

Abstract
The vascular endothelial cadherin (VE-cad)-based complex is involved in the maintenance of vascular endothelium integrity. Using immunoprecipitation experiments, we have demonstrated that, in confluent human umbilical vein endothelial cells, the VE-cad-based complex interacts with annexin 2 and that annexin 2 translocates from the cytoplasm to the cell-cell contact sites as cell confluence is established. Annexin 2, located in cholesterol rafts, binds to both the actin cytoskeleton and the VE-cad-based complex so the complex is docked to cholesterol rafts. These multiple connections prevent the lateral diffusion of the VE-cad-based complex, thus strengthening adherens junctions in the ultimate steps of maturation. Moreover, we observed that the down-regulation of annexin 2 by small interfering RNA induces a delocalization of VE-cad from adherens junctions and consequently a destabilization of these junctions. Furthermore, our data indicate that the decoupling of the annexin 2/p11 complex from the VE-cad-based junction, triggered by vascular endothelial growth factor treatment, facilitates the switch from a quiescent to an immature state.
Vascular endothelium consists of a monolayer of endothelial cells which lines the whole vascular tree. It forms an active boundary between the bloodstream and the underlying tissues, thus controlling the movement of circulating white cells between blood and inflamed tissues. The endothelium is also at the origin of the extension of preexisting vasculature through formation of neo-vessels, a process named angiogenesis (43). Adherens and tight junctions which hold together endothelial cells modulate leukocyte traffic and angiogenesis. Without diminishing the importance of tight junctions, adherens junctions are particularly crucial in controlling the formation and maintenance of interendothelial adhesion.
Endothelial cells express a cell-specific cadherin designated vascular endothelial cadherin (VE-cad) which constitutes the main component of interendothelial adherens junctions (4, 12, 30). This transmembrane adhesive protein plays a crucial role in the maintenance of endothelium integrity and in the modulation of its permeability (3, 20). As for other members of the cadherin receptor family, VE-cad links endothelial cells together by homophilic interactions mediated by its extracellular part and associates intracellularly with β- or γ-catenin in a mutually exclusive fashion and with p120. Despite their sequence similarities, β/γ-catenins and p120 bind to distinct sites on the cytoplasmic tail of VE-cad. While β-catenin or γ-catenin links to the distal part of the VE-cad cytoplasmic tail, p120 interacts with the membrane-proximal domain. These three catenins also exhibit different biological cellular roles. Whereas p120 stabilizes VE-cad at the plasma membrane (46, 47), β/γ-catenins interact with α-catenin. Subsequently in this article, the complex formed by VE-cad and p120 α-, β-, and γ-catenins will be designated as the “VE-cad-based complex” or “VE-cad complex.”
Until recently, it was commonly believed that α-catenin binding to β- or γ-catenins promotes connections between the VE-cad-based complex and the actin cytoskeleton, thus strengthening the VE-cad ectodomain-based interactions (8, 35, 38). However, this concept has been challenged since it was recently demonstrated that α-catenin cannot simultaneously bind to β-catenin and the actin cytoskeleton (48).
Although it would be premature to dismiss a role for α-catenin, it is possible that some other actin-binding proteins that bind to the different components of adherens junctions might also be involved in the connection of the VE-cad complex with the actin cytoskeleton. In fact, during the cadherin-mediated cell-cell adhesion, the actin cytoskeleton undergoes a drastic reorganization. Thus, it was recently reported that, upon cadherin liganding, two actin populations with different spatial distributions are clearly distinguishable at early epithelial cell-cell contacts (49). The first population is composed of thin circumferential actin bundles, and the second is localized at cell-cell junctions. These two actin populations are regulated by distinct mechanisms. Indeed, the actin bundles are formed by reorganization of preexisting actin filaments and the junctional filaments by de novo actin polymerization. To ensure the dynamic coordination between cadherin homophilic liganding and the remodeling of both actin populations, more regulatory proteins are required than assumed previously (33).
To shed some light on the connection between the VE-cad-based complex and the actin cytoskeleton, we examined the composition of the junctional VE-cad-based complex extracted from confluent endothelial cell monolayers by combining anti-VE-cad immunoprecipitation (IP) with proteomic tools, thereby identifying novel VE-cad partners (unpublished data). In these experiments, annexin 2 (A2) was identified as one of the most abundant VE-cad partners.
The superfamily of annexins forms a Ca2+-dependent regulated class of proteins able to dock in a reversible manner with the inner leaflet of the plasma membrane by interacting with acidic phospholipids (19). A2 interacts, via its N-terminal domain, with p11 (also called S100A10) to form a heterotetramer in which a central p11 dimer connects two A2 monomers (14, 26, 32). Moreover, A2 exhibits an F-actin binding site localized within the last 9 amino acid residues of its C terminus (17). A2 not only is able to bind to actin but also can bundle actin filaments (22). It participates in the regulation of membrane organization and more particularly in the assembly of cholesterol rafts (2). These A2-containing cholesterol rafts are highly dynamic membrane domains that serve as F-actin assembly platforms. The precise role of A2 in the dynamic remodeling of these platforms remains to be elucidated. Nevertheless, Hayes et al. recently demonstrated that A2 regulates actin polymerization by interfering with the barbed ends of growing actin filaments (23).
In the present article, we examine the role played by A2 in the regulation and/or stabilization of endothelial adherens junctions. We demonstrate by IP that A2 interacts directly with the junctional VE-cad-based complex. In addition, our results prove that A2 is absolutely required to maintain VE-cad at cell-cell junctions. Our data also indicate that A2 docks the VE-cad-based complex to cholesterol rafts and binds actin fibers to the complex as cells reach confluence. Thus, the connection of the VE-cad-based complex to both cholesterol rafts and to A2-anchored actin filaments strengthens adherens junctions in the ultimate step of maturation.
Reciprocally, it can be assumed that the opening of the adherens junction may result from A2 and VE-cad-based complex decoupling. To test this hypothesis, we evaluated effects of the proangiogenic vascular endothelial growth factor (VEGF) on A2/VE-cad-based complex organization in human umbilical vein endothelial cells (HUVECs). We showed that VEGF induced a drastic perturbation of VE-cad localization, release of A2 from the plasma membrane to the cytoplasm, and decoupling of A2 from the VE-cad-based complex. This dissociation leads to the disconnection of the VE-cad complex from the actin cytoskeleton, an event that may constitute one of the earliest steps in adherens junction destabilization favoring the increase of vascular permeability.
MATERIALS AND METHODS
Reagents and antibodies.
The monoclonal anti-VE-cad antibody BV9 was from Santa Cruz Biotechnologies. The polyclonal antibody directed against human VE-cad anti-Cad3 was produced as previously described (24). Anti-A2, anti-A1, anti-A5, anti-α-, β-, and γ-catenins, and anti-PECAM-1 (platelet/endothelial cell adhesion molecule 1) antibody were purchased from BD Biosciences. Anti-N-cadherin antibody and rabbit nonimmune immunoglobulin G (IgG) were purchased from DakoCytomation. Anti-β-actin, fluorescein isothiocyanate (FITC)- and tetramethyl rhodamine isocyanate (TRITC)-phalloidin, methyl-β-cyclodextrin (MβCD), and latrunculin B (La B) were from Sigma-Aldrich. Cy2- or Cy3-labeled goat anti-rabbit antibodies and Cy2- or Cy3-labeled goat anti-mouse antibodies were purchased from Immunotech. Cy5-labeled goat anti-mouse and Cy5-labeled goat anti-rabbit antibodies were provided by Amersham Biosciences. Alexa 488-labeled goat anti-IgG1 and Cy3-labeled goat anti-IgM were purchased from Molecular Probes. Recombinant VEGF was from PeproTech, Inc.
Cell culture.
HUVECs were isolated and cultured as previously described (30). Only cells on passage 2 were used.
Immunofluorescence microscopy.
After treatment, cells cultured on fibronectin-coated glass coverslips were fixed and permeabilized in 85% methanol, 15 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid); pH 7.7], and 2 mM MgCl2 for 10 min at −20°C and blocked with 0.5% bovine serum albumin in phosphate-buffered saline (PBS). After several washing steps in PBS, cells were incubated with primary antibodies and then with the adequate secondary antibodies. The coverslips were mounted onto slides with 4′,6′-diamidino-2-phenylindole (DAPI)-containing Mowiol 40-88 (Sigma-Aldrich). Cells were then observed using an Axioplan 2 microscope (Zeiss) equipped with an Achroplan ×50 objective. Images were captured with an AxioCam MR camera using Axiovision software.
When specified, cells were observed on a confocal microscope TCS-SP2 (Leica) with a ×63/1.4 objective. For image acquisition (1,024 by 1,024, 8 bits), Alexa 488, TRITC, Cy5, and DAPI fluorescences were excited and collected sequentially (400 Hz line by line) by using the 488-nm wavelength of an argon laser for Alexa 488, the 543-nm wavelength of a helium-neon laser for TRITC, the 633-nm wavelength of a helium-neon laser for Cy5 excitation, and the 405-nm wavelength of a photodiode for DAPI. Fluorescence emission was collected from 498 to 540 nm for Alexa 488, from 573 to 630 nm for TRITC, from 644 to 720 nm for Cy5, and from 410 to 460 nm for DAPI.
IP.
Affinity beads were prepared as follows. Sixty micrograms of affinity-purified anti-Cad 3 or 6 μg of anti-A2 antibodies was mixed with 30 μl of a suspension of protein A- or protein G-Sepharose beads, respectively (Sigma-Aldrich). After a 2-h incubation at 4°C, centrifugation at 200 × g for 5 min, and several washes in PBS, grafted beads were equilibrated in the lysis buffer L (10 mM PIPES [pH 7], 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EDTA, 2 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 2 μg/ml aprotinin) containing 0.5% Igepal CA-630. HUVEC monolayers were washed twice with PBS and lysed in 1 ml of 0.5% Igepal-containing buffer L for 20 min at 4°C. After lysate centrifugation at 15,000 × g for 10 min, supernatants were incubated overnight at 4°C under continuous mixing with antibody-grafted protein A/G-Sepharose beads. IPs were performed each time on a 78-cm2 cell monolayer. To elute immunoprecipitated proteins, beads were washed three times in lysis buffer and then boiled in 50 μl Laemmli buffer for 10 min. Whole elutions were loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel for Western blot analysis.
Cell treatments.
After a brief washing in PBS, HUVEC monolayers were incubated in culture medium containing either 3.8 mM MβCD for 30 min or 0.5 μM La B for 5 min. To determine whether VE-cad was internalized following La B treatment, La B-treated cells were incubated with trypsin-EDTA at 0.5 g/liter for 2 min prior to cell lysis and Western blot analysis.
HUVECs were starved between 3 and 6 h before a 30-min stimulation with VEGF (50 ng/ml) in serum-free medium.
Subcellular fractionations.
To separate cytoplasm from membrane, HUVEC monolayers were lysed in detergent-free buffer L. After centrifugation at 1,000 × g for 3 min at 4°C to remove unbroken cells, supernatants were collected and recentrifuged at 150,000 × g for 30 min at 4°C. The resulting supernatants corresponded to the nonmembrane fractions, designated as the cytoplasmic fraction hereafter, and the pellets, resuspended in 0.5% Igepal-containing buffer L, corresponded to the membrane fractions. To isolate cholesterol raft-associated proteins, membrane pellets were resuspended in buffer L containing 0.01% digitonin. After a 20-min incubation on ice, homogenates were centrifuged at 150,000 × g for 30 min at 4°C. The resulting supernatants contained cholesterol raft-associated proteins, while the pellets contained the other membrane-associated proteins.
Liquid chromatography-tandem mass spectrometry.
Cholesterol raft-associated proteins were separated on 4 to 12% gradient gel (Criterion XT Precast; Bio-Rad), and gels were stained with Coomassie blue (Bio-Safe Coomassie G250 stain; Bio-Rad). The Coomassie-blue stained protein bands were in-gel digested with trypsin, and the recovered peptides were separated by liquid chromatography on silica C18 column (Dionex) prior to analysis on an integrated nano-liquid chromatography-tandem mass spectrometry system. Peptide identification from the resulting tandem mass spectrometry data set was achieved using an in-house MASCOT server (Matrix Sciences, London, United Kingdom).
Western blotting.
Cell extracts were analyzed under reducing conditions by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using either 10% Tris-glycine or 15% Tris-Tricine homemade gels or 4 to 12% gradient Criterion XT gels and electrotransferred onto a pure nitrocellulose membrane (Bio-Rad). After blocking with 5% nonfat dry milk, proteins were detected by specific primary antibodies and horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibodies (Sigma-Aldrich). The immunoreactive bands were revealed using the ECL enhanced chemiluminescence Western blotting detection kit (Amersham Biosciences). Signals recorded on autoradiography films (Amersham Biosciences) were quantified using a Gel Doc EQ apparatus and Quantity One software (Bio-Rad).
siRNA interference.
Twenty-one-nucleotide small interfering RNA (siRNA) duplex 5′-UCAUCCACACCUUUGGUCUUU-3′ and 5′-UCAGCAUCAAAGUUAGUAUUU-3′ targeting sequences for human A2 were purchased from Dharmacon. HUVECs were plated a day before siRNA transfection in medium without antibiotics. Seventy-five picomoles of siRNA was transfected with Oligofectamine (Invitrogen) according to the manufacturer's instructions. Transfection was performed twice with an interval of 24 h. Then, siRNA-transfected HUVECs were plated on glass coverslips in complete medium and used for various experiments 24 and 48 h after plating. Cells were then fixed at 4 or 5 days posttransfection.
RESULTS
Cell-density-dependent localization of A2 to endothelial cell-cell junctions.
By combining IP with proteomic studies, we recently determined the protein composition of confluent HUVEC adherens junctions (unpublished data). Numerous actin-binding proteins, not previously thought to localize at adherens junctions, were thus revealed. Among them, A2 was found. Here, we examined the possible role of A2 in the regulation and/or stabilization of VE-cad-based adherens junctions.
First, the subcellular localization of A2 was explored in density-increasing HUVEC monolayers. To do so, the membrane and cytoplasmic fractions were separated from subconfluent and confluent HUVECs by ultracentrifugation and analyzed by Western blotting (Fig. 1A). As expected, in subconfluent as well as in confluent cells, VE-cad was found in the membrane fraction (Fig. 1A, lanes 2 and 4). The absence of VE-cad in the cytoplasm established that this fraction was not contaminated by membranes (Fig. 1A, lanes 1 and 3). On the contrary, it was observed that A2, which was exclusively found in the cytoplasmic fraction in subconfluent cells, was distributed between the cytoplasm and the membranes in confluent cells (Fig. 1A). These results showed that A2 partially translocated from cytoplasm to membranes as cell confluence increased.
FIG. 1.

Cell density-dependent accumulation of A2 at cell-cell junctions. (A) Redistribution of A2 to membranes in confluent HUVECs. Subcellular fractionation of subconfluent (lanes 1 and 2) and confluent (lanes 3 and 4) HUVECs was performed as described in Materials and Methods. Proteins isolated from cytoplasm (lanes 1 and 3) and membrane (lanes 2 and 4) fractions were analyzed on 10% Tris-glycine gels prior to immunoblotting for VE-cad, and A2. Molecular mass markers (kDa) are given at the left margin of each blot. (B) Subcellular distribution of A2, VE-cad, and actin. HUVECs were plated on glass coverslips at two different densities and cultured for 3 days. The density of confluent cells was 8 times higher than that of subconfluent cells. Cells were then stained with anti-A2 monoclonal antibody, anti-Cad3 polyclonal antibody, Cy5-labeled goat anti-mouse and Cy3-labeled goat anti-rabbit antibodies, FITC-labeled phalloidin, and DAPI. The right panels are magnified images of the areas boxed in the left panels (Magn.). Bars, 20 μm. (C) Confocal analysis of A2 and VE-cad in confluent HUVECS. Cells were plated on glass coverslips, cultured for 3 days, and then stained with anti-A2 monoclonal antibody, anti-Cad3 polyclonal antibody, and Alexa 488-labeled goat anti-mouse and Cy5-labeled goat anti-rabbit antibodies. Colocalization was determined with the CF2D software from Leica. Images correspond to an optical section (≈650 nm). The diagram (Co-localization) represents the relative intensity of the pixels in the red and the green images. Dots gated in the region of interest (yellow), corresponding to the colocalization, are represented in white in the images. Bar, 20 μm.
To demonstrate A2 translocation, an immunocytochemical localization of A2 was performed on HUVECs plated at different densities. Figure 1B shows that A2 is exclusively cytoplasmic in subconfluent cells and accumulates at the plasma membrane as cell confluence increases. To assess whether VE-cad and A2 colocalize at cell-cell contacts, confocal microscopy analysis was performed on 650-nm optical sections (Fig. 1C; larger areas also shown in Fig S1 in the supplemental material). This revealed that VE-cad and A2 effectively colocalized along the whole cell-cell junction (Fig. 1C). Nevertheless, some minor restricted areas exhibit juxtaposed red and green dots, thus indicating that a small amount of A2, even though localized at the plasma membrane, was not associated with the VE-cad-based junction (Fig. 1C). Moreover, at cell-cell contacts, A2 and VE-cad colocalize with the actin cytoskeleton (Fig. 1B).
It is notable that the adherens junction architecture changes with increasing cell density. Actually, subconfluent cells possess an elongated shape and adherens junctions appear immature, exhibiting a thin, discontinuous, and zigzag staining for VE-cad (Fig. 1B). As the cell density increased, cells adopted a cobblestone morphology and adherens junctions presented an intense linear VE-cad staining (Fig 1B). Moreover, phalloidin staining indicated that parallel actin stress fibers visible in subconfluent HUVECs were progressively reoriented to constitute the actin cortical ring in confluent cells (Fig. 1B). This indicates that, as cell confluence is established, the actin cytoskeleton undergoes a major remodeling which correlates with the arrival of A2 at cell-cell junctions.
Connection of A2 to the VE-cad-based complex.
To assess whether A2 is associated with the VE-cad/catenin complex, A2 immunoprecipitates were analyzed by Western blotting. As reported in Fig. 2A, antibodies directed against VE-cad, p120, and α-, β-, and γ-catenins recognized bands of 150, 120 to 100, 110, 93, and 83 to 80 kDa, respectively. Besides VE-cad and catenins, p11, the protein known to form a heterotetrameric complex with A2 at the plasma membrane (39), was also detected in the anti-A2 immunoprecipitates (Fig. 2A). Conversely, anti-VE-cad IP showed association with A2 only when cells have reached confluence (Fig. 2B). Other annexins expressed in endothelial cells, such as A1 and A5 (Table 1), were not detected in anti-VE-cad IPs (Fig. 3A). Moreover, VE-cad was detected in neither anti-A1 (Fig. 3B) nor anti-A5 (Fig. 3C) IPs performed on confluent HUVECs. Altogether, these data indicate that the A2/p11 complex is associated with the VE-cad/catenin complex at adherens junction sites in confluent cells. It is noteworthy that N-cadherin, the other cadherin expressed by endothelial cells, and PECAM-1, a transmembrane receptor that localizes at interendothelial cell-cell contacts, were not detected in anti-A2 IPs (Fig. 2A). Altogether, the results established that A2 interacts specifically with the VE-cad-based complex.
FIG. 2.

Association of A2 to the VE-cad-based complex. (A) Anti-A2 IPs (IP A2), performed on confluent HUVECs, were resolved on 10% Tris-glycine gels, electrotransferred, and probed for VE-cad, N-cadherin (N-cad), PECAM-1, p120, α-, β-, and γ-catenins (α-, β-, and γ-cat), and actin and on 15% Tris-Tricine gels and probed for A2 and p11. For comparison, aliquots of whole-cell lysates (Lys) were analyzed in parallel. (B) Anti-VE-cad IPs were performed on subconfluent (−) and confluent (Conf; +) HUVECs and resolved on 4 to 12% gradient precast Criterion XT gels. As negative controls, whole-cell lysates and IPs performed on confluent HUVEC lysate using rabbit nonimmune IgG (NI) were analyzed in parallel. Parallel blots were probed for VE-cad and A2. Molecular mass markers (kDa) are given at the left margin of each panel.
TABLE 1.
Examples of proteins identified in cholesterol rafts
| Proteina | Accession no. | pI | Score | Mass (kDa) | Coverage (%) | No. of peptides |
|---|---|---|---|---|---|---|
| Cytoplasmic proteins | ||||||
| α-Actin | gi 178027 | 5.24 | 328.07 | 42,081 | 22.28 | 6 |
| β-Actin | gi 4501885 | 5.29 | 757.83 | 41,710 | 37.33 | 10 |
| α1-Actinin | gi 4501891 | 5.25 | 423.84 | 102,993 | 9.19 | 6 |
| α4-Actinin | gi 2804273 | 5.27 | 621.67 | 102,204 | 13.01 | 8 |
| Actin-related protein 3 | gi 27806335 | 5.61 | 32.64 | 47,341 | 1.91 | 1 |
| AHNAK nucleoprotein isoform 1 | gi 61743954 | 5.80 | 180.05 | 628,699 | 1.04 | 4 |
| A1 | gi 442631 | 7.77 | 311.53 | 35,018 | 24.52 | 5 |
| A2, isoform 2 | gi 12655075 | 7.57 | 274.61 | 38,580 | 22.12 | 5 |
| A5 | gi 809190 | 4.94 | 104.72 | 35,783 | 8.46 | 2 |
| A6, isoform 2 | gi 71773415 | 5.46 | 81.98 | 75,229 | 4.35 | 2 |
| α-Catenin | gi 1092190 | 6.06 | 47.65 | 100,018 | 1.32 | 1 |
| β1-Catenin | gi 4503131 | 5.53 | 258.73 | 85,442 | 9.48 | 4 |
| Moesin | gi 4505257 | 6.08 | 717.50 | 67,778 | 26.34 | 11 |
| α-Tubulin | gi 340021 | 4.94 | 318.10 | 50,120 | 12.86 | 4 |
| β-Tubulin, class I | gi 4580988 | 4.78 | 327.73 | 49,639 | 17.57 | 6 |
| Vinculin isoform VCL | gi 4507877 | 5.83 | 544.83 | 116,649 | 10.60 | 10 |
| Membrane proteins | ||||||
| PECAM-1 | gi 129747 | 6.55 | 91.59 | 82,484 | 3.25 | 2 |
| β1-Integrin (isoform 1B) | gi 19743815 | 5.35 | 62.12 | 87,389 | 2.15 | 1 |
| Raft markers | ||||||
| Clathrin heavy chain 1 | gi 4758012 | 5.48 | 191.18 | 191,493 | 2.87 | 4 |
| Caveolin 1 | gi 4972627 | 5.85 | 46.24 | 13,066 | 8.85 | 1 |
Cholesterol raft-associated proteins, isolated as previously described in Materials and Methods, were separated by electrophoresis prior to being in-gel digested by trypsin. Identification of the trypsin peptides was achieved by tandem mass spectrometry. Only proteins relevant for this study are listed. Complete results are available from the corresponding author on request.
FIG. 3.

Specificity of the association between the VE-cad-based complex and the endothelial annexins. Anti-VE-cad (A), anti-A1 (B), and anti-A5 (C) IPs were performed on confluent HUVECs and were resolved on 12% Tris-glycine gels. Anti-VE-cad IPs were immunoblotted for VE-cad and A1, A2, and A5. Anti-A1 and anti-A5 IPs were probed for VE-cad and either A1 or A5, respectively. For comparison, aliquots of whole-cell lysates (Lys) were analyzed in parallel. Molecular mass markers (kDa) are given at the left margin of each blot.
Based on these results, A2 and the VE-cad-based complex may be either directly connected or associated via actin filaments. To resolve this question, the actin cytoskeleton was depolymerized in confluent HUVECs with La B (11). Once the membrane and cytoplasm fractions were separated from cells either treated or not with La B, the subcellular localization of A2, actin, and VE-cad was analyzed by Western blotting (Fig. 4A). Quantification of the blotted bands indicated that the amount of membrane-associated A2 remained approximately constant before and after La B treatment (Fig. 4A). In contrast, a loss of 40% of the amount of membrane-associated actin was observed after La B treatment (Fig. 4A). This indicated that actin depolymerization did not induce a release of A2 from membranes. This is in agreement with confocal and phase-contrast microscopy analyses performed on untreated and La B-treated HUVECs (Fig. 4B). The drug-induced depolymerization of actin filaments led to cell retraction and cell-cell junction disruption (Fig. 4B). In spite of this drastic treatment, A2 remained colocalized with VE-cad at the cell edge, suggesting that VE-cad and A2 interact without requiring actin filaments. Furthermore, anti-VE-cad and anti-A2 IP experiments were performed on HUVECs treated or not with La B (Fig. 4C) and the blotted bands were quantified (data not shown). Similar amounts of A2 were detected in VE-cad immunoprecipitates isolated from either La B-treated or untreated cells. Conversely, in A2 immunoprecipitates, the amount of VE-cad coimmunoprecipitated remained unchanged between treated and untreated cells (Fig. 4C). These experiments revealed that, after actin cytoskeleton depolymerization, A2 did not decouple from the VE-cad-based complex. It could be concluded that, in confluent endothelial cells, the association of the A2/p11 complex with the junctional VE-cad-based complex does not require actin filaments.
FIG. 4.

No requirement for actin filaments in the connection between A2 and the VE-cad-based complex. (A) Confluent HUVECs were treated or not with La B for 5 min. Membrane (M) and cytoplasm (C) fractions were separated from La B-treated (+) or untreated (−) cells. Proteins isolated from M and C fractions were analyzed on 10% Tris-glycine gels and immunoblotted for VE-cad, actin, and A2. Molecular mass markers (kDa) are given at the left margin of the blot. The amounts of A2 and actin associated with membrane were quantified after scanning of immunoreactive bands (n = 3 ± standard error). The histograms represent the mean percentages of A2 (gray bars) and actin (black bars) in treated compared to untreated cells (100%; white bars). (B) Confocal microscopy analysis of VE-cad, A2, and actin in HUVECs before (Control) and after La B treatment. Cells plated on glass coverslips were stained with anti-Cad3 polyclonal antibody, anti-A2 monoclonal antibody, Cy2-labeled goat anti-rabbit and Cy5-labeled goat anti-mouse antibodies, TRITC-labeled phalloidin, and DAPI. Images correspond to an ≈650-nm confocal optical section. The efficiency of La B treatment was verified by phase-contrast microscopy (PC). In the merged panels, arrowheads point to areas where VE-cad and A2 perfectly colocalize. Bars, 20 μm. (C) Anti-VE-cad and anti-A2 IPs were performed on lysates extracted from confluent La B-treated (+) or untreated (−) HUVECs. Anti-VE-cad and anti-A2 IPs were separated on 4 to 12% gradient precast Criterion XT and 10% Tris-glycine gels, respectively, and immunoblotted for VE-cad and A2. Molecular mass markers (kDa) are given at the left margin of the blot.
Contribution of cholesterol raft-associated A2 to the stability of adherens junctions.
To evaluate the contribution of A2 to the localization of VE-cad at cell-cell junctions, A2 was knocked down in HUVECs with a pool of two selected siRNAs. Practically, 1 day after the second siRNA transfection, HUVECs were plated on glass coverslips and allowed to grow for additional 24- or 48-h periods prior to fixation. Four days after A2 siRNA transfection, immunoblot analysis indicated that 50 to 70% of A2 expression was abrogated while a scrambled siRNA had no effect (Fig. 5A). The effect observed with the A2 siRNA was specific, since the expression of proteins such as actin, VE-cad, and A1 and A5 was not down-regulated following A2 siRNA transfection (Fig. 5A).
FIG. 5.

Delocalization of VE-cad from adherens junctions induced by siRNA-mediated A2 down-regulation. (A) siRNAs were used to knock down the A2 level in HUVECs. Western-blot analysis was performed to determine VE-cad, actin, A1, A2, and A5 levels in HUVECs transfected with control siRNA (lane 1) or A2 siRNA (lane 2). Lysates were resolved on 10% Tris-glycine gels and probed for VE-cad, actin, A1, A2, and A5. Molecular mass markers (kDa) are given at the left margin of the blot. (B) One day after the second siRNA transfection, HUVECs were plated on glass coverslips and cultured for 24 h (left panel; 4 days after transfection) or 48 h (right panel; 5 days after transfection) prior to fixation. Cells were then labeled with anti-A2 monoclonal antibody, anti-Cad3 polyclonal antibody, Cy3-labeled goat anti-mouse and Cy2-labeled goat anti-rabbit antibodies, and DAPI. Empty arrowheads indicate adherens junctions where A2 and VE-cad colocalized, and full arrowheads indicate areas where adherens junctions are missing. Bars, 20 μm.
siRNA transfection induced no cell retraction, as established by phase-contrast microscopy (data not shown). The effect of A2 down-regulation on VE-cad localization was then analyzed by immunofluorescence microscopy 24 and 48 h after cell seeding on glass coverslips. At 24 or 48 h post-seeding time, the immunofluorescence signal for A2 was undetectable in some cells while unaffected in others. This heterogeneity reflected the difficulty of transfecting siRNA in HUVECs. As expected, transfection of the scrambled siRNA had no effect on A2 and VE-cad localization. At 24 h post-cell seeding time, A2 siRNA induced a drastic loss of VE-cad at adherens junctions between adjacent transfected cells (Fig. 5B). At 48 h post-cell seeding time, VE-cad began to reappear at cell-cell junctions exhibiting a zigzag staining pattern, a characteristic feature of immature junctions. Thus, it could be concluded that A2 down-regulation significantly disturbs the localization of VE-cad at cell-cell junctions.
Knockdown by siRNA affected the whole pool of cellular A2. Consequently, it was difficult to state whether either membrane-associated A2 or cytoplasmic A2 was involved in the observed phenotype. To discriminate between these two possibilities, the cholesterol raft-disrupting drug MβCD (10) was used to release A2 from the plasma membrane. Subcellular fractionation combined with immunoblotting analysis indicated that, following MβCD treatment, 60% of the membrane-bound A2 was released to the cytoplasm, whereas, as expected, in confluent untreated HUVECs, A2 was mainly associated with membranes (Fig. 6A and B). Eighty-two percent of actin remained associated with membranes after MβCD treatment, indicating that actin was weakly affected by the drug (Fig. 6A and B).
FIG. 6.

Disruption of adherens junctions induced by MβCD treatment. (A) Confluent HUVECs were treated with MβCD (+) for 30 min or untreated (−). Membrane (M) and cytoplasm (C) fractions were separated from MβCD-treated or untreated cells. Proteins isolated from M and C fractions were analyzed on 10% Tris-glycine gels and probed for VE-cad, actin, and A2. Molecular mass markers (kDa) are given at the left margin of the blot. (B) Quantification of the membrane-associated A2 and actin after scanning of immunoreactive bands in panel A (n = 3 ± standard error). To consider any variation in loading, the amounts of actin and A2 were normalized to the corresponding VE-cad immunoreactive bands of the same lanes. The histograms represent the percentages of A2 (gray bars) and actin (black bars) in treated compared to untreated cells (100%; white bars). (C) HUVECs plated on glass coverslips were treated with MβCD for 30 min or not treated (Control). VE-cad, A2, and nuclei were labeled as previously described in the legend to Fig. 5B. Asterisks indicate gaps in the cell monolayers. Bar, 20 μm.
As revealed by immunofluorescence microscopy, treatment of confluent HUVECs with MβCD provoked a disruption of adherens junctions leading to the formation of large gaps in the monolayer (Fig. 6C). Some cell-cell junctions resisted this treatment, possibly due to poor accessibility of MβCD to cholesterol rafts. This cell-cell contact destabilization might result from adherens junction disruption possibly caused by VE-cad internalization or degradation. To determine whether VE-cad internalization took place subsequently to MβCD treatment, HUVECs were trypsinized prior to lysis. This trypsinization was used to remove cell surface-expressed VE-cad molecules while leaving intact internalized ones. Western blot analysis revealed that A2 and β-catenin, the loading control, showed no appreciable variation in protein levels (Fig. 7A). In contrast, trypsinization degraded the totality of VE-cad in both MβCD-treated and untreated HUVECs, establishing that VE-cad remained at the cell surface following MβCD treatment (Fig. 7A, lanes 2 and 4). This experiment also revealed that following MβCD treatment, VE-cad did not undergo degradation (Fig. 7A, compare lanes 1 and 3). It could be concluded that destabilization of adherens junctions observed subsequently to MβCD treatment was due to neither the loss of membrane-associated VE-cad nor its degradation.
FIG. 7.

Decoupling of A2 from the VE-cad-complex induced by MβCD treatment. (A) Confluent HUVECs were treated with MβCD for 30 min (lanes 3 and 4) or not treated (lanes 1 and 2) and then submitted (lanes 2 and 4) or not (lanes 1 and 3) to trypsin digestion for 2 min prior to lysis. Cell lysates were separated on 4 to 12% gradient precast Criterion XT gels and immunoblotted for VE-cad, β-catenin (β-cat), and A2. Molecular mass markers (kDa) are given at the left margin of the blot. (B) Anti-VE-cad and anti-A2 IPs were performed on confluent HUVECs treated for 30 min with MβCD (+) or untreated (−). Immunoprecipitates were separated on 10% Tris-glycine gels and probed for VE-cad; α-, β-, and γ-catenins (α-, β-, γ-cat); actin; and A2. Molecular mass markers are given at the left margin of the blot. (C) HUVECs were plated on glass coverslips, treated with MβCD for 30 min (MβCD), or untreated (Control) and stained for anti-Cad3 polyclonal antibody, anti-A2 monoclonal antibody, Cy2-labeled goat anti-rabbit and Cy3-labeled goat anti-mouse antibodies, FITC-labeled phalloidin, and DAPI. The right panels are magnified images of the areas boxed in the left panels (Magn.). Bars, 20 μm.
To search for the intracellular event involved in such a phenotype, the VE-cad-based complex was isolated by IP from either MβCD-treated or untreated HUVECs and its composition was analyzed by Western blotting (Fig. 7B). Anti-VE-cad IP showed that following MβCD treatment, the VE-cad/catenin complex remained intact. On the contrary, a partial release of A2 from the VE-cad-based complex was observed (Fig. 7B). Anti-A2 IP confirmed the partial decoupling of A2 from the VE-cad/catenin complex in MβCD-treated cells. Consequently, it could be assumed that the MβCD-induced destabilization of adherens junctions resulted from the release of cholesterol raft associated-A2 and its disconnection from the VE-cad-based complex.
Actin was detected in both anti-VE-cad and anti-A2 IP isolated from confluent endothelial cells (Fig. 7B). However, after MβCD treatment, actin association was decreased in both VE-cad and A2 IPs; therefore, A2 exclusion from cholesterol rafts paralleled VE-cad-actin dissociation (Fig. 7 B). Moreover, a drastic reshaping of actin filaments was observed in MβCD-treated cells by immunofluorescence microscopy (Fig. 7C). Indeed, in control untreated cells, cortical actin fibers were parallel to the cell-cell junctions as defined by A2 and VE-cad labeling. On the contrary, after MβCD treatment, which is known to induce actin fiber reorganization (28), A2 disappeared from most cell-cell contact sites and is thus unable to link actin fibers to VE-cad-based complex (Fig. 7C, magnified boxed area). Consequently to MβCD treatment, actin fibers adopted a radial orientation and VE-cad exhibited a zigzag pattern staining at the cell-cell junction reminiscent of the immature junction pattern.
Altogether, these results suggest that by connecting the VE-cad-based complex, A2 is a major actor in the maintenance of endothelial adherens junction integrity.
Partners of cholesterol raft-associated A2 potentially involved in the connection with VE-cadherin.
To determine whether VE-cad and the other proteins constituting the interendothelial adherens junctions were associated with cholesterol rafts, cholesterol-associated proteins were extracted from confluent HUVEC membranes with digitonin, a detergent able to bind exclusively to membrane-linked cholesterol (15). Identification of proteins present within cholesterol rafts was achieved after in-gel digestion by trypsin and analysis of the resulting peptides by mass spectrometry (Table 1 [complete results available from the corresponding author on request]). As expected, clathrin and caveolin-1, two markers of cholesterol rafts, were detected, thus establishing that the fractionation was correct. Although this protein extraction method enabled the solubilization of transmembrane proteins such as PECAM-1 and integrin β-1, VE-cad was not detected in the cholesterol rafts (Table 1). β-Catenin, α-catenin, vinculin, and α-actinin were the only proteins belonging to adherens junctions extracted by digitonin treatment (Table 1). Actin, tubulin, and cytoskeleton-associated proteins such as actin-related protein 3 were also detected. In addition to A2, A1, A5, and A6 were also found, as well as AHNAK and moesin, two proteins known to belong to cholesterol rafts in other cell types (18, 21).
Western blot analysis mostly confirmed most of the proteomic study: i.e., the presence of A2, moesin, α-actinin, β-catenin, and PECAM-1 and the absence of VE-cad, γ-catenin, and p120 in cholesterol rafts (Fig. 8). Additionally, the phosphatase SHP2 and the A2 partner p11 were also detected with specific antibodies. The only divergence observed between proteomic and Western blotting analyses concerned α-catenin that was detected in cholesterol rafts by proteomic study (Table 1) and not by Western blotting (Fig. 8). Other relevant proteins not present in the cholesterol raft fraction (lane 4) were found either in the cytoplasmic (lane 1) and total membrane fractions (lane 2) or in the fraction containing all of the membrane-associated proteins not solubilized by digitonin (lane 3).
FIG. 8.
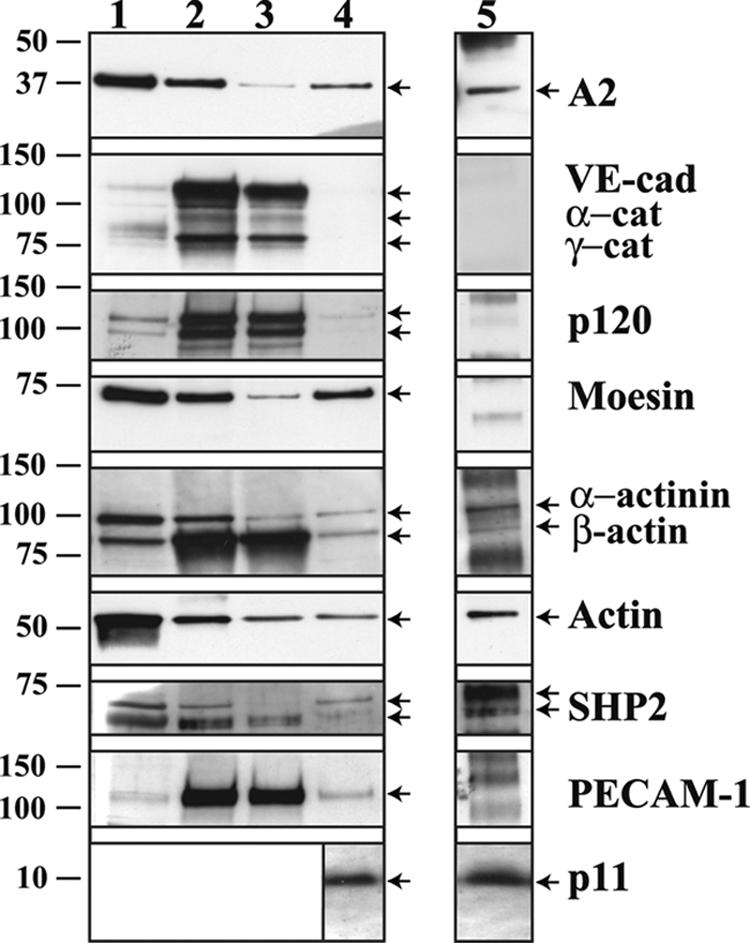
Identification of the proteins associated with cholesterol rafts. Subcellular fractionations performed on confluent HUVECs allowed the isolation of proteins contained in the cytoplasm (lane 1), in the total membranes (lane 2), in the digitonin-extracted cholesterol rafts (lane 4), and in the digitonin-insoluble membranes (lane 3). Proteins of each fraction were separated on 4 to 12% gradient precast Criterion XT gels and probed for A2; VE-cad; α-, γ-, and β-catenins (α-, β-, and γ-cat, respectively); p120; moesin; α-actinin; actin; SHP2; and PECAM-1. Samples of each fraction were also resolved on 15% Tris-Tricine gels and probed for p11. Anti-A2 IPs were performed on the digitonin-extracted cholesterol raft fraction (lane 5). After separation on 4 to 12% gradient precast Criterion XT gels, immunoprecipitates were probed for A2; VE-cad; α-, γ-, and β-catenins; p120; moesin; α-actinin; actin; SHP2; and PECAM-1. IPs were also resolved on 15% Tris-Tricine gels and probed for p11. Proteins unambiguously identified were labeled with arrows. Molecular mass markers (kDa) are given at the left margin of the blots.
Furthermore, the fractionation method illustrated in Fig. 8 allowed a quantitative comparison of the amounts of proteins present in each fraction. It could be determined by Western blot analysis that approximately 70% of membrane-tethered A2 and moesin were concentrated in the cholesterol rafts. As expected, VE-cad and α- and γ-catenins essentially appeared to be present in the membrane fractions (Fig. 8, lanes 2 and 3), whereas p120 was distributed between the membrane and the cytoplasm fractions in an 80:20 ratio.
IPs of cholesterol raft-associated A2 were performed to assess whether, within rafts, the cholesterol-associated proteins interacted with A2. Among the cholesterol raft-associated proteins, only α-actinin, β-catenin, SHP2, actin, and p11 appeared to be connected to cholesterol-associated A2 (Fig. 8, lane 5). Moreover, compared to the raft fraction, the A2 immunoprecipitates were enriched in α-actinin and SHP2 (Fig. 8, compare lanes 4 and 5). These last proteins constitute partners of A2 potentially able to establish the connection between the VE-cad-based complex and the cholesterol rafts.
Effects of VEGF on A2 localization at cell-cell contacts.
VEGF is a cytokine known for its capacity to increase vascular permeability. We examined the localization of VE-cad and A2 in HUVECs in the presence or absence of VEGF. After a 30-min treatment with VEGF, VE-cad staining was greatly disturbed compared to the control (Fig. 9A). At some cell-cell junction sites (Fig. 9; VEGF), VE-cad labeling exhibited a spike pattern reminiscent of what was observed in MβCD-treated cells (Fig. 7C). Furthermore, at these particular sites, no A2 was detected at cell-cell contacts. These data suggest that treatment of HUVECs by physiological agonists of vascular permeability such as VEGF may induce the decoupling of the A2/p11 complex from the VE-cad-based junction. To verify this hypothesis, anti-VE-cad IPs were performed on confluent HUVECs treated or not with VEGF (Fig. 9B). It appeared that, following VEGF treatment, A2 was totally released from the VE-cad-based complex. Concomitantly, the amount of actin coimmunoprecipitated with VE-cad was drastically reduced. Altogether, these data indicated that VEGF treatment results in a VE-cad-based complex/A2 decoupling leading to a partial disconnection of the VE-cad-based complex from the actin cytoskeleton.
FIG. 9.

VEGF-induced delocalization of A2 and VE-cad from adherens junctions. (A) HUVECs plated on glass coverslips were treated with VEGF or not treated (control). VE-cad, A2, and nuclei were labeled as previously described in Fig. 4B. Empty arrowheads point to adherens junctions where A2 and VE-cad colocalized, and full arrowheads point to missing or perturbed junctions. Bar, 20 μm. (B) Anti-VE-cad IPs were performed on confluent HUVECs treated for 30 min with VEGF (+) or untreated (−). Immunoprecipitates were separated on 10% Tris-glycine gels and probed for VE-cad, actin, and A2. Molecular mass markers (kDa) are given at the left margin of the blot. For comparison, aliquots of whole-cell lysates (Lys) were analyzed in parallel.
DISCUSSION
Until recently, the VE-cad-based adherens junction was thought to be basically composed of the transmembrane adhesive receptor VE-cad, β-catenin, and α-catenin bound directly to actin filaments (12, 36). This model was elaborated only by considering indirect evidence. Recently, it was established that α-catenin cannot bind simultaneously to the cadherin/catenin complex and to actin filaments, demolishing the idea that α-catenin constitutes the direct link between the actin cytoskeleton and adherens junctions (48). Nevertheless, the actin cytoskeleton appears to play a crucial role in the regulation of adherens junction stability (33, 44). In fact, the interactions between the VE-cad-based complex and the underlying actin cytoskeleton are very dynamic and probably modulated by several intermediate proteins (45). Obviously, it is of great interest to determine the nature of the proteins involved in this controversial connection. Using proteomic tools, we recently discovered that the VE-cad-based complex is associated with several actin-binding proteins. Among them, A2 constituted one of the most abundant proteins. In the present article, we have established the role played by A2 in the stabilization of interendothelial adherens junctions.
First, we observed that A2 translocates from the cytoplasm to the plasma membrane and more particularly to cell-cell contact sites as HUVECs become confluent (Fig. 1A and B). This correlates with the fact that A2 docks with these specific sites when VE-cad-mediated adherens junctions reach maturity. Although the mechanism underlying the A2 targeting to membrane as cell confluence is established remains to be elucidated, it probably requires the formation of the heterotetramer (A2)2(p11)2 (14). Indeed, immunofluorescent labeling of p11 and A2 perfectly overlapped with that of VE-cad at cell-cell junctions (data not shown).
Second, using IP experiments, we demonstrated that both A2 and p11 subunits interact with the VE-cad-based complex composed at least of VE-cad; α-, β-, and γ-catenins; and p120. This interaction is independent of actin filament binding, since it is not abolished by drug-induced actin depolymerization treatment (Fig. 4). In contrast, no interaction between A2 and N-cadherin, the other cadherin present in endothelium, was detected. Similarly, A2 does not interact with PECAM-1, a receptor expressed at endothelial adherens cell-cell junctions. Moreover, among annexins expressed in the endothelium, only A2 possesses the ability to interact with the VE-cad-based complex.
The fact that the A2/p11 complex interacts with the VE-cad complex when cells reach confluence suggests that it may be involved in the maturation of interendothelial adherens junctions. This hypothesis is supported by the fact that abrogation of the expression of A2 in HUVECs by specific siRNA (Fig. 5A) drastically disturbs the localization of VE-cad at cell-cell junctions (Fig. 5B).
Furthermore, our experiments performed on confluent endothelial cells pretreated with MβCD, a cholesterol raft-disrupting drug (10, 28), established that approximately 60% of the membrane-tethered pool of A2 is released into the cytoplasm (Fig. 6B). This results in a destabilization of adherens junctions with formation of intercellular gaps at some cell-cell contact sites (Fig. 6C). This destabilization is due to neither internalization nor degradation of VE-cad, as established by trypsin digestion experiments (Fig. 7A), but results from a delocalization of VE-cad from cell-cell junctions (Fig. 6C). siRNA down-regulation of A2 and MβCD-mediated depletion of membrane-associated A2 led to very similar cellular effects: i.e., partial disappearance of VE-cad from cell-cell junctions and a corollary destabilization of adherens junctions. The mechanism by which the disconnection of A2 from the VE-cad complex is induced remains unknown. It cannot be excluded that this event may result from the release of signaling proteins usually located in the cholesterol rafts (36). This signaling pathway needs further investigation.
As previously observed, the actin cytoskeleton is absolutely needed to maintain the adherens junction architecture (44). Nevertheless, the connection between the VE-cad-based complex and actin fibers remains elusive. Our experiments showed that the MβCD-induced disruption of A2 from the VE-cad complex was accompanied by a partial loss of actin in the IP (Fig. 7C). Considering, furthermore, that A2 is known to strongly interact with actin fibers (23), we assume that the connection of the VE-cad-based complex might be mediated in part by A2.
In confluent HUVECs, approximately 70% of membrane-associated A2 is localized in cholesterol rafts (Fig. 8). Therefore, connection with the A2/p11 complex indirectly anchors the VE-cad complex to cholesterol rafts. Due to its stoichiometry, the AII/p11 complex may dock with several VE-cad-based complexes. Using electron microscopy and biochemical studies, we recently established that each VE-cad complex is formed of hexameric structures in which six molecules of VE-cad associate in an antiparallel manner (5, 25, 31). The multiplicity of interactions between the self-associated VE-cad molecules and the A2/p11 complex may lead to the elaboration of a supercomplex embedded in cholesterol rafts. Furthermore, at membrane cholesterol raft sites, the A2/p11 complex mediates in part interactions with the actin cytoskeleton (17, 34, 37). Consequently, once connected to the cholesterol raft-anchored pool of A2, the VE-cad-based complexes may be indirectly bound to actin cytoskeleton. We propose a model of VE-cad-actin interaction through A2. In subconfluent endothelial cells, the VE-cad-based complex freely diffuses laterally in the plasma membrane. Simultaneously with the arrival of A2 at the membrane, the VE-cad-based complex is progressively docked with the A2-containing cholesterol rafts and indirectly linked to the actin cytoskeleton. This entrapment provides a rigid structure that considerably restricts lateral diffusion of the VE-cad-based complex, thus strengthening and stabilizing adherens junctions in confluent endothelial cells (Fig. 10). This correlates with observations made in myoblasts in which N-cadherin lateral diffusion is increased when raft structures are disrupted (9).
FIG. 10.

Potential mechanism of stabilization of VE-cad-mediated adherens junctions. In subconfluent HUVECs, adherens junctions are immature and poorly associated with the actin cytoskeleton. The VE-cad-based complex is free to move laterally in the plasma membrane. Once HUVECs reach confluence and adherens junctions become mature, the A2/p11 complex, which accumulates underneath the plasma membrane at cholesterol raft sites (CR), associates with the VE-cad-based complex. Consequently, the complex is docked to cholesterol rafts. Thus, by providing a link between the VE-cad/catenin complex and the actin cytoskeleton, the cholesterol raft-embedded A2 stabilizes the adherens junctions, thereby allowing the development of stable intercellular connections.
To determine how the VE-cad-based complex and A2 are interconnected, the cholesterol raft-associated proteins were isolated by digitonin extraction of cholesterol-bound proteins combined with ultracentrifugation. Identification of these proteins was obtained after their in-gel digestion by trypsin and analysis of the resulting peptides by mass spectrometry (Table 1 and Fig. 8). Strikingly, VE-cad was not detected within the cholesterol raft-associated protein fraction, whereas A2 was found. This suggests that the connection between the VE-cad-based complex and A2 is mediated through intermediate proteins possibly identified as those associated with A2 within cholesterol rafts. Our proteomic analysis reveals that, in cholesterol rafts, A2 interacts with actin, β-catenin, α-actinin, p11, and SHP2 (Fig. 8, lane 5). This suggests that the last three proteins constitute candidates able to potentially connect A2 to the VE-cad/β-catenin complex.
Among the identified proteins, the phosphatase SHP2 is an interesting candidate. Indeed, on the one hand, it was demonstrated to coimmunoprecipitate with the VE-cad-based complex by selectively interacting with β-catenin in confluent quiescent HUVECs (40). This interaction maintains β-catenin under its dephosphorylated form (6, 27). On the other hand, IP experiments performed on confluent cow pulmonary aortic endothelial cells revealed that SHP2 coimmunoprecipitates with A2 (7). Moreover, tyrosine phosphorylation of SHP2 promoted its dissociation from the VE-cad-based complex when HUVECs were activated by agonists able to increase vascular permeability, such as thrombin (40). This correlates with a drastic increase in tyrosine phosphorylation of β-catenin. Consequently, the ability to associate with the VE-cad-based complex in a tyrosine phosphorylation-dependent manner may confer the capacity to regulate the interaction between A2 and the VE-cad-based complex with the cholesterol raft-tethered pool of SHP2.
During angiogenesis, endothelial cell-cell junctions become leaky and endothelial cells acquire the capacity to migrate, two events leading to the constitution of new vessels (41, 43). The increase in vascular permeability might be correlated with the disconnection of A2 from the VE-cad-based complex. To test this hypothesis, we submitted confluent HUVEC monolayers to VEGF, a cytokine able to induce in vivo and in vitro angiogenesis (13, 29). Following this treatment, the junctional localization of A2 was dramatically disturbed (Fig. 9A). In fact, we showed that A2 and actin are decoupled from the VE-cad-based complex subsequently to VEGF treatment (Fig. 9B). This suggests that the VEGF-induced increase in vascular permeability might be a consequence of the disconnection of the actin cytoskeleton from the VE-cad-based complex.
It could be assumed that, consecutively to MβCD or VEGF treatments, VE-cad and the catenins become phosphorylated (16), either by protein kinases such as Src (1, 42) or by the loss of SHP2 from the VE-cad-based complex, thus initiating the junction disabling. Indeed, results in the literature indicated that membrane cholesterol depletion from confluent endothelial cells with MβCD induces the phosphorylation of p120 and γ-catenin (10). In both cases, phosphorylation of the adherens junction components is induced by loss of SHP2 or destabilization of cholesterol rafts, two phenomena that implicate the plasma membrane-associated pool of A2. Whether the VE-cad/A2 disconnection is linked to the phosphorylation status of VE-cad-based complex remains to be explored.
In the present article, we have provided evidence for the existence of a novel mechanism able to promote the maturation of the interendothelial adherens junctions. By connecting the VE-cad-based complex to the cholesterol raft-tethered pool of A2, stabilization of adherens interendothelial junctions occurs, allowing the switch from an immature to a mature state.
Supplementary Material
Acknowledgments
We are indebted to staff of the maternity hospital from Hôpital Nord (Grenoble, France) for kindly collecting umbilical cords for these experiments. We thank Y. Wallez and T. Mannic for help with VEGF use, E. Hewat for critical reading of the manuscript, and D. Grunwald for taking all confocal microscopy images.
S.H. is a recipient of a fellowship from Association de Recherche sur la Polyarthrite. This work was supported by grants from CNRS (ACI PCV and DRAB), Association pour la Recherche sur le Cancer (ARC no. 4447 and 3775), and Alliance des Recherche sur le Cancer (ARECA-ARC).
Footnotes
Published ahead of print on 26 December 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Angelini, D. J., S. W. Hyun, D. N. Grigoryev, P. Garg, P. Gong, I. S. Singh, A. Passaniti, J. D. Hasday, and S. E. Goldblum. 2006. TNFα increases tyrosine phosphorylation of vascular endothelial-cadherin and opens the paracellular pathway through Fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell Mol. Physiol. 291L1232-L1245. [DOI] [PubMed] [Google Scholar]
- 2.Babiychuk, E. B., and A. Draeger. 2000. Annexins in cell membrane dynamics. Ca(2+)-regulated association of lipid microdomains. J. Cell Biol. 1501113-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84869-901. [DOI] [PubMed] [Google Scholar]
- 4.Bazzoni, G., E. Dejana, and M. G. Lampugnani. 1999. Endothelial adhesion molecules in the development of the vascular tree: the garden of forking paths. Curr. Opin. Cell Biol. 11573-581. [DOI] [PubMed] [Google Scholar]
- 5.Bibert, S., M. Jaquinod, E. Concord, C. Ebel, E. Hewat, C. Vanbelle, P. Legrand, M. Weidenhaupt, T. Vernet, and D. Gulino-Debrac. 2002. Synergy between extracellular modules of vascular endothelial cadherin promotes homotypic hexameric interactions. J. Biol. Chem. 27712790-12801. [DOI] [PubMed] [Google Scholar]
- 6.Biswas, P., J. Zhang, J. D. Schoenfeld, D. Schoenfeld, D. Gratzinger, S. Canosa, and J. A. Madri. 2005. Identification of the regions of PECAM-1 involved in beta- and gamma-catenin associations. Biochem. Biophys. Res. Commun. 3291225-1233. [DOI] [PubMed] [Google Scholar]
- 7.Burkart, A., B. Samii, S. Corvera, and H. S. Shpetner. 2003. Regulation of the SHP-2 tyrosine phosphatase by a novel cholesterol- and cell confluence-dependent mechanism. J. Biol. Chem. 27818360-18367. [DOI] [PubMed] [Google Scholar]
- 8.Butz, S., J. Stappert, H. Weissig, and R. Kemler. 1992. Plakoglobin and beta-catenin: distinct but closely related. Science 2571142-1144. (Letter.) [DOI] [PubMed] [Google Scholar]
- 9.Causeret, M., N. Taulet, F. Comunale, C. Favard, and C. Gauthier-Rouviere. 2005. N-cadherin association with lipid rafts regulates its dynamic assembly at cell-cell junctions in C2C12 myoblasts. Mol. Biol. Cell 162168-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corvera, S., C. DiBonaventura, and H. S. Shpetner. 2000. Cell confluence-dependent remodeling of endothelial membranes mediated by cholesterol. J. Biol. Chem. 27531414-31421. [DOI] [PubMed] [Google Scholar]
- 11.Coue, M., S. L. Brenner, I. Spector, and E. D. Korn. 1987. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213316-318. [DOI] [PubMed] [Google Scholar]
- 12.Dejana, E. 2004. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5261-270. [DOI] [PubMed] [Google Scholar]
- 13.Dejana, E., R. Spagnuolo, and G. Bazzoni. 2001. Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration. Thromb. Haemost. 86308-315. [PubMed] [Google Scholar]
- 14.De Seranno, S., C. Benaud, N. Assard, S. Khediri, V. Gerke, J. Baudier, and C. Delphin. 2006. Identification of an AHNAK binding motif specific for the Annexin2/S100A10 tetramer. J. Biol. Chem. 28135030-35038. [DOI] [PubMed] [Google Scholar]
- 15.Elias, P. M., J. Goerke, D. S. Friend, and B. E. Brown. 1978. Freeze-fracture identification of sterol-digitonin complexes in cell and liposome membranes. J. Cell Biol. 78577-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esser, S., M. G. Lampugnani, M. Corada, E. Dejana, and W. Risau. 1998. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1111853-1865. [DOI] [PubMed] [Google Scholar]
- 17.Filipenko, N. R., and D. M. Waisman. 2001. The C terminus of annexin II mediates binding to F-actin. J. Biol. Chem. 2765310-5315. [DOI] [PubMed] [Google Scholar]
- 18.Foster, L. J., C. L. De Hoog, and M. Mann. 2003. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. USA 1005813-5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerke, V., C. E. Creutz, and S. E. Moss. 2005. Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6449-461. [DOI] [PubMed] [Google Scholar]
- 20.Gulino, D., E. Delachanal, E. Concord, Y. Genoux, B. Morand, M. O. Valiron, E. Sulpice, R. Scaife, M. Alemany, and T. Vernet. 1998. Alteration of endothelial cell monolayer integrity triggers resynthesis of vascular endothelium cadherin. J. Biol. Chem. 27329786-29793. [DOI] [PubMed] [Google Scholar]
- 21.Harder, T., R. Kellner, R. G. Parton, and J. Gruenberg. 1997. Specific release of membrane-bound annexin II and cortical cytoskeletal elements by sequestration of membrane cholesterol. Mol. Biol. Cell 8533-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayes, M. J., U. Rescher, V. Gerke, and S. E. Moss. 2004. Annexin-actin interactions. Traffic 5571-576. [DOI] [PubMed] [Google Scholar]
- 23.Hayes, M. J., D. Shao, M. Bailly, and S. E. Moss. 2006. Regulation of actin dynamics by annexin 2. EMBO J. 251816-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermant, B., S. Bibert, E. Concord, B. Dublet, M. Weidenhaupt, T. Vernet, and D. Gulino-Debrac. 2003. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 27814002-14012. [DOI] [PubMed] [Google Scholar]
- 25.Hewat, E. A., C. Durmort, L. Jacquamet, E. Concord, and D. Gulino-Debrac. 2007. Architecture of the VE-cadherin hexamer. J. Mol. Biol. 365744-751. [DOI] [PubMed] [Google Scholar]
- 26.Hubaishy, I., P. G. Jones, J. Bjorge, C. Bellagamba, S. Fitzpatrick, D. J. Fujita, and D. M. Waisman. 1995. Modulation of annexin II tetramer by tyrosine phosphorylation. Biochemistry 3414527-14534. [DOI] [PubMed] [Google Scholar]
- 27.Ilan, N., and J. A. Madri. 2003. PECAM-1: old friend, new partners. Curr. Opin. Cell Biol. 15515-524. [DOI] [PubMed] [Google Scholar]
- 28.Kwik, J., S. Boyle, D. Fooksman, L. Margolis, M. P. Sheetz, and M. Edidin. 2003. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. USA 10013964-13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambeng, N., Y. Wallez, C. Rampon, F. Cand, G. Christe, D. Gulino-Debrac, I. Vilgrain, and P. Huber. 2005. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 96384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lampugnani, M. G., M. Resnati, M. Raiteri, R. Pigott, A. Pisacane, G. Houen, L. P. Ruco, and E. Dejana. 1992. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J. Cell Biol. 1181511-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Legrand, P., S. Bibert, M. Jaquinod, C. Ebel, E. Hewat, F. Vincent, C. Vanbelle, E. Concord, T. Vernet, and D. Gulino. 2001. Self-assembly of the vascular endothelial cadherin ectodomain in a Ca2+-dependent hexameric structure. J. Biol. Chem. 2763581-3588. [DOI] [PubMed] [Google Scholar]
- 32.Lewit-Bentley, A., S. Rety, J. Sopkova-de Oliveira Santos, and V. Gerke. 2000. S100-annexin complexes: some insights from structural studies. Cell Biol. Int. 24799-802. [DOI] [PubMed] [Google Scholar]
- 33.Mege, R. M., J. Gavard, and M. Lambert. 2006. Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18541-548. [DOI] [PubMed] [Google Scholar]
- 34.Oliferenko, S., K. Paiha, T. Harder, V. Gerke, C. Schwarzler, H. Schwarz, H. Beug, U. Gunthert, and L. A. Huber. 1999. Analysis of CD44-containing lipid rafts: recruitment of annexin II and stabilization by the actin cytoskeleton. J. Cell Biol. 146843-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozawa, M., H. Baribault, and R. Kemler. 1989. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 81711-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pokutta, S., and W. I. Weis. 2000. Structure of the dimerization and beta-catenin-binding region of alpha-catenin. Mol. Cell 5533-543. [DOI] [PubMed] [Google Scholar]
- 37.Rescher, U., D. Ruhe, C. Ludwig, N. Zobiack, and V. Gerke. 2004. Annexin 2 is a phosphatidylinositol (4,5)-bisphosphate binding protein recruited to actin assembly sites at cellular membranes. J. Cell Sci. 1173473-3480. [DOI] [PubMed] [Google Scholar]
- 38.Rimm, D. L., E. R. Koslov, P. Kebriaei, C. D. Cianci, and J. S. Morrow. 1995. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc. Natl. Acad. Sci. USA 928813-8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schafer, B. W., and C. W. Heizmann. 1996. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem. Sci. 21134-140. [DOI] [PubMed] [Google Scholar]
- 40.Ukropec, J. A., M. K. Hollinger, S. M. Salva, and M. J. Woolkalis. 2000. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J. Biol. Chem. 2755983-5986. [DOI] [PubMed] [Google Scholar]
- 41.Vila, E., and M. Salaices. 2005. Cytokines and vascular reactivity in resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 288H1016-H1021. [DOI] [PubMed] [Google Scholar]
- 42.Wallez, Y., F. Cand, F. Cruzalegui, C. Wernstedt, S. Souchelnytskyi, I. Vilgrain, and P. Huber. 2006. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 261067-1077. [DOI] [PubMed] [Google Scholar]
- 43.Wallez, Y., I. Vilgrain, and P. Huber. 2006. Angiogenesis: the VE-cadherin switch. Trends Cardiovasc. Med. 1655-59. [DOI] [PubMed] [Google Scholar]
- 44.Waschke, J., F. E. Curry, R. H. Adamson, and D. Drenckhahn. 2005. Regulation of actin dynamics is critical for endothelial barrier functions. Am. J. Physiol. Heart Circ. Physiol. 288H1296-H1305. [DOI] [PubMed] [Google Scholar]
- 45.Weis, W. I., and W. J. Nelson. 2006. Re-solving the cadherin-catenin-actin conundrum. J. Biol. Chem. 28135593-35597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao, K., D. F. Allison, M. D. Kottke, S. Summers, G. P. Sorescu, V. Faundez, and A. P. Kowalczyk. 2003. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J. Biol. Chem. 27819199-19208. [DOI] [PubMed] [Google Scholar]
- 47.Xiao, K., J. Garner, K. M. Buckley, P. A. Vincent, C. M. Chiasson, E. Dejana, V. Faundez, and A. P. Kowalczyk. 2005. p120-catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol. Biol. Cell 165141-5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamada, S., S. Pokutta, F. Drees, W. I. Weis, and W. J. Nelson. 2005. Deconstructing the cadherin-catenin-actin complex. Cell 123889-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang, J., M. Betson, J. Erasmus, K. Zeikos, M. Bailly, L. P. Cramer, and V. M. Braga. 2005. Actin at cell-cell junctions is composed of two dynamic and functional populations. J. Cell Sci. 1185549-5562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.