunoprecipitation (ChIP).
Kc cells were harvested, pelleted, and lysed using standard lysis buffer. β-Galactosidase and luciferase assays were performed using Galacto-Star and Luc-Screen systems from Tropix (Bedford, MA). Transfections were performed in duplicate on 2 × 106 cells using 300 ng of reporter construct, 20 ng of Actin-LacZ as a transfection control, 50 ng of Actin-Arm* to stimulate the Wg pathway, and pAc5.1 as required to normalize the amount of DNA (13).
ChIP was performed according a protocol from Upstate (catalog no. 17-295) with the following modifications. A total of 3 × 106 Kc cells/pull-down were used, and volumes were scaled up as required. Prior to formaldehyde cross-linking, cells were cross-linked with 10 mM DTBP (dimethyl 3,3′-dithiobispropionimidate-2HCl) in phosphate-buffered saline (PBS) on ice (catalog no. 20665; Pierce) (16). For ChIP from embryonic extracts, embryos were collected 4 to 10 h after egg laying at 29°C. After embryos were subjected to dechorination in 50% bleach for 2 min and washed with water and 0.7% NaCl-0.1% Triton X-100, 100 μl of embryos was fixed in 2 ml of 2% formaldehyde (in 100 mM NaPO4 buffer, pH 7.2). Heptane (6 ml) was then added, and the tube was inverted for 15 min at room temperature, which was followed by a heptane wash. Fixation was stopped by resuspending embryos in 4 ml of 125 mM glycine-0.1% Triton X-100 in PBS. Embryos were then pelleted at 500 × g and washed in 4 ml of ice-cold PBS-0.1% Triton X-100 and stored in PBS-0.1% Triton X-100. For ChIP, 70 μl of embryos was resuspended in 400 μl of sodium dodecyl sulfate lysis buffer (1% sodium dodecyl sulfate, 10 mM EDTA, 50 mM Tris, pH 8.1) plus protease inhibitor cocktail (catalog no. 11836153001; Roche). Embryos were then ground with disposable pestles and sonicated, and the lysate was then processed in the same way as ChIP in Kc cells. Ten percent of the 400-μl lysate was used for each pull-down.
Immunoprecipitates were analyzed using quantitative PCR. ChIP oligonucleotide sequences are available upon request. The rabbit anti-TCF antibodies were generated in the Cadigan laboratory (13). The guinea pig anti-CBP antibody was obtained from M. Mannervik (38). Other antibodies were purchased from Upstate: acetylated H3K9/K18 (AcH3K9/K18; catalog no. 07-593), AcH4K5/K8/K12/K16 (06866), and H3K4Me3 (07-473).
RNA extraction, quantitative reverse transcription-PCR, and Western blotting.
Cells were harvested and pelleted, and total RNA was prepared using RNAwiz (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA was quantified using UV absorbance at 260 nm. cDNA was made by reverse transcription using oligo(dT) and equal amounts of total RNA (1 to 2 μg). Quantitative PCR was performed using an iCycler IQ real-time detection system (Bio-Rad, Hercules, CA). The following oligonucleotides were used: 5′-TAAAATTCTCGGCGGCTACAA-3′ and 5′-CGCACCTGGTGGTACATCAG-3′ for nkd, 5′-AGAGCAGCAGAAGCGTTAGC-3′ and 5′-AAAGCCGGAGAAGCTACAAA-3′ for Notum, 5′-AGACCTACTGCATCGACAAC-3′ and 5′-GACAAGATGGTTCAGGTCAC-3′ for β-tubulin56D (β-tub56D), 5′-CGTTTCGGACTAGATCAACAGAG-3′ and 5′-CATCTTCGTCGTCATCACTTAGC-3′ for TCF, and 5′-GATGAGTTACATGCCAGCCCA-3′ and 5′-GATACCATCGGCGGCAGAT-3′ for Arm.
Western blot analysis with a mouse monoclonal Arm antibody was performed as previously described (13, 35).
RESULTS
Wg signaling causes an increase in TCF binding and histone acetylation at a region containing a WRE in the nkd locus.
Previous work from our laboratory identified the Wg pathway feedback antagonist naked cuticle (nkd) as a direct target of Wg signaling in Kc167 (Kc) cells (13, 35). A schematic representation of the nkd locus is shown in Fig. 1A. The major TSS of nkd in Kc cells was confirmed by primer extension (data not shown). Stimulation of Kc cells with WCM resulted in a dramatic increase in nkd mRNA levels that reached steady state by 5 h (Fig. 1B and data not shown). For the rest of this report, WCM treatments of 5.5 h were used unless otherwise indicated. RNAi depletion of TCF, arm, or the coactivator pygopus (pygo) significantly reduced the response of nkd to WCM (Fig. 1C).
FIG. 1.

Wg signaling induces TCF recruitment and histone acetylation at the nkd locus. (A) Cartoon of the nkd locus. Black boxes represent nkd exons. The gray box represents the predicted ORF Acp76A. We were unable to detect Acp76A expression in Kc cells (data not shown). White boxes represent clusters of TCF binding sites. Arrows represent TSSs. Dashed lines show locations of primer sets used in ChIP analysis, with the WRE and the ORF control positions indicated. (B) nkd expression is rapidly activated by Wg signaling in Kc cells. Cells were treated with WCM for the indicated times and quantitative reverse transcription-PCR was then used to measure the transcript levels of nkd in reference to β-tub56D. (C) nkd activation requires TCF, Arm, and Pygo. Kc cells were treated for 5.5 h with WCM following 6 days of treatment with control dsRNA or with dsRNA corresponding to TCF, arm, or pygo. TCF, arm, or pygo RNAi significantly blocks WCM activation of nkd transcripts. (D) TCF is recruited to the WRE region in response to Wg signaling. ChIP with antiserum directed against the N terminus (N-term Ab) of TCF indicates significant TCF binding to the WRE region in unstimulated cells, which dramatically increases after WCM treatment for 5.5 h. TCF RNAi demonstrates that the signal is specific, and arm RNAi shows that the Wg-dependent increase in TCF binding is dependent upon Arm. (E) WCM also increases TCF binding to the WRE region when antiserum against the C terminus (C-term Ab) of TCF is used. (F and G) WCM induces histone acetylation at the WRE. ChIP analysis for AcH3 (F) and AcH4 (G) indicates that these histones are acetylated in response to Wg pathway activation. TCF or arm RNAi significantly blocks the acetylation levels of both histones. Each bar represents the mean of two independent samples, with the error bars indicating standard deviations. All experiments were performed at least three times, and the data shown are representative. Cont, control.
The nkd locus spans approximately 40 kb and contains a large 30-kb intron (Fig. 1A). We previously identified a WRE in this intron about 5.5 kb downstream of the nkd TSS (13, 35). As reported, TCF is preferentially bound to the region containing the intronic WRE compared to the nkd open reading frame (ORF), as judged by ChIP (Fig. 1D). This TCF binding is authentic, since RNAi knockdown of TCF reduced the signal to the background level seen at the ORF (Fig. 1D). Interestingly, a highly reproducible increase in TCF binding at the WRE region was observed upon stimulation with WCM treatment (Fig. 1D). This increased recruitment was blocked by arm RNAi (Fig. 1D), indicating that Arm is required for the Wg-dependent increase in TCF binding.
The increase in TCF binding to the WRE region occurs rapidly and is detectable within 1 h after WCM treatment (data not shown). It is not due to increased expression of TCF, since protein and transcript levels are unchanged by WCM treatment (10) (see Fig. 8F). The levels of nuclear TCF were also not changed by Wg signaling (10). It is possible that the increase in the TCF ChIP signal is caused by a Wg-induced change in TCF conformation leading to increased epitope availability. However, increased TCF binding was observed with two anti-TCF antisera, one directed against the N terminus of TCF (Fig. 1D) and one to the C terminus (Fig. 1E), making this possibility less likely.
FIG. 8.

CBP is required for Wg-dependent activation of nkd and Notum but not for TCF recruitment to WREs. Cells were treated for 4 days with control dsRNA or with a RNA duplex corresponding to CBP and then incubated for 5.5 h with WCM. (A and B) Transcript levels were measured by Quantitative reverse transcription-PCR in reference to β-tub56D. Depletion of CBP significantly reduces WCM induced activation of nkd (A) and Notum (B) expression. (C) ChIP analysis shows that CBP RNAi treatment does not affect WCM-induced TCF recruitment to the nkd WRE region. (D to F) The expression of β-tub56D (D), arm (E), and TCF (F) is not affected by CBP RNAi. β-tub56D levels were normalized to total RNA, and arm and TCF levels were normalized to β-tub56D. Each bar represents the mean of two independent samples, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative. Cont, control.
Previous work indicates that histones are acetylated at WREs in response to Wnt signaling (4, 14, 29, 57). We used ChIP to ask if histones around the nkd WRE are acetylated in response to WCM treatment. In the absence of signaling, acetylated H3K9/K18 (AcH3) (Fig. 1F) and acetylated H4K5/K8/K12/K16 (AcH4) (Fig. 1G) levels at the WRE are slightly higher than at the ORF. WCM treatment induces a fourfold increase of H3 and H4 acetylation (Fig. 1F and G). In contrast, acetylation levels at the ORF remain low. TCF or arm RNAi treatment significantly blocked the increase in AcH3 and AcH4 by WCM (Fig. 1F and G). Taken together, these data show that Wg signaling can induce an increase in nkd transcription concomitant with TCF recruitment and histone acetylation at the WRE.
Upon Wg stimulation, TCF, Arm and H3K4Me3 are found at distinct locations in the nkd locus, while histone acetylation is widespread.
Previously, we reported that TCF was bound preferentially at the WRE region in the nkd locus and was not found at several other locations that contained TCF binding sites (13). To examine the nkd locus more systematically, we identified 11 clusters of putative TCF binding sites (three sites/150 bp) (Fig. 2A, open boxes) with a web-based algorithm called Target Explorer (59). Primer sets for ChIP were designed to target many of these sites, in addition to other sites spanning the nkd locus (Fig. 2A, vertical dashed lines).
FIG. 2.

Wg signaling induces the recruitment of TCF and Arm to a single major site in the nkd locus. (A) ChIP analysis across the nkd locus indicates that TCF is present at the region containing the WRE in the absence of signaling (dashed line) and that TCF binding to this region is increased by Wg pathway activation (black line). Vertical dashed lines indicate the locations of primers sets in relation to the nkd exons. The location of the WRE is indicated. (B) The profile of Arm binding at the nkd locus mirrors that of TCF. ChIP analysis shows that Arm is preferentially recruited to the same region containing the WRE as TCF. Each data point represents the mean of two independent ChIP samples, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative. Cont, control.
Although Target Explorer judged the quality of the TCF sites in the 11 clusters to be roughly equal, TCF is primarily bound to the region containing the WRE (Fig. 2A). The profile of Arm binding across the nkd locus is very similar to that of TCF, with a large peak around 5.5 kb downstream of the TSS (Fig. 2B). The smaller peak 30 kb downstream of the TSS was not reproducibly observed. We estimate that the resolution of the ChIP analysis is approximately 1.0 to 1.5 kb, so these experiments suggest that TCF and Arm are predominately found at a single location in nkd, though we cannot rule out that TCF/Arm binding occurs in regions lying in between the primer sets used.
The levels of AcH3 and AcH4 were also examined across the entire nkd locus. In the absence of Wg signaling, histone acetylation levels were not uniform (Fig. 3A and B, broken lines). This is especially evident for AcH4, which displayed several peaks in the unstimulated state (Fig. 3B). These differences cannot be attributed to primer efficiency, as similar peaks are not seen with TCF or Arm ChIP (Fig. 2). The largest peak for both AcH3 and AcH4 in unstimulated cells occurs 11 kb downstream of the TSS, and its significance is unclear.
FIG. 3.

In contrast to housekeeping genes, histones are acetylated over a broad region at the nkd locus in response to Wg, whereas H3K4Me3 is restricted to the proximal promoter. (A) AcH3 levels rise across the locus when nkd is activated by WCM (compare black and dashed lines). Note that the peak of AcH3 is roughly centered at the WRE. (B) AcH4 levels also rise across the nkd locus when the Wg pathway is activated. AcH3 and AcH4 levels in unstimulated cells are somewhat irregular (dashed lines), although the positions of the peaks were consistent in multiple experiments. (C) H3K4Me3 is restricted to the nkd proximal promoter. Wg induces a spike of H3K4Me3 that is localized to the region containing the TSS. (D to F) AcH3 and AcH4 are found with sharp peaks at the TSSs of pygo/rough deal (D), TCF (E), and β-tub56D (F). For each locus, primer sets upstream and downstream of the TSS are indicated as A and C, respectively, with primer set B covering the TSS. Each data point represents the mean of two independent ChIP samples, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative. Cont, control.
Upon WCM treatment, AcH3 and AcH4 levels are dramatically increased over a region of approximately 50 kb (Fig. 3A and B; compare black and dashed lines). In general, the highest levels for both modifications surround the WRE and tail off in both directions (with the exception of a spike 22 kb downstream of the TSS). The levels of total H3 and H4 across the nkd locus were not increased by WCM treatment (data not shown), ruling out an increase in histone density as the cause for the elevated AcH3 and AcH4 levels. The increased histone acetylation is detectable within 1 h of WCM treatment and is not observed in cells depleted of arm RNAi (data not shown). Thus, recruitment of TCF/Arm to a single location in the nkd intron results in a widespread effect on chromatin throughout the locus.
Histone acetylation often occurs in sharp peaks at the proximal promoters of active genes (3, 25, 54, 55). To confirm that this is also true in Kc cells, H3/H4 acetylation of several constitutively active genes was examined in WCM treated cells. Histone acetylation was markedly higher at the promoter regions of pygo and rough deal (Fig. 3D), TCF (Fig. 3E), and β-tub56D (Fig. 3F) compared with regions 3 to 4 kb away. Similar results were observed in Kc cells without WCM treatment (data not shown). These results indicate that the histone acetylation patterns at these housekeeping genes are distinct from the broad acetylation observed at nkd after Wg stimulation.
It was recently reported that β-catenin can interact with mixed-lineage leukemia (MLL1/MLL2)/SET1-type histone methyltransferase complexes and that β-catenin promotes H3K4Me3 at a region containing a WRE 1 kb upstream of the 5′ end of c-Myc, a Wnt target gene (57). However, we found no detectable levels of this histone methylation at the nkd WRE region in the absence or presence of Wg signaling (Fig. 3C). Rather, a sharp increase in H3K4Me3 was observed at the nkd proximal promoter, consistent with other studies that observed an H3K4Me3 spike at TSSs (3, 37).
Wg-dependent widespread histone acetylation of nkd occurs independently of transcription.
While a large portion of the increased AcH3/AcH4 is observed within the nkd intron, it is also elevated in the 15 kb upstream of the nkd TSS and is not high in the ORF (Fig. 3A and B). This suggests that the effect is not simply due to transcription of the nkd gene. To confirm this, cells were pretreated with the RNA polymerase II inhibitor α-amanitin (67) before being challenged with WCM. As predicted, the drug blocks the increase in nkd expression normally observed in WCM-treated cells (Fig. 4A). However, the Wg-dependent increase in TCF binding to the nkd WRE region still occurs in cells pretreated with α-amanitin (Fig. 4B). This indicates that blocking transcription does not affect Wg-dependent events upstream of TCF recruitment to the nkd locus.
FIG. 4.

Wg-induced histone acetylation occurs independently of transcription. Kc cells were pretreated with 10 μg/ml α-amanitin for 2 h before a 4-h incubation with WCM. (A) α-Amanitin blocks activation of nkd expression by WCM. nkd transcripts were measured by quantitative reverse transcription-PCR normalized to β-tub56D transcripts, which were unchanged over the 6-h α-amanitin treatment. (B) WCM increases TCF binding to the nkd WRE region independent of transcription. (C) α-Amanitin blocks the Wg-dependent increase in H3K4Me3 at the nkd TSS. H3K4Me3 levels in control cells treated with α-amanitin are not shown but were similar to control cells without α-amanitin. (D and E) WCM increases AcH3 (D) and AcH4 (E) across the nkd locus in the absence of transcription. Each data point represents the mean of two independent RNA or ChIP samples, with the error bars indicating standard deviations. All experiments were performed multiple times and the data shown are representative. Cont, control.
RNA polymerase II is known to recruit SET1 and Trithorax histone methyltransferases to promoters in yeast and flies, respectively, where they promote H3K4Me3 upon transcription elongation (46, 58). Consistent with this, α-amanitin pretreatment prevents the Wg-dependent increase in H3K4Me3 at the nkd TSS (Fig. 4C). However, α-amanitin has no effect on the ability of WCM to promote widespread acetylation of H3 (Fig. 4D) and H4 (Fig. 4E). These results demonstrate that stimulation of widespread histone acetylation at the nkd locus by Wg signaling is not a by-product of transcription initiation/elongation.
Does histone acetylation spread from the nkd WRE?
The ChIP experiments described above suggest that localized recruitment of TCF and Arm to the nkd WRE region causes an increase in histone acetylation over the entire locus. However, time course experiments have not been able to detect an obvious spread of AcH3 or AcH4 from the WRE in response to WCM treatment; i.e., the increased acetylation appears to occur at roughly the same rate along the whole nkd gene (data not shown). One alternative to the spreading hypothesis is that other TCF site clusters throughout the locus recruit functionally important TCF/Arm protein at levels not detected by ChIP. These lower levels of TCF/Arm could then cause the Wg-dependent increase of AcH3/AcH4 in those regions.
The multiple sites of the TCF/Arm recruitment model predicts that the nkd WRE is not required for the Wg signaling-dependent histone acetylation observed in other parts of the locus. Unfortunately, we have not been able to successfully perform AcH3 or AcH4 ChIP on transfected nkd WRE reporter constructs to test this idea directly. However, it is possible to address whether the nkd WRE is required to confer a Wg transcriptional response. To this end, ∼7 kb of genomic DNA surrounding the WRE was cloned upstream of the hsp70 promoter and a luciferase reporter gene. In addition to the WRE, this stretch of DNA contains three other TCF site clusters identified by the Target Explorer program that are comparable in quality to the TCF sites found in the WRE (Fig. 5A, open boxes).
FIG. 5.
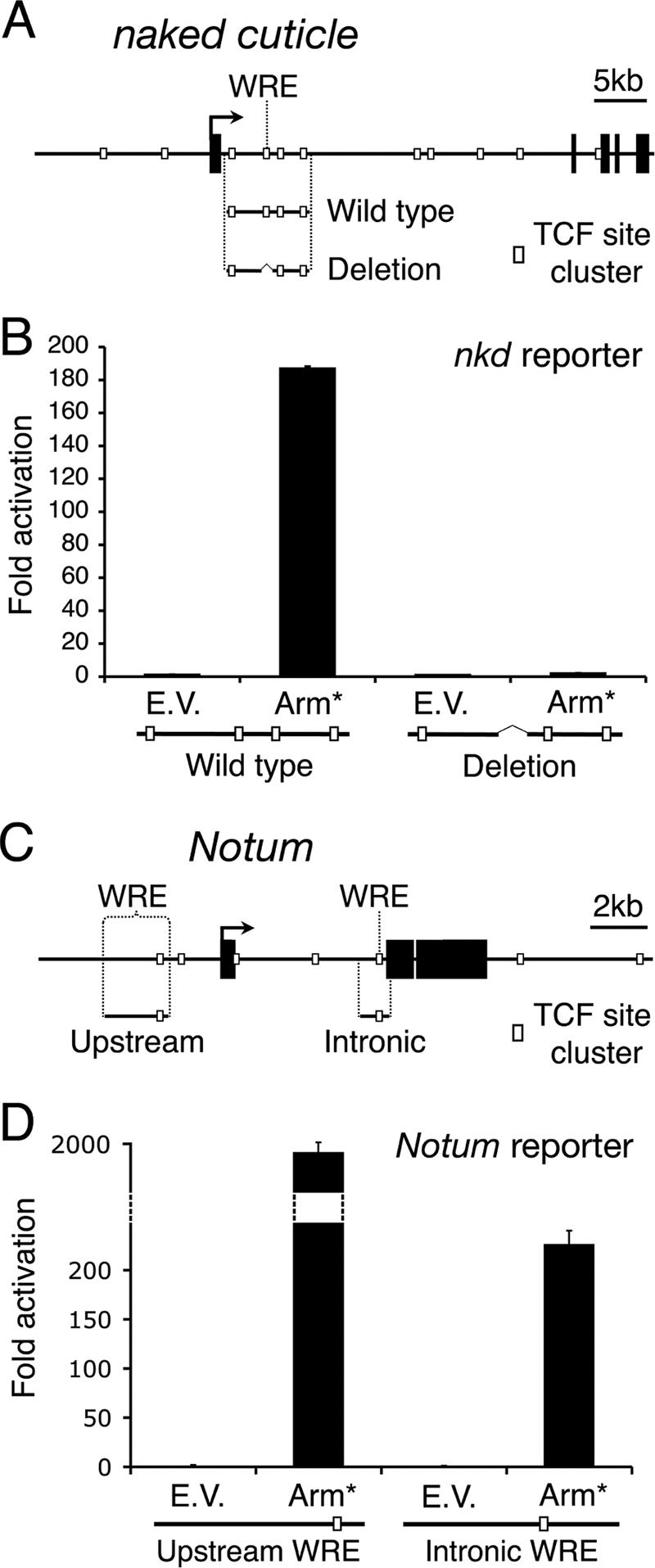
The regions bound by TCF in Wg targets contain functional WREs. (A) Cartoon depicting the fragments used in the nkd reporter gene constructs. The wild-type construct is 7.1 kb in length, and the WRE deletion construct lacks 1 kb surrounding the WRE. White boxes indicate TCF site clusters. (B) Kc cells were transfected with empty vector (E.V.) or Arm* and an hsp70-luciferase reporter plasmid containing the wild-type or WRE deletion construct. At 72 h after transfection, reporter gene activity was measured. The wild-type construct is activated 180-fold by Arm*, whereas the deletion construct is inactive. (C) Cartoon depicting the Notum upstream and intronic WRE reporter gene fragments. (D) Regulation of reporter constructs by Arm* was carried out as for nkd. Both upstream and intronic WREs are highly responsive to activation of Wg signaling. Each bar represents the mean of two independent transfections, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative.
WREs can be activated by cotransfection with a constitutively active form of Arm (Arm*) (13, 35). Consistent with this, the 7-kb nkd intronic reporter gene was stimulated almost 200-fold by Arm* (Fig. 5B). When a 1-kb region containing the nkd WRE was deleted from this construct, activation by Arm* was completely blocked (Fig. 5B). This result demonstrates that the WRE is absolutely required to respond to Wg signaling. Furthermore, these findings imply that, despite containing high-quality consensus TCF sites, the other TCF clusters in this 7-kb fragment are unable to respond to the Wg pathway. Taken together with the ChIP data, these results support a model where TCF/Arm act solely through the WRE to increase histone acetylation throughout the nkd locus and activate transcription.
Wg-dependent widespread histone acetylation also occurs at the Notum locus.
To extend of our findings beyond nkd, we turned to another Wg target gene, Notum (also called wingful). The locus is shown schematically in Fig. 5C (see also Fig. 6A). Notum is a secreted negative-feedback inhibitor of the pathway that is expressed in response to Wg signaling throughout Drosophila development (18, 20). A 2.2-kb WRE has previously been identified upstream of the Notum TSS, which was activated by WCM in Drosophila S2 cells (26). This reporter was also robustly activated by Arm* in Kc cells (Fig. 5D). In addition, a 1-kb fragment from the first Notum intron, which contained a TCF site cluster identified by Target Explorer, was also highly activated by Arm* (Fig. 5D). Consistent with the idea that Notum is a bona fide Wg target gene in Kc cells, endogenous Notum transcript levels increased significantly upon WCM treatment in a TCF-, arm-, and pygo-dependent manner (Fig. 6B).
FIG. 6.

TCF binding to the Notum locus and histone acetylation in response to Wg signaling. (A) Cartoon of the Notum locus. Black boxes represent Notum exons. Gray boxes represent CG5895 exons. White boxes represent clusters of consensus TCF binding sites. Arrows represent the predicted TSSs. The locations of the WREs are indicated. Vertical dashed lines show locations of primer sites used in ChIP analysis. (B) Notum is a target of Wg signaling in Kc cells. Cells were treated for 5.5 h with WCM following 6 days of treatment with control dsRNA or with dsRNA corresponding to TCF, arm, or pygo. Quantitative reverse transcription-PCR was then used to measure the transcript levels of Notum in reference to β-tubulin. WCM treatment induces Notum transcript levels approximately 250-fold over uninduced levels. RNAi of TCF, arm, or pygo significantly blocks this effect. (C) TCF ChIP analysis shows that TCF is present at the intronic WRE region in the absence of signaling (dashed line) and that TCF is recruited to this site and a WRE-containing region upstream of the TSS upon WCM stimulation (black line). (D and E) Histones are acetylated over a broad region at the Notum locus in response to Wg. Cells treated with control and WCM were subjected to AcH3 (D) and AcH4 (E) ChIP analysis. In both cases, histone acetylation is increased in response to Wg in a broad manner across the loci, with the highest levels roughly corresponding to TCF recruitment sites. Each bar or data point represents the mean of two independent samples, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative. Cont, control.
A Target Explorer search revealed the presence of seven TCF site clusters around the Notum locus. The previously identified upstream WRE (which contains a TCF site cluster at its 3′ end) showed no significant TCF binding above background in the absence of WCM (Fig. 6C, dashed line). However, TCF binding increased three- to fourfold at this location upon pathway activation, and significant binding was also observed in the 5′ region of the upstream Notum WRE (approximately 4 kb upstream of the TSS) (Fig. 6C). In addition to this upstream binding, the region containing the intronic Notum WRE recruits TCF in the absence of pathway activation (Fig. 6C, dashed line), which is increased by WCM treatment (Fig. 6C, black line). These data indicate that in Kc cells, there are at least two WREs at the Notum locus that recruit TCF in response to Wg. Wg signaling activates CG5895, the gene just downstream of Notum (Fig. 6A), in S2 cells (44). The two TCF site clusters nearest to CG5895 are not bound by TCF in Kc cells (Fig. 6C), though it is possible that this gene is activated by one or more of the WREs identified in the Notum locus.
In the absence of Wg signaling, AcH3 and AcH4 levels are mostly low at the Notum locus (Fig. 6D and E, dashed lines). Like nkd, however, there are unexplained peaks, one in particular approximately 6 kb downstream of the Notum gene. This high signal is not due to primer efficiency, as no such peak is observed in the TCF ChIP (Fig. 6C), and acetylation at this site was not significantly affected by Wg treatment (Fig. 6D and E).
As was found with nkd, stimulation of Wg signaling results in a widespread increase in histone acetylation over the entire Notum locus (Fig. 6D and E, black lines). As with nkd, nucleosome density was not increased across the locus upon Wg signaling (data not shown). There are two peaks of acetylation, the larger one in the Notum intron and a smaller one in the region of the upstream WRE. Acetylation of the Notum core promoter does not appear to be the main focus of this modification. In contrast to the nkd core promoter, Wg signaling did not cause an increase in H3K4Me3 levels at the Notum TSS (data not shown).
To determine whether this Wg signaling-dependent, widespread histone acetylation occurs in other contexts, we performed ChIP on Drosophila embryonic extracts. Daughterless (Da)-Gal4 (a ubiquitous driver) was used to express Wg or a dominant negative form of the Wg receptor Frizzled-2 (GPI-dFz2) to block Wg signaling (6). As in Kc cells, TCF was bound at both the 5′ and 3′ ends of the upstream Notum WRE region, and this binding is increased when the pathway was activated (Fig. 7A). In contrast to the data obtained with Kc cells (Fig. 6C), the intronic WRE region (approximately 5.5 kb downstream of the TSS) does not display detectable TCF binding. Consistent with the results in Kc cells, induction of Wg signaling results in widespread AcH3 (Fig. 7B), suggesting that endogenous Wg target genes in vivo are subject to types of transcriptional regulation similar to those seen in cell culture.
FIG. 7.
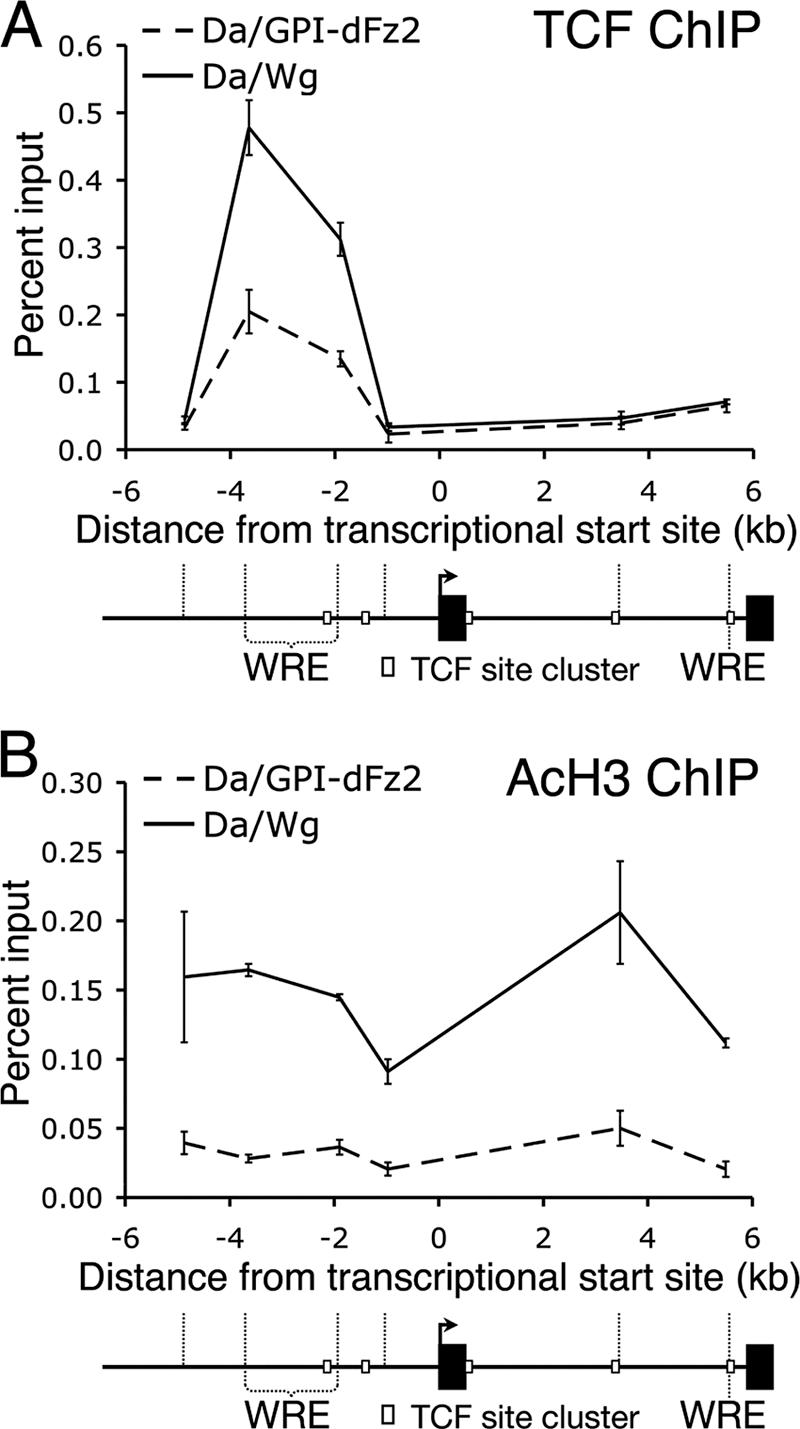
The Notum locus is widely acetylated in response to Wg signaling in vivo. Da-Gal4 was used to drive expression of Wg (Da/Wg) or GPI-dFz2 (Da/GPI-dFz2) to ubiquitously activate or inhibit the pathway in embryos, respectively. (A) ChIP analysis indicates that TCF binds to the upstream Notum WRE region when the pathway is activated (black line) and inhibited (dashed line) but not the region containing the intronic WRE. (B) AcH3 ChIP analysis shows widespread increased acetylation across the Notum locus in response to Wg signaling (compare dashed and black lines). Each data point represents the mean of two independent ChIP samples, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative.
CBP is required for widespread histone acetylation at Wg target loci.
Which HAT(s) is responsible for the Wg-induced acetylation at Wg targets? An attractive candidate is CBP, a HAT with specificity for both H3 and H4 N termini tails (1, 48). We have recently reported that CBP is required for Wg signaling in Kc cells and wing imaginal discs. CBP binds directly to Arm and is recruited to the nkd WRE region in a Wg- and Arm-dependent manner (35). The requirement for CBP in mediating the widespread increase in AcH3/AcH4 at Wg transcriptional targets can be directly tested using Kc cells depleted of CBP via RNAi.
One complication with RNAi inhibition of CBP is that it compromises cell growth and health (35; also data not shown). However, the proper dose of CBP dsRNA can compromise Wg activation of nkd and Notum expression (Fig. 8A and B) without affecting the expression of the β-tub56D, arm, or TCF genes (Fig. 8D to F). Under these conditions, CBP RNAi does not affect Wg-dependent Arm stabilization (Fig. 8G) and TCF recruitment to the nkd WRE region (Fig. 8C). These controls indicate that the CBP RNAi treatment employed does not affect the Wg pathway upstream of TCF, but transcriptional activation of Wg targets is greatly reduced.
Depletion of CBP had a dramatic effect on the ability of WCM to promote histone acetylation at the nkd and Notum loci (Fig. 9). In the case of nkd, depletion of CBP resulted in AcH3 and AcH4 levels below those observed in unstimulated cells (Fig. 9A and B). At the Notum locus, CBP RNAi almost completely blocked the ability of WCM to increase histone acetylation (Fig. 9C and D). For both targets, the effect on AcH3/AcH4 was observed at several locations, indicating that CBP is required for the widespread histone acetylation at both loci. Interestingly, Wg signaling promotes CBP recruitment primarily to the WRE region of nkd and not to other locations in the locus (Fig. 10).
FIG. 9.

CBP is required for widespread histone acetylation across the nkd and Notum loci in response to Wg. Cells were treated as described in the legend of Fig. 8 and assayed for AcH3 and AcH4 using ChIP with a select subset of primer sets for nkd (A and B) and Notum (C and D). In each case, acetylated histone levels increased in response to Wg (compare broad dashed and black lines). Pretreatment of the cells with CBP dsRNA (fine dashed lines) blocked the Wg-dependent increase of histone acetylation, resulting in levels close to or below unstimulated levels. Each data point represents the mean of two independent ChIP samples, with the error bars indicating standard deviation. All experiments were performed multiple times, and the data shown are representative. CM, control medium; Cont, control.
FIG. 10.

CBP is localized to the nkd WRE in WCM-treated cells. Kc cells were treated with control medium or with WCM for 5.5 h and processed for ChIP with anti-CBP antiserum. CBP binding was dramatically elevated at the region containing the WRE but not at other locations in nkd. Each data point represents the mean of two independent ChIP samples, with the error bars indicating standard deviations. All experiments were performed multiple times, and the data shown are representative. Cont, control.
DISCUSSION
TCF recruitment to select binding sites in Wg targets.
TCF, the sole member of the TCF/LEF1 family in flies, contains an HMG domain that is sufficient for sequence-specific binding to DNA (64). DNA-protein interaction assays have defined the sequences that are preferentially bound by TCF in vitro (23, 64). There is abundant evidence that these sites are important for the activity of WREs in vivo (2).
In this study, we used a combination of Target Explorer-assisted searching and ChIP to determine where TCF is recruited in two well-characterized Wg targets. TCF binding at these loci is restricted to certain regions of DNA that contain clusters of consensus TCF binding sites (Fig. 2A and 6C). Interestingly, other regions of chromatin that contain qualitatively similar TCF binding site clusters do not recruit TCF in vivo and cannot activate reporter genes when cloned upstream of a naive promoter (Fig. 5B). These results strongly suggest that TCF sites require additional contextual information to function as WREs. The nature of these additional cues is under investigation.
While there is agreement between our ChIP data for TCF binding and reporter gene results for the nkd gene (Fig. 2A and 5B), there is a minor discrepancy for Notum. The upstream WRE of Notum is more active in the reporter assay than the intronic Notum WRE (Fig. 5D), but TCF binding is more robust at the region containing the intronic WRE in the endogenous gene (Fig. 6C). Despite these minor inconsistencies, the combination of ChIP and reporter gene analysis has proven to be an excellent way to identify bona fide WREs in Wg targets.
A comparison between the ChIP data from Kc cells and embryos reveals an additional complication in the search for WREs. In the cultured cell line, TCF is bound both upstream of the Notum TSS and in the first intron (Fig. 6C), but in embryos only the upstream region is bound (Fig. 7A). In addition, binding of TCF at the nkd WRE was not detected in embryos (data not shown). A difference in TCF binding to target genes between several human cell lines has also been reported (69). These results illustrate that cell/tissue-specific factors play an important role in determining TCF recruitment.
Wg signaling induces widespread histone acetylation of target genes.
While TCF was localized to distinct areas of target loci, Wg signaling had a widespread effect on histone acetylation. The Wg-mediated increase in AcH3/AcH4 spanned over 45 kb and 15 kb for nkd and Notum, respectively. This histone acetylation occurs both within and outside of the transcriptional units of the targets (Fig. 3A and B and 6D and E). In addition, robust Wg-dependent histone acetylation was also observed in cells when RNA polymerase II was inhibited (Fig. 4). This demonstrates that these modifications are not a by-product of transcription.
Several large-scale studies have revealed relatively sharp peaks of histone acetylation surrounding the core promoters of active genes (3, 25, 55, 56). Likewise, several housekeeping genes in Kc cells had peaks of AcH3/AcH4 at their TSSs (Fig. 3D to F). In contrast, the pattern of histone acetylation at Wg targets does not suggest that the core promoter is a primary target of the modification. Rather, the peaks of histone acetylation for nkd and Notum are generally centered on and around the WREs, suggesting that the effect radiates from the localized sites of TCF/Arm bound to the chromatin.
Widespread chromatin modifications originating from a nucleation site have been observed in several other cases, with the best examples involving transcriptional repression. For example, heterochromatin protein 1 is thought to mediate heterochromatin formation from specific binding sites through widespread methylation via Suppressor of variegation 3-9, a methyltransferase (11). In addition, localized recruitment of Polycomb group proteins caused widespread H3K27Me3 of histones across the Ultrabithorax gene (27, 49). While the spread of histone methylation is linked to gene silencing, our results suggest that a similar spread of histone acetylation is important in mediating gene activation of Wg targets.
The acetylation of an entire locus upon gene activation is not typically observed. In a study of resting and activated human T cells, more than 45,000 regions of AcH3 from >20,000 genes were identified (55). These “acetylation islands,” which were found at both promoter and nonpromoter regions, ranged in size from 44 to 7,827 bp (55). These data are consistent with similar studies in other human cell lines (3, 25). There are examples of specific loci where broader areas of AcH3 or AcH4 are observed, most notably the β-globin locus in erythroid cells (5, 15), the Ifng gene in differentiated T cells and natural killer cells (9, 73), and the human growth hormone (hGH) locus in placental cells (28). The cis and/or trans-acting factors that distinguish the histone acetylation patterns of these genes (and the Wg targets identified in this report) from the vast majority of active genes remain to be determined.
Although nkd and Notum are not the only genes that display widespread histone acetylation, they are unique in terms of the dynamic nature of their chromatin modifications. Increased AcH3/AcH4 at these loci is detectable within 1 h of the addition of extracellular Wg and is saturated by 5.5 h. In contrast, naïve T cells treated with antigen and interleukin-12 undergo widespread histone acetylation at the Ifng gene after 2 to 3 days, as they differentiate into cytotoxic T cells (73). At the β-globin locus, changes in histone acetylation occur between fetal and adult erythroblasts (36). In these cases, the alterations in chromatin modification are occurring during differentiation, so it is likely that they are the result of several events. The rapidity of the effect we have identified suggests that the widespread modification is due solely to activation of Wg signaling and originates from the TCF/Arm binding sites in the target genes.
The role of CBP in Wg-dependent chromatin modification.
CBP is required for Wg to activate target genes in cell culture (Fig. 8A and B) as well as in fly tissues (35). This requirement has also been reported in several vertebrate systems (12, 40, 61). CBP can directly bind to β-catenin/Arm and positively regulate Wnt signaling (24, 35, 43, 60, 61). Furthermore, CBP or its close relative p300 are recruited to WRE regions upon activation of Wnt signaling in vertebrate (29, 40, 57) or Drosophila (35) cells. These data argue that CBP plays a direct and evolutionarily conserved role in mediating Wnt/β-catenin transcriptional activation.
The Wg signaling-dependent widespread increase in AcH3/AcH4 at target loci requires CBP (Fig. 9), supporting a model where CBP's classic function as a HAT plays a major role in the process. However, CBP recruitment at nkd is localized to the WRE region (Fig. 10). This is reminiscent of the situation with the Polycomb group complexes, which are highly localized but promote H3K27Me3 over large areas (27, 49). DNA looping that brings histones to the localized K27 methylase has been suggested as a possible mechanism for this phenomenon (28). While a looping model is possible for Wg targets, it should be noted that CBP and p300 recruitment to specific regions of the human genome has been correlated with elevated AcH3/AcH4 at hundreds of locations, but the extent of histone acetylation is not widespread (17, 25). This suggests the existence of additional factors at Wg targets that cooperate with CBP to achieve longer-range chromatin modification.
TCF recruitment at WREs is stimulated by Wg signaling.
TCF recruitment at WREs is stimulated by Wg signaling. The prevailing model for the role of TCF family members in the regulation of Wnt/β-catenin targets is that they act as passive anchors on the DNA through which corepressors or coactivators are recruited (50, 65). This view implies that the amount of TCF bound to WREs is similar in the absence or presence of pathway stimulation. However, we find that Wg stimulation causes a significant increase in TCF binding to WRE regions. This phenomenon was observed at several WREs in Kc cells (Fig. 1D and 6C) and in embryos (Fig. 7A). This increase in the TCF ChIP signal does not appear to be due to an increase in nuclear TCF levels (10) and was observed with two antibodies directed against different portions of the TCF protein (Fig. 1D and E). This makes it unlikely that the increase in TCF binding is due to a Wg-induced conformational change in the protein.
While the mechanism of the increase in TCF binding is not clear, it is worth noting that it still occurs in cells where histone acetylation was low (Fig. 8C) and when transcription is blocked (Fig. 4B). Therefore, these processes do not indirectly lead to the increase in TCF binding. However, the effect on TCF binding is dependent on Arm (Fig. 1D). Interestingly, this result is consistent with a previous study that showed that β-catenin greatly enhanced binding of LEF1 to chromatinized TCF sites in vitro (63). This raises the possibility that in flies, Arm has the same effect on TCF.
Another potential role for CBP in activating the Wg pathway is through acetylation of β-catenin, which increases its affinity for TCF (32, 34, 70). However, when CBP was depleted in our cell culture system, Wg signaling still caused an increase in TCF binding to the nkd WRE region (Fig. 8C). This suggests that Arm is still able to bind to TCF when CBP levels are inhibited by RNAi.
While our results are inconsistent with the prevailing model of TCF binding to WREs, it should be pointed out that there is very little experimental evidence addressing this issue. It was found that LEF1 and TCF4 binding to WRE regions in several genes were unchanged by increased Wnt/β-catenin signaling levels (57, 69). The discrepancy could reflect functional differences between the TCF and LEF1 proteins, or differences in the organisms or cell types used. Further analysis of more WREs in several systems is needed to determine whether either example is generally applicable to TCF/LEF1 binding to Wnt targets.
Acknowledgments
We thank especially R. Nusse for the pTubwg S2 cells from which WCM was prepared and Kimberly Harnish for providing additional WCM at a critical juncture during this project. We also thank past and present members of the Cadigan laboratory for encouragement and constructive criticism.
This work was supported by NIH grant RO1 CA95869 to K.M.C. D.S.P. was supported by the University of Michigan Organogenesis Training Grant and a Rackham Predoctoral Fellowship.
Footnotes
Published ahead of print on 26 December 2007.
REFERENCES
- 1.Bannister, A. J., and T. Kouzarides. 1996. The CBP co-activator is a histone acetyltransferase. Nature 384641-643. [DOI] [PubMed] [Google Scholar]
- 2.Barolo, S. 2006. Transgenic Wnt/TCF pathway reporters: all you need is Lef? Oncogene 257505-7511. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein, B. E., M. Kamal, K. Lindblad-Toh, S. Bekiranov, D. K. Bailey, D. J. Huebert, S. McMahon, E. K. Karlsson, E. J. Kulbokas III, T. R. Gingeras, S. L. Schreiber, and E. S. Lander. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120169-181. [DOI] [PubMed] [Google Scholar]
- 4.Billin, A. N., H. Thirlwell, and D. E. Ayer. 2000. Beta-catenin-histone deacetylase interactions regulate the transition of LEF1 from a transcriptional repressor to an activator. Mol. Cell. Biol. 206882-6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulger, M., D. Schubeler, M. A. Bender, J. Hamilton, C. M. Farrell, R. C. Hardison, and M. Groudine. 2003. A complex chromatin landscape revealed by patterns of nuclease sensitivity and histone modification within the mouse beta-globin locus. Mol. Cell. Biol. 235234-5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cadigan, K. M., M. P. Fish, E. J. Rulifson, and R. Nusse. 1998. Wingless repression of Drosophila frizzled 2 expression shapes the Wingless morphogen gradient in the wing. Cell 93767-777. [DOI] [PubMed] [Google Scholar]
- 7.Cadigan, K. M., and Y. I. Liu. 2006. Wnt signaling: complexity at the surface. J. Cell Sci. 119395-402. [DOI] [PubMed] [Google Scholar]
- 8.Cadigan, K. M., and R. Nusse. 1997. Wnt signaling: a common theme in animal development. Genes Dev. 113286-3305. [DOI] [PubMed] [Google Scholar]
- 9.Chang, S., and T. M. Aune. 2005. Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proc. Natl. Acad. Sci. USA 10217095-17100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, J. L., H. V. Lin, T. A. Blauwkamp, and K. M. Cadigan. 30 November 2007. Spenito and Split ends act redundantly to promote Wingless signaling. Dev. Biol. [Epub ahead of print.] doi: 10.1016/j.ydbio.2007.11.023. [DOI] [PubMed]
- 11.Danzer, J. R., and L. L. Wallrath. 2004. Mechanisms of HP1-mediated gene silencing in Drosophila. Development 1313571-3580. [DOI] [PubMed] [Google Scholar]
- 12.Emami, K. H., C. Nguyen, H. Ma, D. H. Kim, K. W. Jeong, M. Eguchi, R. T. Moon, J. L. Teo, H. Y. Kim, S. H. Moon, J. R. Ha, and M. Kahn. 2004. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc. Natl. Acad. Sci. USA 10112682-12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang, M., J. Li, T. Blauwkamp, C. Bhambhani, N. Campbell, and K. M. Cadigan. 2006. C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. EMBO J. 252735-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng, Y., N. Lee, and E. R. Fearon. 2003. TIP49 regulates beta-catenin-mediated neoplastic transformation and T-cell factor target gene induction via effects on chromatin remodeling. Cancer Res. 638726-8734. [PubMed] [Google Scholar]
- 15.Forsberg, E. C., K. M. Downs, H. M. Christensen, H. Im, P. A. Nuzzi, and E. H. Bresnick. 2000. Developmentally dynamic histone acetylation pattern of a tissue-specific chromatin domain. Proc. Natl. Acad. Sci. USA 9714494-14499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujita, N., and P. A. Wade. 2004. Use of bifunctional cross-linking reagents in mapping genomic distribution of chromatin remodeling complexes. Methods 3381-85. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Bassets, I., Y. S. Kwon, F. Telese, G. G. Prefontaine, K. R. Hutt, C. S. Cheng, B. G. Ju, K. A. Ohgi, J. Wang, L. Escoubet-Lozach, D. W. Rose, C. K. Glass, X. D. Fu, and M. G. Rosenfeld. 2007. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 128505-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerlitz, O., and K. Basler. 2002. Wingful, an extracellular feedback inhibitor of Wingless. Genes Dev. 161055-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giles, R. H., J. H. van Es, and H. Clevers. 2003. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 16531-24. [DOI] [PubMed] [Google Scholar]
- 20.Giraldez, A. J., R. R. Copley, and S. M. Cohen. 2002. HSPG modification by the secreted enzyme Notum shapes the Wingless morphogen gradient. Dev. Cell 2667-676. [DOI] [PubMed] [Google Scholar]
- 21.Gregorieff, A., and H. Clevers. 2005. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 19877-890. [DOI] [PubMed] [Google Scholar]
- 22.Grewal, S. I., and D. Moazed. 2003. Heterochromatin and epigenetic control of gene expression. Science 301798-802. [DOI] [PubMed] [Google Scholar]
- 23.Hallikas, O., K. Palin, N. Sinjushina, R. Rautiainen, J. Partanen, E. Ukkonen, and J. Taipale. 2006. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 12447-59. [DOI] [PubMed] [Google Scholar]
- 24.Hecht, A., K. Vleminckx, M. P. Stemmler, F. van Roy, and R. Kemler. 2000. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 191839-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heintzman, N. D., R. K. Stuart, G. Hon, Y. Fu, C. W. Ching, R. D. Hawkins, L. O. Barrera, S. Van Calcar, C. Qu, K. A. Ching, W. Wang, Z. Weng, R. D. Green, G. E. Crawford, and B. Ren. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39311-318. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmans, R., R. Stadeli, and K. Basler. 2005. Pygopus and legless provide essential transcriptional coactivator functions to Armadillo/beta-catenin. Curr. Biol. 151207-1211. [DOI] [PubMed] [Google Scholar]
- 27.Kahn, T. G., Y. B. Schwartz, G. I. Dellino, and V. Pirrotta. 2006. Polycomb complexes and the propagation of the methylation mark at the Drosophila ubx gene. J. Biol. Chem. 28129064-29075. [DOI] [PubMed] [Google Scholar]
- 28.Kimura, A. P., S. A. Liebhaber, and N. E. Cooke. 2004. Epigenetic modifications at the human growth hormone locus predict distinct roles for histone acetylation and methylation in placental gene activation. Mol. Endocrinol. 181018-1032. [DOI] [PubMed] [Google Scholar]
- 29.Kioussi, C., P. Briata, S. H. Baek, D. W. Rose, N. S. Hamblet, T. Herman, K. A. Ohgi, C. Lin, A. Gleiberman, J. Wang, V. Brault, P. Ruiz-Lozano, H. D. Nguyen, R. Kemler, C. K. Glass, A. Wynshaw-Boris, and M. G. Rosenfeld. 2002. Identification of a Wnt/Dvl/beta-catenin → Pitx2 pathway mediating cell-type-specific proliferation during development. Cell 111673-685. [DOI] [PubMed] [Google Scholar]
- 30.Kleber, M., and L. Sommer. 2004. Wnt signaling and the regulation of stem cell function. Curr. Opin. Cell Biol. 16681-687. [DOI] [PubMed] [Google Scholar]
- 31.Knirr, S., and M. Frasch. 2001. Molecular integration of inductive and mesoderm-intrinsic inputs governs even-skipped enhancer activity in a subset of pericardial and dorsal muscle progenitors. Dev. Biol. 23813-26. [DOI] [PubMed] [Google Scholar]
- 32.Labalette, C., C. A. Renard, C. Neuveut, M. A. Buendia, and Y. Wei. 2004. Interaction and functional cooperation between the LIM protein FHL2, CBP/p300, and beta-catenin. Mol. Cell. Biol. 2410689-10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee, H. H., and M. Frasch. 2000. Wingless effects mesoderm patterning and ectoderm segmentation events via induction of its downstream target sloppy paired. Development 1275497-5508. [DOI] [PubMed] [Google Scholar]
- 34.Levy, L., Y. Wei, C. Labalette, Y. Wu, C. A. Renard, M. A. Buendia, and C. Neuveut. 2004. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol. Cell. Biol. 243404-3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, J., C. Sutter, D. S. Parker, T. Blauwkamp, M. Fang, and K. M. Cadigan. 2007. CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 262284-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li, Q., G. Barkess, and H. Qian. 2006. Chromatin looping and the probability of transcription. Trends Genet. 22197-202. [DOI] [PubMed] [Google Scholar]
- 37.Liang, G., J. C. Lin, V. Wei, C. Yoo, J. C. Cheng, C. T. Nguyen, D. J. Weisenberger, G. Egger, D. Takai, F. A. Gonzales, and P. A. Jones. 2004. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. USA 1017357-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lilja, T., H. Aihara, M. Stabell, Y. Nibu, and M. Mannervik. 2007. The acetyltransferase activity of Drosophila CBP is dispensable for regulation of the Dpp pathway in the early embryo. Dev. Biol. 305650-658. [DOI] [PubMed] [Google Scholar]
- 39.Logan, C. Y., and R. Nusse. 2004. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20781-810. [DOI] [PubMed] [Google Scholar]
- 40.Ma, H., C. Nguyen, K. S. Lee, and M. Kahn. 2005. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene 243619-3631. [DOI] [PubMed] [Google Scholar]
- 41.Margueron, R., P. Trojer, and D. Reinberg. 2005. The key to development: interpreting the histone code? Curr. Opin. Genet. Dev. 15163-176. [DOI] [PubMed] [Google Scholar]
- 42.Matsubayashi, H., S. Sese, J. S. Lee, T. Shirakawa, T. Iwatsubo, T. Tomita, and S. Yanagawa. 2004. Biochemical characterization of the Drosophila wingless signaling pathway based on RNA interference. Mol. Cell. Biol. 242012-2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyagishi, M., R. Fujii, M. Hatta, E. Yoshida, N. Araya, A. Nagafuchi, S. Ishihara, T. Nakajima, and A. Fukamizu. 2000. Regulation of Lef-mediated transcription and p53-dependent pathway by associating beta-catenin with CBP/p300. J. Biol. Chem. 27535170-35175. [DOI] [PubMed] [Google Scholar]
- 44.Mosimann, C., G. Hausmann, and K. Basler. 2006. Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with beta-catenin/Armadillo. Cell 125327-341. [DOI] [PubMed] [Google Scholar]
- 45.Narlikar, G. J., H. Y. Fan, and R. E. Kingston. 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108475-487. [DOI] [PubMed] [Google Scholar]
- 46.Ng, H. H., S. Dole, and K. Struhl. 2003. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11709-719. [DOI] [PubMed] [Google Scholar]
- 47.Nightingale, K. P., L. P. O'Neill, and B. M. Turner. 2006. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 16125-136. [DOI] [PubMed] [Google Scholar]
- 48.Ogryzko, V. V., R. L. Schiltz, V. Russanova, B. H. Howard, and Y. Nakatani. 1996. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87953-959. [DOI] [PubMed] [Google Scholar]
- 49.Papp, B., and J. Muller. 2006. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 202041-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parker, D., T. Blauwkamp, and K. M. Cadigan. 2007. Wnt/β-catenin-mediated transcriptional regulation, p. 1-61. In S. Sokol (ed.), Wnt signaling in embryonic development. Advances in developmental biology, vol. 17. Elsevier, San Diego, CA. [Google Scholar]
- 51.Piepenburg, O., G. Vorbruggen, and H. Jackle. 2000. Drosophila segment borders result from unilateral repression of hedgehog activity by wingless signaling. Mol. Cell 6203-209. [PubMed] [Google Scholar]
- 52.Polakis, P. 2000. Wnt signaling and cancer. Genes Dev. 141837-1851. [PubMed] [Google Scholar]
- 53.Reya, T., and H. Clevers. 2005. Wnt signalling in stem cells and cancer. Nature 434843-850. [DOI] [PubMed] [Google Scholar]
- 54.Robyr, D., Y. Suka, I. Xenarios, S. K. Kurdistani, A. Wang, N. Suka, and M. Grunstein. 2002. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109437-446. [DOI] [PubMed] [Google Scholar]
- 55.Roh, T. Y., S. Cuddapah, and K. Zhao. 2005. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 19542-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roh, T. Y., G. Wei, C. M. Farrell, and K. Zhao. 2007. Genome-wide prediction of conserved and nonconserved enhancers by histone acetylation patterns. Genome Res. 1774-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sierra, J., T. Yoshida, C. A. Joazeiro, and K. A. Jones. 2006. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 20586-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith, S. T., S. Petruk, Y. Sedhov, E. Cho, S. Tillib, E. Canaani, and A. Mazo. 2004. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat. Cell Biol. 6162-167. [DOI] [PubMed] [Google Scholar]
- 59.Sosinsky, A., C. P. Bonin, R. S. Mann, and B. Honig. 2003. Target Explorer: an automated tool for the identification of new target genes for a specified set of transcription factors. Nucleic Acids Res. 313589-3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun, Y., F. T. Kolligs, M. O. Hottiger, R. Mosavin, E. R. Fearon, and G. J. Nabel. 2000. Regulation of beta-catenin transformation by the p300 transcriptional coactivator. Proc. Natl. Acad. Sci. USA 9712613-12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takemaru, K. I., and R. T. Moon. 2000. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 149249-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tolwinski, N. S., and E. Wieschaus. 2004. Rethinking WNT signaling. Trends Genet. 20177-181. [DOI] [PubMed] [Google Scholar]
- 63.Tutter, A. V., C. J. Fryer, and K. A. Jones. 2001. Chromatin-specific regulation of LEF-1-beta-catenin transcription activation and inhibition in vitro. Genes Dev. 153342-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van de Wetering, M., R. Cavallo, D. Dooijes, M. van Beest, J. van Es, J. Loureiro, A. Ypma, D. Hursh, T. Jones, A. Bejsovec, M. Peifer, M. Mortin, and H. Clevers. 1997. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88789-799. [DOI] [PubMed] [Google Scholar]
- 65.van Es, J. H., N. Barker, and H. Clevers. 2003. You Wnt some, you lose some: oncogenes in the Wnt signaling pathway. Curr. Opin. Genet. Dev. 1328-33. [DOI] [PubMed] [Google Scholar]
- 66.Waltzer, L., and M. Bienz. 1998. Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature 395521-525. [DOI] [PubMed] [Google Scholar]
- 67.Wieland, T., and H. Faulstich. 1991. Fifty years of amantin. Experientia 471186-1191. [DOI] [PubMed] [Google Scholar]
- 68.Willert, K., and K. A. Jones. 2006. Wnt signaling: is the party in the nucleus? Genes Dev. 201394-1404. [DOI] [PubMed] [Google Scholar]
- 69.Wöhrle, S., B. Wallman, and A. Hecht. 2007. Differential control of Wnt target genes involves epigenetic mechanisms and selective promoter occupancy by T-cell factors. Mol. Cell. Biol. 278164-8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolf, D., M. Rodova, E. A. Miska, J. P. Calvet, and T. Kouzarides. 2002. Acetylation of beta-catenin by CREB-binding protein (CBP). J. Biol. Chem. 27725562-25567. [DOI] [PubMed] [Google Scholar]
- 71.Yang, X., M. van Beest, H. Clevers, T. Jones, D. A. Hursh, and M. A. Mortin. 2000. decapentaplegic is a direct target of dTcf repression in the Drosophila visceral mesoderm. Development 1273695-3702. [DOI] [PubMed] [Google Scholar]
- 72.Yu, X., J. Riese, S. Eresh, and M. Bienz. 1998. Transcriptional repression due to high levels of Wingless signalling. EMBO J. 177021-7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou, W., S. Chang, and T. M. Aune. 2004. Long-range histone acetylation of the Ifng gene is an essential feature of T cell differentiation. Proc. Natl. Acad. Sci. USA 1012440-2445. [DOI] [PMC free article] [PubMed] [Google Scholar]