

Abstract
Tumor necrosis factor (TNF) is a prototypic proinflammatory cytokine that contributes significantly to the development of immunopathology in various disease states. A complication of TNF blockade therapy, which is used increasingly for the treatment of chronic inflammatory diseases, is the reactivation of latent tuberculosis. This study used a low-dose aerogenic model of murine tuberculosis to analyze the effect of TNF neutralization on disease progression in mice with chronic tuberculous infections. Histological, immunohistochemical, and flow cytometric analyses of Mycobacterium tuberculosis-infected lung tissues revealed that the neutralization of TNF results in marked disorganization of the tuberculous granuloma, as demonstrated by the dissolution of the previously described B-cell-macrophage unit in granulomatous tissues as well as by increased inflammatory cell infiltration. Quantitative gene expression studies using laser capture microdissected granulomatous lung tissues revealed that TNF blockade in mice chronically infected with M. tuberculosis leads to the enhanced expression of specific proinflammatory molecules. Collectively, these studies have provided evidence suggesting that in the chronic phase of M. tuberculosis infection, TNF is essential for maintaining the structure of the tuberculous granuloma and may regulate the granulomatous response by exerting an anti-inflammatory effect through modulation of the expression of proinflammatory mediators.
A total of one-third of the world's population is infected with the tubercle bacillus (42). More important, tuberculosis remains a principal infectious cause of mortality worldwide, resulting in approximately 1.8 million deaths annually (42). The recent emergence of extensively drug-resistant strains of Mycobacterium tuberculosis is yet another reminder that the tubercle bacillus is a serious threat to public health (22). The vast majority of those infected with M. tuberculosis do not manifest disease, though it is generally thought that a significant portion of this population harbors a latent infection. As a result, this population is a major obstacle to disease eradication, as reactivation of latent tuberculosis contributes significantly to the pathogenesis of the tubercle bacillus (12). Understanding the mechanisms underlying the establishment of latent infection and subsequent reactivation is critical to the control of M. tuberculosis.
With the inhalation of the tubercle bacillus, a cascade of cellular migratory events is triggered that culminates in the formation of the tuberculous granuloma, the hallmark of tuberculosis infection (15, 17, 38, 40). The tuberculous granuloma is a highly structured and yet dynamic entity comprised of a wide array of immune cells. It is generally accepted that the granuloma represents a component of the protective immune response against M. tuberculosis. While the precise mechanisms underlying the formation and maintenance of the granuloma remain to be determined, experimental evidence exists that specific cytokines and chemokines play an important role in regulating the granulomatous response triggered by M. tuberculosis infection (1, 12).
Tumor necrosis factor alpha (TNF-α), a cytokine whose protean biological functions include the regulation of immune cell trafficking (35) and the mycobacterial granulomatous response (2, 3, 13, 21, 30), plays a significant role in the control of acute and chronic tuberculosis as well as Mycobacterium bovis BCG infection in the mouse (13, 21, 30, 34). Recently, the relevance of TNF in the control of persistent human tuberculous infection has been demonstrated by epidemiological evidence that individuals treated with TNF blockade therapy for a variety of inflammatory diseases exhibit increased risks for the development of reactivation tuberculosis (20). Indeed, this phenomenon has recently been recapitulated in computational biology virtual trials (27). However, the precise mechanisms by which TNF contains M. tuberculosis in the latent phase of infection remain to be characterized.
It has previously been shown that TNF neutralization in the chronic phase of tuberculous infection results in histopathological features indicative of enhanced inflammation in the lungs of infected mice; these features are associated with increased cellularity, squamous metaplasia, and disorganization of the granuloma, suggesting that this cytokine might possess anti-inflammatory effects (30). In the study by Mohan et al., the histopathological analysis of tuberculous lung tissues was carried out at 2 months after initiation of TNF blockade, at which time the pulmonic bacterial burden in mice treated with the TNF-neutralizing MP6-XT22 antibody was about 1.0 log higher than that in animals receiving control rat immunoglobulin G (IgG). Therefore, it remains possible that the granulomatous phenotype observed in the TNF-neutralized mice could be due to increased bacterial burdens secondary to MP6-XT22 treatment. In addition, that study used a murine experimental tuberculosis model involving a low-dose intravenous challenge with virulent M. tuberculosis Erdman. We have recently used the low-dose aerogenic murine tuberculosis model, a system that is generally thought to better mimic the respiratory mode of dissemination of the tubercle bacillus in humans, to show that TNF blockade in chronic tuberculous infection alters the production of specific chemokines by CD11b+ cells (3). Neutralization of TNF by the administration of MP6-XT22 at 4 months after low-dose aerogenic infection caused an exacerbation of the disease (3). This disease recrudescence is associated with significantly increased bacterial burdens in TNF-neutralized mice compared to the burdens in rat IgG-treated controls beginning at about 18 days after the initiation of MP6-XT22 treatment (3). Using this low-dose model of persistent murine tuberculosis, the present study focuses on examining the early effect of MP6-XT22 on the tuberculous granuloma during the initial phase of treatment (within the first 9 days postneutralization), when pulmonic bacterial burdens between the TNF-neutralized group and the control group are comparable. This approach avoids the introduction of organ bacterial load as a confounding factor in data interpretation. The results have shown that upon TNF depletion, structural integrity of the tuberculous granuloma is disrupted, as evident by the dissolution of the previously described B-cell-macrophage units that exist in M. tuberculosis-infected lung tissues (38). In addition, TNF blockade in mice chronically infected with the tuberculous bacillus results in increased expression of proinflammatory cytokines and chemokines in the lungs. The latter observation suggests that TNF may exert an anti-inflammatory effect on the tuberculous granulomatous response in the lungs of mice infected with M. tuberculosis during the chronic phase of infection.
(Data presented here are part of a thesis [for S. D. Chakravarty] submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Sue Golding Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.)
MATERIALS AND METHODS
Animals.
Female C57BL/6 mice (8 to 10 weeks old; Charles River Laboratories, Rockland, MA) were used for all experiments. All infected mice were maintained in our biosafety level 3 animal laboratories and routinely monitored for murine pathogens through serological and histological examinations. All animal protocols utilized in this study were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine.
Mycobacteria and infection of mice.
To maintain virulence, bacterial stocks of M. tuberculosis strain Erdman (Trudeau Institute, Saranac Lake, NY) were generated by being passaged through mice as described previously (14). Mice were infected via the aerosol route and placed in a closed-air aerosolization system (In-Tox Products, Albuquerque, NM) to deliver the desired CFU (3, 38). Inocula of about 50 to 200 CFU/lung were employed for the mouse studies presented here. Confirmation of the accuracy of inoculum delivery was determined by assessing lung bacterial burden at 24 h postinfection (30).
Antibodies and treatment of mice.
The mouse TNF-neutralizing antibody MP6-XT22 (DNAX and National Cell Culture Center) and normal rat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) were used as described previously (30). Beginning 4 to 6 months postinfection, the neutralization of TNF was begun through intraperitoneal injection with 0.5 mg MP6-XT22 twice weekly for the duration of the experiment. At appropriate intervals after the commencement of in vivo TNF neutralization, tissue bacillary loads were quantified by plating serial dilutions of lung, spleen, and liver homogenates onto Middlebrook 7H10 agar (Difco Laboratories, Detroit, MI) as described previously (30).
Histopathology and immunohistochemical staining.
Tissue samples for histopathological studies were fixed in 10% normal buffered formalin, followed by paraffin embedment. For histopathological examination, 6-μm sections were stained with hematoxylin and eosin (H&E). Immunohistochemical detection of different leukocyte subsets was carried out as described previously (38). Briefly, 6-μm cryosections of lung tissue were fixed in cold acetone for 5 min, followed by 80% ethanol for an additional 4 min. Upon subsequent quenching of endogenous peroxidase activity and blocking for endogenous biotin (Avidin/Biotin blocking kit; Vector Laboratories, Burlingame, CA), sections were blocked in 5% goat serum in phosphate-buffered saline (PBS) to prevent nonspecific binding before incubation with primary antibodies. The antibodies used for cell surface markers of interest were rat anti-mouse CD4 (CD4+ T cells), CD8 (CD8+ T cells), CD19 (B cells), and Ly6G (neutrophils) from BD Pharmingen; rat anti-mouse F4/80 (macrophages) from Serotec; and rabbit anti-human CD3 (T cells) (cross-reactive with murine CD3) from Dako. Sections were washed with PBS prior to reaction with appropriate biotinylated secondary antibodies (Vector Labs). The avidin-biotin complex-based signal amplification was achieved using the Vectastain Elite ABC kit, and diaminobenzidine (both from Vector) was used for signal development. Images were viewed on a Zeiss Axioskop 2 (Thornwood, NY) light microscope using a 10× 0.30 numerical aperture Plan Neofluar objective.
Flow cytometry.
CD45+ cells from lungs of M. tuberculosis-infected mice treated with rat IgG and MP6-XT22 antibodies were obtained by preparing single-cell suspensions with subsequent immunomagnetic separation using CD45+ microbeads and MACS MS separation columns (Miltenyi Biotec, Auburn, CA) as described previously (7, 38). Dead cells were identified and excluded from analysis by means of staining with the Live/Dead reduced biohazard cell viability kit number 2 (green staining) or number 4 (blue staining) (Molecular Probes, Eugene, OR) (38). Three- or four-color flow cytometric analyses of live cells were completed using the following fluorochrome-conjugated antibodies: anti-CD3-PerCP, anti-CD45-allophycocyanin, anti-CD19- phycoerythrin (BD Pharmingen), and anti-F4/80-phycoerythrin (Serotec Laboratories). An appropriate isotype control monoclonal antibody for each fluorochrome (BD Pharmingen) was included in all analyses. CD45+ cells (1 × 106) were stained using the above antibodies in fluorescence-activated cell sorter buffer (PBS containing 5% mouse serum, 10% fetal bovine serum, and 0.01% NaN3) for 40 min at 4°C. Following additional washes in fluorescence-activated cell sorter buffer and PBS, the cells were fixed in 4% paraformaldehyde for 30 min at 4°C. One milliliter of PBS was added to the stained cells, which were then collected using a FACSCalibur (BD Biosciences, San Jose, CA) cytometer. Analysis was performed using CellQuest software (BD Biosciences).
LCM-derived RNA procurement and isolation.
Procurement of laser capture microdissection (LCM)-derived RNA and its isolation were carried out as described previously (45). Briefly, upon H&E staining of lung sections and identification of granulomatous tissue using microscopy, a 15-mm or 30-mm laser beam was used for LCM by using the Pix-Cell II apparatus (Arcturus Engineering, Mountain View, CA). Capturing caps were used in the LCM procedure to procure granulomatous tissues. The nature of the granulomatous tissues dissected for the study and their location within the infected lungs have been described previously (45). These tissues represent areas in infected mouse lungs that have been infiltrated with a variety of immune cells (45). For our experiments, sections prepared from the entire left lung of each mouse studied were used for LCM procurement of granulomatous tissues. RNA was prepared from pooled granulomatous tissues thus prepared. For RNA extraction of LCM-procured granulomas, thermosensitive membranes with procured granulomas were peeled off from the capturing caps and transferred into an Eppendorf tube containing 390 μl of a preincubation buffer (350 μl 1× PBS, pH 7.4, 30 ml 10% sodium dodecyl sulfate, and 10 μl proteinase K [20 mg/ml] for up to 20 membranes per tube). Before the transfer of the thermosensitive film, this buffer was preincubated at 37°C for 30 min to minimize any contaminating RNase activity. When introducing the procured granulomas into the preincubation buffer, an additional 10 μl of fresh proteinase K (20 mg/ml) was added. Four to 16 h later, RNA was extracted with 2 ml of TRIzol (Invitrogen) reagent. Two micrograms of yeast (Saccharomyces cerevisiae) tRNA was then added, and extraction was carried out according to the manufacturer's protocol. RNA pellets were resuspended into 100 μl of diethyl pyrocarbonate-treated water. The RNA was treated with RNase-free DNase I (Roche, Indianapolis, IN) for 2 h at 37°C. The samples were subjected to a second round of TRIzol extraction. The RNA was resuspended in 10 μl of diethyl pyrocarbonate-treated water; 1 μl of which was put aside for the negative control reaction (the no-reverse transcription [RT] reaction) to exclude the possibility that signals obtained are due to DNA contamination.
Real-time RT-PCR.
Reverse transcription of LCM-derived RNA was completed using the First Strand cDNA synthesis kit for RT-PCR (avian myeloblastosis virus) (Roche) according to the manufacturer's protocol and using random primers. For real-time PCR, molecular beacons (synthesized by Integrated DNA Technologies, Coralville, IA) designed for genes of interest were utilized. The relative gene expression method was employed, and subsequent calculations were carried out as described previously (45). The levels of gene expression measured were normalized to the housekeeping gene GAPDH (glyceraldehyde-3-phosphate dehydrogenase). The GAPDH real-time PCR was always run in parallel with that of the other genes of interest. The PCR protocol used was as follows: a hold of 10 min at 95°C and 40 cycles of 20 s at 95°C, 20 s at 55°C, and 30 s at 72°C. The real-time PCR was completed on an ABI Prism 7700 (Applied Biosystems, Foster City, CA), with data collection and analysis performed using its sequence detection system software, version 1.9.
Statistical analysis.
Where appropriate, data points were subjected to the Mann-Whitney t test (nonparametric conditions) to determine statistical significance using GraphPad Prism 4 software. A P value of <0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Disorganization of the structure of murine granulomas subsequent to TNF neutralization in the persistent phase of tuberculous infection: histological studies.
To begin assessing the early effects of TNF blockade on the structure of pulmonic granulomas during the chronic phase of M. tuberculosis infection, histological examinations were carried out to examine the lungs of chronically infected mice treated with MP6-XT22 at 9 days after the initiation of treatment. At this early time post-TNF blockade, the bacterial burdens of infected lungs of MP6-XT22-treated mice and those of the rat IgG-treated controls were comparable (data not shown). Prior to antibody treatment, the lung granulomas of mice chronically infected with M. tuberculosis Erdman consisted of discrete lymphoid aggregates situated among a diffuse infiltration of lymphocytes and histiocytic cells (Fig. 1), as described previously (30). By day 9 after initiation of MP6-XT22 treatment, the dissolution of the lymphoid nodules became apparent (Fig. 1). In contrast, the lymphoid aggregates were well maintained in the rat IgG-treated control group (Fig. 1). The TNF neutralization-induced disaggregation of the lymphoid nodules continued to be apparent at 21 days posttreatment with MP6-XT22 (Fig. 1). These results indicate that one effect of TNF blockade on the murine pulmonary granulomatous response during the chronic phase of infection is the dissolution of the lymphoid aggregates. These data confirm the observation derived from the intravenous model of tuberculosis that TNF modulates the architecture of the lymphoid aggregates in lungs of mice chronically infected with the tubercle bacillus (30). In addition, the present results extend the previous observation to the aerogenic infection model and provide evidence that the effect of TNF blockade on the organization of the tuberculous granulomas occurs rapidly upon initiation of MP6-XT22 treatment when the lung bacterial burdens of the TNF-neutralized and rat IgG-treated mice are comparable.
FIG. 1.

Lung histopathology of MP6-XT22 and control rat IgG-treated C57BL/6 mice chronically infected with M. tuberculosis. Lungs were harvested from mice at 6 months postinfection prior to the administration of the TNF-neutralizing MP6-XT22 and then at 9 days (9d) and 21 days (21d) posttreatment (Tx); lung samples were formalin fixed, paraffin embedded, sectioned, and H&E stained. Total magnification, ×100. Samples are representative of sections from three or four mice per treatment group per time point (three sections per mouse). The experiment was repeated twice with similar results.
Dissolution of the B-cell-macrophage units in lung granulomas of mice with chronic tuberculosis upon TNF neutralization: immunohistochemical and flow cytometric studies.
It has recently been shown that the lymphoid aggregates present in the lungs of mice with chronic tuberculosis consist mainly of B cells (15, 38). Emerging evidence suggests that these B-cell nodules exhibit characteristics of germinal centers, suggesting the occurrence of tertiary lymphoneogenesis in the lungs of mice chronically infected with the tubercle bacillus (26, 40). In mice, these B-cell aggregates are encircled by F4/80+ macrophages (38). Therefore, these B-cell-macrophage units represent a well-defined and microscopically apparent feature that reflects the structural organization of the tuberculous granuloma. The histological studies described above suggest the possibility that upon TNF blockade, the structure of the B-cell-macrophage units may be disrupted. It has previously been shown that virtually all the lymphoid aggregates in the lungs of mice with chronic tuberculosis represent B-cell nodules (i.e., the majority of the cells that make up the lymphoid nodules revealed histologically by H&E staining of M. tuberculosis-infected mouse lungs are CD19 immunoreactive) (26, 38). As these B-cell aggregates are conspicuously circumscribed by macrophages (38), the B-cell-macrophage units can be readily detected by immunohistochemical staining targeting F4/80+ macrophages. Results obtained using this immunohistochemical approach revealed that the administration of MP6-XT22 to mice chronically infected with M. tuberculosis led to dissociation of the B-cell-macrophage units in the infected lungs (Fig. 2). Dissociation of the spatial relationship between B cells and macrophages occurred as early as 9 days after the initiation of MP6-XT22 treatment and continued until at least 21 days after the start of TNF blockade (Fig. 2). By contrast, treatment of mice with chronic tuberculosis using control rat IgG had no effect on the structure of the B-cell-macrophage units. That TNF neutralization in mice with chronic tuberculosis leads to the dissolution of B-cell aggregates is further supported by the results of immunohistochemical studies evaluating CD19 immunoreactivity in the lung granulomatous tissues of infected mice (Fig. 3). The results of these studies have shown that by 9 days after the initiation of treatment with MP6-XT22, there is conspicuous dissolution of the CD19 immunoreactive nodules, with a marked decrease in the number of CD19+ cells in the sections studied (Fig. 3). Dissolution of the B-cell aggregates continues to be apparent 21 days after the start of TNF neutralization (Fig. 3). In contrast, the administration of control rat IgG to mice with chronic tuberculosis has no effect on the structure of the B-cell nodules (Fig. 3). The dissolution of the B-cell nodules and the disruption of the relationship between B cells and macrophages are not due to a decrease in the number of these leukocyte subsets, as flow cytometric analyses of lung cells revealed that these immune cells are present in comparable quantities in the MP6-XT22- and the rat IgG-treated groups (Fig. 4). While TNF neutralization has no effect on the number of B lymphocytes and macrophages in the lungs of the treated mice, it is still possible that MP6-XT22 might affect processes (such as apoptosis and cellular recruitment) that regulate the turnover kinetics of these cells. The flow cytometry results exclude the possibility that the disorganization of the B-cell-macrophage structure is due to the attenuation of F4/80 or CD19 immunoreactivity as a result of TNF neutralization. This is because TNF neutralization has no significant effect on the mean fluorescence index of F4/80- and CD19-expressing cells and in vitro treatment of M. tuberculosis-infected, bone-marrow-derived macrophages from C57BL/6 mice with MP6-XT22 or rat IgG does not lead to altered F4/80 immunoreactivity of these cells (data not shown). Together, the data suggest that the disorganization of the spatial arrangement of B lymphocytes and macrophages is due to aberrant trafficking of these immune cells.
FIG. 2.

Immunohistochemical analysis of the effects of TNF neutralization on the B-cell aggregates-macrophage units in the lungs of C57BL/6 mice persistently infected with M. tuberculosis. Lungs were obtained from mice at 6 months postinfection prior to the administration of MP6-XT22, and subsequently at 9 days (9d) and 21 days (21d) posttreatment (Tx), optimal cutting temperature embedded, cryosectioned, and stained for macrophages (F4/80+) to identify the B-cell aggregate-macrophage subunit. Control mice received rat IgG treatment. Virtually all lymphoid aggregates in the lungs of mice with chronic tuberculosis are B-cell nodules (38). “B” denotes aggregates of B lymphocytes; macrophages are designated by “M.” Total magnification, ×100. Samples are representative of sections from three or four mice per treatment group per time point (three sections per mouse). The study was repeated once with similar results.
FIG. 3.

Disaggregation of CD19+ B-cell aggregates (brown staining) in the lungs of mice with chronic M. tuberculosis upon TNF neutralization by the MP6-XT22-neutralizing antibody at days 9 and 21 after initiation of treatment. Robust CD19+ B-cell nodules (arrowheads) are seen in the lungs of mice treated with control rat IgG. The lungs of mice treated with MP6-XT22 for 9 and 21 days demonstrate the disaggregation of CD19+ B-cell aggregates but still display remnants of B cells (arrowheads). Photomicrographs are representative and are at ×200 total magnification of 6-μm frozen sections immunoreacted to anti-CD19 antibodies.
FIG. 4.
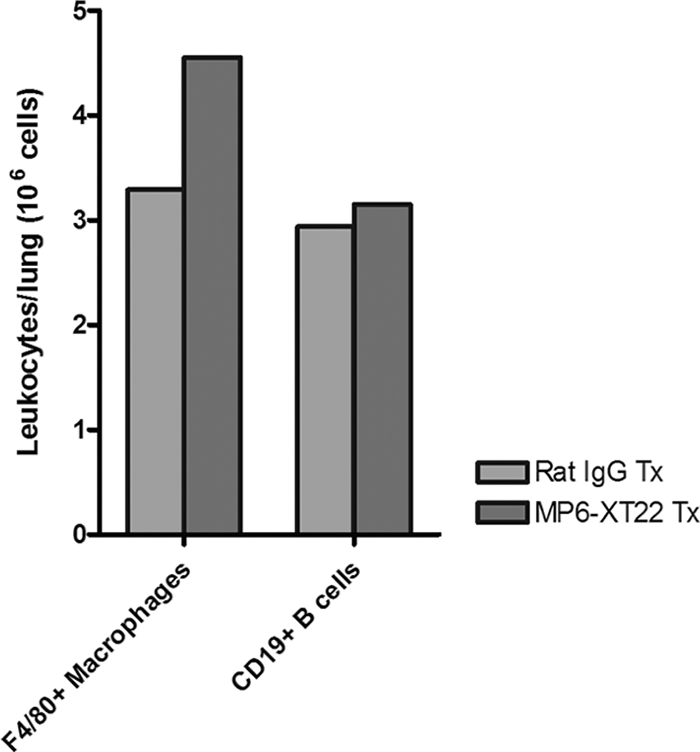
Quantification of F4/80+ macrophages and CD19+ B cells among CD45+ leukocytes obtained from lungs of MP6-XT22- and control rat IgG-treated mice with chronic tuberculosis through flow cytometry. Dead cells were identified and excluded from analysis by means of staining with the Live/Dead reduced biohazard cell viability kit number 2 (green staining) or number 4 (blue staining). Positively enriched live CD45+ cells were obtained and subjected to staining using antibodies for cell surface markers of interest. Absolute numbers of CD19+ B cells and F4/80+ macrophages at 9 days (9d) posttreatment are shown. Proportions of CD45+ cells analyzed were comparable between the two groups. Bars represent data from lung cells pooled from four or five mice. The experiments were repeated once with similar results.
In contrast to the conspicuous effect of MP6-XT22 on the spatial arrangement of the B-cell aggregates and macrophages, implementation of TNF blockade does not alter the distribution of CD4+ T cells, CD8+ T cells, or Ly6G+ neutrophils in the lungs of mice with chronic tuberculosis, as assessed by immunohistochemical analysis (Fig. 5). The latter observation is not unexpected, as previous studies have shown that T cells and neutrophils do not exhibit any specific discernible pattern of distribution in tuberculous lungs of mice during the chronic phase of infection (38). Collectively, these results suggest that during chronic tuberculosis infection, TNF is essential for the structural organization of the tuberculous granuloma. Specifically, TNF regulates aggregation of the lymphoid nodules of B cells and their association with macrophages. The fact that the dissolution of the B-cell-macrophage units occurs as early as 9 days after the initiation of TNF blockade (the equivalent of three doses of MP6-XT22) strongly suggests that the spatial arrangement of these two leukocyte subsets requires active maintenance by TNF, either directly or indirectly. Although the immunohistochemical approach did not reveal any microscopically apparent alteration in the distribution of neutrophils and T cells in the lungs of mice with chronic tuberculosis upon TNF neutralization, it remains possible that the method used may not have the level of sensitivity required to detect subtle, yet important, changes in the localization of these immune cells that might translate into functional alteration in the tuberculous granulomatous response.
FIG. 5.

Immunohistochemical staining of cryosections for T-lymphocyte subsets and neutrophils in M. tuberculosis-infected C57BL/6 mice undergoing TNF blockade therapy. Lungs were obtained from mice at 6 months postinfection prior to the administration of MP6-XT22 and subsequently at 9 days posttreatment (Tx) (the control group received rat IgG). Lungs were sectioned and stained for CD4+ and CD8+ T cells as well as neutrophils (Ly6G+). Total magnification, ×100. Samples are representative of sections from three or four mice per treatment group per time point (three sections per mouse). The experiment was repeated once with similar results.
The effect of neutralization of TNF on gene expression in granulomatous tissues procured from the lungs of mice chronically infected with M. tuberculosis: comparison of MP6-XT22-treated and rat IgG-treated groups.
As discussed above, results derived from an intravenous model of murine tuberculosis have provided evidence suggesting that TNF neutralization leads to increased tissue inflammation (30). The present study used the aerogenic tuberculosis model to further characterize the putative anti-inflammatory effect of TNF during the chronic phase of infection. To begin addressing the inflammation-enhancing effect of TNF blockade, real-time PCR analysis was employed to study gene expression in the tuberculous granuloma. The tissues studied were procured through LCM to ensure that the effect is granuloma specific. Precautions were exercised to avoid sampling error; specifically, RNAs used for the gene expression studies were extracted from pooled granulomatous tissues microdissected from the entire left lung of each individual mouse examined. As in the immunohistochemical and flow cytometric studies described above, lung tissues were analyzed in the early phase post-MP6-XT22 treatment, when the tissue bacterial burdens of the experimental and control groups are comparable. As a result, any alterations in granuloma gene expression observed in the MP6-XT22-treated mice compared to that in the controls receiving rat IgG can be ascribed specifically to the effect of TNF neutralization.
In the first gene expression study, mice with chronic tuberculosis at 6 months postinfection with ∼150 CFU of M. tuberculosis Erdman were used. The experimental group received MP6-XT22, while the control mice were treated with rat IgG. There were seven and three mice in the experimental and control groups, respectively. Gene expression was carried out using LCM-procured pulmonic granulomatous tissues 6 days after initiation of TNF blockade, at which time the mice had received two doses of either MP6-XT22 or rat IgG and the bacterial burdens in the lungs of the experimental and control groups, as expected, were comparable (Fig. 6A). Based on the observation that TNF blockade in mice with chronic tuberculosis leads to increased pulmonic inflammation (30), inflammatory chemokines, chemokine receptors that direct the migration of Th1 cells (the leukocyte subset that plays a critical role in engendering the protective host response to M. tuberculosis), monocytic phagocytes (12) as well as proinflammatory cytokines were targeted for evaluation. The expression of chemokines and chemokine receptors that direct B-cell migration, BLC/CXCL13 and CXCR5, respectively (31, 33, 46), was evaluated because of the observations that TNF blockade causes disaggregation of the B lymphoid nodules (Fig. 1 and 3) and that B lymphocytes from murine tuberculous lungs express CXCR5 and migrate toward CXCL13 (26). In addition, the expression of NOS2 and Toll-like receptor 2 (TLR2) (the former is expressed in immunologically activated macrophages [8, 25, 36] and the latter is a molecule that plays an important role in signaling the interaction of M. tuberculosis components with host cells [23, 28, 37, 41]) was evaluated.
FIG. 6.

Host gene expression analysis of LCM-procured granulomatous lung tissues from control rat IgG-treated and MP6-XT22-treated mice using real-time PCR. (A) Bacillary load in the lungs of the control rat IgG-treated and MP6-XT22-treated C57BL/6 mice infected with M. tuberculosis. Mice were aerogenically infected with a relatively low inoculum of the virulent Erdman strain of M. tuberculosis (∼150 CFU). Following 6 months postinfection, the mice were sacrificed 6 days after initiation of rat IgG or MP6-XT22 treatment. Lungs were homogenized and plated on 7H10 plates, and colonies were enumerated after 21 days. Data depicted represent the mean CFU obtained from each treatment group (the group treated with control IgG contained three mice, and the group treated with MP6-XT22 contained seven mice), and error bars represent standard deviations. (B) RNA prepared from granulomatous tissues in the lungs of mice at 6 days posttreatment (with control rat IgG or MP6-XT22) was used for cDNA synthesis and subjected to real-time PCR for genes of interest. Data presented represent the mean of results obtained from each treatment group (as described for panel A), and error bars represent standard errors of the means. Genes of interest shown with no P value displayed indicate that the differences observed between the two treatment groups were not statistically significant.
A total of 21 immunologic factors were analyzed (Fig. 6B). Studies targeting the proinflammatory cytokines revealed that the expression of interferon-γ (IFN-γ) and interleukin-12p40 (IL-12p40), a component of the heterodimeric IL-12, was up-regulated in the lung granulomas of TNF-neutralized mice compared to levels in the rat IgG-treated controls (P values were 0.0333 for IFN-γ and 0.0476 for IL-12p40) (Fig. 6B). The enhanced expression of NOS2 in the lung granulomas of TNF-neutralized mice (P = 0.0119) (Fig. 6B) paralleled the MP6-XT22-induced up-regulation of proinflammatory IFN-γ and IL-12p40. Interestingly, the expression of IL-10 was increased in the TNF-neutralized mice compared to that in IgG-treated controls (P = 0.0083) (Fig. 6B). The mechanism underlying the latter observation is unclear, but may be a reflection of the host's attempt to counter the TNF blockade-induced increase in lung inflammation via the immunosuppressive effect of IL-10 (5, 10). Additionally, C57BL/6 transgenic mice producing increased amounts of IL-10 under control of the IL-2 promoter were shown, in a previous study, to have disorganized granuloma structures and reactivated persistent chronic tuberculosis when aerogenically infected (39). That study suggests that increased pulmonic expression of IL-10 in MP6-XT22-treated mice could adversely affect disease outcome. Finally, TLR2, the pattern recognition receptor that plays a major role in signaling the interaction of M. tuberculosis components with host antigen-presenting cells, was up-regulated in the lung granulomas of TNF-neutralized mice compared to the levels in control animals (P = 0.0083) (Fig. 6B). Therefore, it is possible that the increased expression of this Toll receptor may contribute to augmenting inflammation during TNF blockade in the chronic phase of tuberculous infection.
When we examined inflammatory chemokine expression, macrophage inflammatory protein 2 (MIP-2)-1α/CCL3, a ligand for CCR1 and CCR5, and MIP-1β/CCL4, a ligand for CCR5 (31, 46), were significantly elevated in TNF-depleted mice compared to levels in controls (P values were 0.0333 for both MIP-1α/CCL3 and MIP-1β/CCL4) (Fig. 6B). The expression levels of MIP-3α/CCL20, a ligand for CCR6, and monocyte chemoattractant protein 3 (MCP-3)/CCL7, a ligand for CCR1, CCR2, and CCR3 were also substantially elevated in response to neutralization of TNF by 6 days posttreatment (P values were 0.0083 for MIP-3α/CCL20 and 0.0179 for MCP-3/CCL7) (Fig. 6B). Thus, upon neutralization of TNF in mice with chronic tuberculosis, there is an increase in the granulomatous expression of specific chemokines that can direct the migration of a wide variety of leukocytes, including subsets of T cells, B cells, monocytes, and dendritic cells (24, 31, 33, 46). Of note, our previous study using the low-dose aerogenic mouse model has shown that TNF neutralization regulates the expression of CCL3, CCL4, CCL5, and CXCL9 by CD11b+ cells at 6 days after MP6-XT22 treatment (3). In agreement with the results of the present study based on quantitative PCR using RNA derived from granulomatous tissues, our previous experiments showed the up-regulation of CCL3 and CCL4 in CD11b+ cells at 6 days post-TNF blockade (3). However, while expression of CCL5 and CXCL9 was shown to be down-regulated in the TNF-neutralized mice in the CD11b+ cell experiments, results of the present study using total granulomatous tissues suggest that MP6-XT22 has no effect on the expression of these two chemokines. This discrepancy is likely due to the fact that whole granulomatous tissues are made up of a wide variety of immune cells, including CD11b+ phagocytes, the focus of our previous study. The results of the present study, obtained from studying whole granulomatous tissues, can be expected to yield information representative of the overall chemokine milieu of the tuberculous granuloma.
Gene expression studies targeting chemokine receptors revealed that relative to rat IgG treatment, the administration of MP6-XT22 to mice with chronic tuberculosis resulted in significantly increased expression levels of CCR6 at 6 days after initiation of TNF blockade (P = 0.0119) (Fig. 6B). CCR6 has been reported to be expressed by most B cells as well as by a subpopulation of T cells and dendritic cells (43). Interestingly, the expression of the sole known ligand for CCR6, MIP-3α/CCL20, was also increased in lung granulomas of mice with chronic tuberculous infection upon TNF neutralization (Fig. 6B). Results of the study targeting BLC/CXCL13, the homeostatic chemokine that has been shown to regulate lymphoneogenesis (4, 11), revealed that the expression levels are lower in TNF-neutralized mice compared to those in rat IgG-treated controls, although the difference did not reach statistical significance (Fig. 6B).
Collectively, data from our quantitative gene expression studies have revealed that upon TNF neutralization, pulmonic granulomatous tissues of mice with chronic tuberculosis exhibit enhanced inflammation. This result is indicated by (i) the increased expression of two primary proinflammatory cytokines, IFN-γ and IL-12p40, (ii) enhanced mRNA levels of specific chemokines and the chemokine receptor CCR6 that can direct migration of a wide variety of immune cells, and (iii) augmented expression of TLR2, which can potentially increase the inflammatory response of the host to mycobacterial components. These data provide evidence suggesting that TNF may exert an anti-inflammatory effect on tuberculous granulomatous tissues during chronic tuberculosis.
The effect of neutralization of TNF on gene expression in granulomatous tissues procured from the lungs of mice chronically infected with M. tuberculosis: comparison of MP6-XT22-treated animals and untreated controls.
To use an experimental system that more closely represents the clinical scenario, studies were carried out to examine the effect of TNF blockade on gene expression in lung granulomas of mice with chronic tuberculosis in which the control group received no treatment, i.e., rat IgG was not administered. Since IFN-γ is the primary inflammatory molecule up-regulated in the first study described above, this cytokine was the target of evaluation in the repeat experiment. In these studies, mice were evaluated at days 3 and 6 after the administration of MP6-XT22; untreated animals (day 0) were used as controls (Fig. 7), thus allowing the kinetics of the TNF-induced up-regulation of IFN-γ to be evaluated. As with the previous set of experiments, we used LCM-procured RNA from the pulmonic granulomas of the experimental and control mice for our real-time PCR analyses. Indeed, similar to the model that uses rat IgG as controls, results of the second experiment revealed up-regulation of IFN-γ in lung granulomas of MP6-XT22 mice with chronic tuberculosis (Fig. 7B). At day 3 after initiation of TNF blockade, when the experimental group had received one dose of MP6-XT22, pulmonic IFN-γ expression levels were comparable among the treated and control mice. By day 6 post-TNF blockade, after two doses of neutralizing antibodies had been administered, IFN-γ was significantly up-regulated in the lungs of MP6-XT22-treated animals (the P value was 0.047 for IFN-γ) (Fig. 7B). A total of three controls, five TNF-neutralized mice at day 3, and four TNF-neutralized mice at day 6, were studied, and the lung bacterial burdens at the time of evaluation among the three groups of animals were comparable (Fig. 7A).
FIG. 7.

Gene expression profiling of LCM-procured granulomatous lung tissue from MP6-XT22-treated or untreated mice through real-time PCR. (A) Lung bacterial burdens in MP6-XT22-treated C57BL/6 mice infected with M. tuberculosis. Mice were aerogenically infected with a relatively low inoculum of the virulent Erdman strain of M. tuberculosis (∼120 CFU). At 6 months postinfection, the mice were sacrificed at day 0 (untreated) and at days 3 and 6 after the administration of MP6-XT22. Lungs were homogenized and plated on 7H10 plates, and colonies were enumerated after 21 days. Data presented represent the mean CFU obtained from each time point (three for day 0, five for day 3, and four for day 6), and error bars represent standard deviations. The lung bacterial loads of mice in all experimental groups were comparable. (B) RNA was prepared from LCM-procured granulomatous tissues in the lungs of mice prior to the administration of MP6-XT22 and at 3 and 6 days posttreatment. The RNA was used for cDNA synthesis and subjected to real-time PCR for the IFN-γ gene. Data presented represent the means of results obtained from each time point group (as described for panel A), and error bars represent standard errors of the means. The difference observed in IFN-γ gene expression levels between time points for the day 0 group and the day 6 group was statistically significant (P = 0.047).
Thus, results of the second gene expression study, using a model designed to more closely mimic TNF blockade in the clinical setting, again provided evidence that neutralization of this cytokine in mice with chronic tuberculosis leads to enhanced expression of IFN-γ. These data highlight the possibility that TNF exerts an anti-inflammatory effect during persistent tuberculous infection. Although TNF is generally thought to be proinflammatory, it has been demonstrated to exert immunosuppressive/anti-inflammatory effects in a number of in vivo animal models. For example, TNF has been shown to have an anti-inflammatory effect in a murine Corynebacterium parvum model in vivo (16). Importantly, in a murine model of acute mycobacterial infection involving BCG and TNF-deficient mice, this cytokine has been shown to negatively regulate the Th1 immune response, which could be detrimental to the host (44). In addition, it has been reported that TNF mediates a tissue-damaging effect in the acute phase of inflammation in the mouse experimental allergic encephalomyelitis model while exerting an immunosuppressive effect as the disease progresses, which eventually leads to protection against tissue damage and remission (18, 19). A protective anti-inflammatory effect of TNF has also been described for the murine nonobese diabetes model (9). With the use of an influenza hemagglutinin transgenic T-cell receptor mouse model, TNF has been shown to suppress a broad range of T-cell responses, including IFN-γ production (9). This in vivo observation of the IFN-γ-attenuating attribute of TNF was recapitulated in an in vitro model of chronic TNF exposure (9). Collectively, these results provide strong evidence that TNF can contextually exert both proinflammatory and anti-inflammatory effects, depending on the pathophysiological states, and that the latter effect may be mediated through the suppression of IFN-γ production.
Conclusions.
For humans, it has been reported that TNF blockade-induced reactivation tuberculosis is associated with increased inflammation and disorganization of the granulomatous architecture (20). The role of IFN-γ in enhancing inflammation in this situation is unclear. In addition, it is not known whether the TNF neutralization-induced enhanced inflammation and the disorganized granulomas observed in the study of humans are the results of severe end-stage tuberculous recrudescence, where the bacterial burden is expected to be high. Results of the present study using a well-established murine model of chronic tuberculosis suggest that increased inflammation and disorganization of the granuloma structure occur in the very early stage of TNF blockade therapy, when the tissue bacterial burdens are comparable among the experimental and control mouse groups. Since excessive chemokine production at the site of inflammation can result in mistrafficking of immune cells (6, 32), it is possible that the enhanced inflammatory response observed in TNF-neutralized mice (a component of which is increased expression of specific chemokines) could lead to disorganization of the granulomatous structure. In the case of the B-lymphoid nodules, it is noteworthy that chemoattraction of CCR6+ B lymphocytes plays a critical role in the formation of intestine-associated B-lymphoid structures (43). While it remains to be evaluated whether the CCR6-CCL20 interaction contributes to the formation of tertiary lymphoid organs, such as the B-lymphocyte aggregates present in the pulmonic granulomatous tissues of mice with chronic tuberculosis (38, 40), it is conceivable that enhanced expression of CCR6 and CCL20 in the lungs of TNF-neutralized mice with chronic tuberculosis (Fig. 6B) may lead to B-cell mistrafficking, therefore resulting in dissolution of the B-cell nodules. Accumulating evidence suggests that the B-cell aggregates in the lungs of mice chronically infected with M. tuberculosis display features of germinal centers (26, 40) and, therefore, are likely products of tertiary lymphogenesis (4, 11). In addition, B cells have been shown to regulate the tuberculous granulomatous inflammatory response (26) and may be required for optimal control of M. tuberculosis. It is thus possible that dissolution of the B-cell nodules may contribute to the dysregulated inflammatory response in the lungs of TNF-neutralized mice with chronic tuberculosis. While the significance of T cells in host immunity to M. tuberculosis has been well established, the role of B lymphocytes in regulating the tuberculous granulomatous response deserves further evaluation.
TNF is required for the effective formation of tuberculous granulomas in the mouse (13, 30). An examination of human tuberculous granulomas in individuals receiving TNF blockade therapy for inflammatory diseases suggests the same (20). TNF has been shown to play an important role in lymphoid organogenesis (29). Whether a direct effect of TNF is essential for the maintenance of the spatial organization of the B-cell-macrophage is presently unclear. It will be of interest to evaluate the mechanisms by which TNF neutralization in mice with chronic tuberculosis can lead to enhanced IFN-γ production and the relative significance of the latter cytokine in mediating the observed inflammation-enhancing effect of TNF blockade. Of additional interest will be an examination of the effect of TNF neutralization-induced altered expression levels of BCL/CXCL13, CCR6, and MIP-3α/CCL20 on the granulomatous reaction in general and of the organization of the B lymphocyte aggregates in the lungs of mice with chronic tuberculosis in particular. Results yielded from these investigations will likely shed light on how the tuberculous granulomatous response is regulated in chronic tuberculosis.
Acknowledgments
This work was supported by National Institutes of Health grants HL71241 (J.C. and J.F.), HL68526 (D.K., J.F., and J.C.), and T32 GM007288 (S.D.C.) and by the Albert Einstein College of Medicine (CFAR P30 AI51519).
We thank Jian Wang and Jiayong Xu for excellent technical assistance and all members of the Chan, Flynn, and Kirschner labs for helpful discussions.
Editor: F. C. Fang
Footnotes
Published ahead of print on 22 January 2008.
REFERENCES
- 1.Algood, H. M., J. Chan, and J. L. Flynn. 2003. Chemokines and tuberculosis. Cytokine Growth Factor Rev. 14467-477. [DOI] [PubMed] [Google Scholar]
- 2.Algood, H. M., P. L. Lin, and J. L. Flynn. 2005. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin. Infect. Dis. 41(Suppl. 3)S189-S193. [DOI] [PubMed] [Google Scholar]
- 3.Algood, H. M., P. L. Lin, D. Yankura, A. Jones, J. Chan, and J. L. Flynn. 2004. TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J. Immunol. 1726846-6857. [DOI] [PubMed] [Google Scholar]
- 4.Aloisi, F., and R. Pujol-Borrell. 2006. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 6205-217. [DOI] [PubMed] [Google Scholar]
- 5.Bogdan, C., Y. Vodovotz, and C. Nathan. 1991. Macrophage deactivation by interleukin 10. J. Exp. Med. 1741549-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bromley, S. K., D. A. Peterson, M. D. Gunn, and M. L. Dustin. 2000. Cutting edge: hierarchy of chemokine receptor and TCR signals regulating T cell migration and proliferation. J. Immunol. 16515-19. [DOI] [PubMed] [Google Scholar]
- 7.Chakravarty, S. D., J. Xu, B. Lu, C. Gerard, J. Flynn, and J. Chan. 2007. The chemokine receptor CXCR3 attenuates the control of chronic Mycobacterium tuberculosis infection in BALB/c mice. J. Immunol. 1781723-1735. [DOI] [PubMed] [Google Scholar]
- 8.Chan, J., Y. Xing, R. S. Magliozzo, and B. R. Bloom. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 1751111-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cope, A. P., R. S. Liblau, X. D. Yang, M. Congia, C. Laudanna, R. D. Schreiber, L. Probert, G. Kollias, and H. O. McDevitt. 1997. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J. Exp. Med. 1851573-1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries, J. E. 1995. Immunosuppressive and anti-inflammatory properties of interleukin 10. Ann. Med. 27537-541. [DOI] [PubMed] [Google Scholar]
- 11.Drayton, D. L., S. Liao, R. H. Mounzer, and N. H. Ruddle. 2006. Lymphoid organ development: from ontogeny to neogenesis. Nat. Immunol. 7344-353. [DOI] [PubMed] [Google Scholar]
- 12.Flynn, J. L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immunol. 1993-129. [DOI] [PubMed] [Google Scholar]
- 13.Flynn, J. L., M. M. Goldstein, J. Chan, K. J. Triebold, K. Pfeffer, C. J. Lowenstein, R. Schreiber, T. W. Mak, and B. R. Bloom. 1995. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2561-572. [DOI] [PubMed] [Google Scholar]
- 14.Flynn, J. L., C. A. Scanga, K. E. Tanaka, and J. Chan. 1998. Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 1601796-1803. [PubMed] [Google Scholar]
- 15.Gonzalez-Juarrero, M., O. C. Turner, J. Turner, P. Marietta, J. V. Brooks, and I. M. Orme. 2001. Temporal and spatial arrangement of lymphocytes within lung granulomas induced by aerosol infection with Mycobacterium tuberculosis. Infect. Immun. 691722-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodge-Dufour, J., M. W. Marino, M. R. Horton, A. Jungbluth, M. D. Burdick, R. M. Strieter, P. W. Noble, C. A. Hunter, and E. Pure. 1998. Inhibition of interferon gamma induced interleukin 12 production: a potential mechanism for the anti-inflammatory activities of tumor necrosis factor. Proc. Natl. Acad. Sci. USA 9513806-13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan, G., F. A. Post, A. L. Moreira, H. Wainwright, B. N. Kreiswirth, M. Tanverdi, B. Mathema, S. V. Ramaswamy, G. Walther, L. M. Steyn, C. E. Barry III, and L. G. Bekker. 2003. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 717099-7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kassiotis, G., and G. Kollias. 2001. TNF and receptors in organ-specific autoimmune disease: multi-layered functioning mirrored in animal models. J. Clin. Investig. 1071507-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassiotis, G., and G. Kollias. 2001. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J. Exp. Med. 193427-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keane, J., S. Gershon, R. P. Wise, E. Mirabile-Levens, J. Kasznica, W. D. Schwieterman, J. N. Siegel, and M. M. Braun. 2001. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 3451098-1104. [DOI] [PubMed] [Google Scholar]
- 21.Kindler, V., A. P. Sappino, G. E. Grau, P. F. Piguet, and P. Vassalli. 1989. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 56731-740. [DOI] [PubMed] [Google Scholar]
- 22.Lancet. 2006. XDR-TB—a global threat. Lancet 368964. [DOI] [PubMed] [Google Scholar]
- 23.Liu, P. T., S. Stenger, H. Li, L. Wenzel, B. H. Tan, S. R. Krutzik, M. T. Ochoa, J. Schauber, K. Wu, C. Meinken, D. L. Kamen, M. Wagner, R. Bals, A. Steinmeyer, U. Zugel, R. L. Gallo, D. Eisenberg, M. Hewison, B. W. Hollis, J. S. Adams, B. R. Bloom, and R. L. Modlin. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 3111770-1773. [DOI] [PubMed] [Google Scholar]
- 24.Mackay, C. R. 2001. Chemokines: immunology's high impact factors. Nat. Immunol. 295-101. [DOI] [PubMed] [Google Scholar]
- 25.MacMicking, J. D., R. J. North, R. LaCourse, J. S. Mudgett, S. K. Shah, and C. F. Nathan. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 945243-5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maglione, P. J., J. Xu, and J. Chan. 2007. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J. Immunol. 1787222-7234. [DOI] [PubMed] [Google Scholar]
- 27.Marino, S., D. Sud, H. Plessner, P. L. Lin, J. Chan, J. L. Flynn, and D. E. Kirschner. 2007. Differences in reactivation of tuberculosis induced from anti-TNF treatments are based on bioavailability in granulomatous tissue. PLoS Comput. Biol. 31909-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Means, T. K., S. Wang, E. Lien, A. Yoshimura, D. T. Golenbock, and M. J. Fenton. 1999. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 1633920-3927. [PubMed] [Google Scholar]
- 29.Mebius, R. E. 2003. Organogenesis of lymphoid tissues. Nat. Rev. Immunol. 3292-303. [DOI] [PubMed] [Google Scholar]
- 30.Mohan, V. P., C. A. Scanga, K. Yu, H. M. Scott, K. E. Tanaka, E. Tsang, M. M. Tsai, J. L. Flynn, and J. Chan. 2001. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect. Immun. 691847-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moser, B., and P. Loetscher. 2001. Lymphocyte traffic control by chemokines. Nat. Immunol. 2123-128. [DOI] [PubMed] [Google Scholar]
- 32.Poznansky, M. C., I. T. Olszak, R. Foxall, R. H. Evans, A. D. Luster, and D. T. Scadden. 2000. Active movement of T cells away from a chemokine. Nat. Med. 6543-548. [DOI] [PubMed] [Google Scholar]
- 33.Proudfoot, A. E. 2002. Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2106-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scanga, C. A., V. P. Mohan, H. Joseph, K. Yu, J. Chan, and J. L. Flynn. 1999. Reactivation of latent tuberculosis: variations on the Cornell murine model. Infect. Immun. 674531-4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sedgwick, J. D., D. S. Riminton, J. G. Cyster, and H. Korner. 2000. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol. Today 21110-113. [DOI] [PubMed] [Google Scholar]
- 36.Shiloh, M. U., and C. F. Nathan. 2000. Reactive nitrogen intermediates and the pathogenesis of Salmonella and mycobacteria. Curr. Opin. Microbiol. 335-42. [DOI] [PubMed] [Google Scholar]
- 37.Stenger, S., and R. L. Modlin. 2002. Control of Mycobacterium tuberculosis through mammalian Toll-like receptors. Curr. Opin. Immunol. 14452-457. [DOI] [PubMed] [Google Scholar]
- 38.Tsai, M. C., S. Chakravarty, G. Zhu, J. Xu, K. Tanaka, C. Koch, J. Tufariello, J. Flynn, and J. Chan. 2006. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cell. Microbiol. 8218-232. [DOI] [PubMed] [Google Scholar]
- 39.Turner, J., M. Gonzalez-Juarrero, D. L. Ellis, R. J. Basaraba, A. Kipnis, I. M. Orme, and A. M. Cooper. 2002. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 1696343-6351. [DOI] [PubMed] [Google Scholar]
- 40.Ulrichs, T., G. A. Kosmiadi, V. Trusov, S. Jorg, L. Pradl, M. Titukhina, V. Mishenko, N. Gushina, and S. H. Kaufmann. 2004. Human tuberculous granulomas induce peripheral lymphoid follicle-like structures to orchestrate local host defence in the lung. J. Pathol. 204217-228. [DOI] [PubMed] [Google Scholar]
- 41.Underhill, D. M., A. Ozinsky, K. D. Smith, and A. Aderem. 1999. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA 9614459-14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.WHO. 2007. Global tuberculosis control: surveillance, planning, financing. WHO/HTM/TB/2007.376. World Health Organization, Geneva, Switzerland.
- 43.Williams, I. R. 2006. CCR6 and CCL20: partners in intestinal immunity and lymphorganogenesis. Ann. N. Y. Acad. Sci. 107252-61. [DOI] [PubMed] [Google Scholar]
- 44.Zganiacz, A., M. Santosuosso, J. Wang, T. Yang, L. Chen, M. Anzulovic, S. Alexander, B. Gicquel, Y. Wan, J. Bramson, M. Inman, and Z. Xing. 2004. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J. Clin. Investig. 113401-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu, G., H. Xiao, V. P. Mohan, K. Tanaka, S. Tyagi, F. Tsen, P. Salgame, and J. Chan. 2003. Gene expression in the tuberculous granuloma: analysis by laser capture microdissection and real-time PCR. Cell. Microbiol. 5445-453. [DOI] [PubMed] [Google Scholar]
- 46.Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity 12121-127. [DOI] [PubMed] [Google Scholar]