

Abstract
Escherichia coli O157:H7 Shiga toxin 2 (Stx2), one of the causative agents of hemolytic-uremic syndrome, is toxic to endothelial cells, including primary cultured human umbilical vein endothelial cells (HUVEC). This sensitivity of cells to Stx2 can be increased with either lipopolysaccharide (LPS) or tumor necrosis factor alpha (TNF-α). The goal of the present study was to identify the intracellular signaling pathway(s) by which LPS and TNF-α sensitize HUVEC to the cytotoxic effects of Stx2. To identify these pathways, specific pharmacological inhibitors and small interfering RNAs were tested with cell viability endpoints. A time course and dose response experiment for HUVEC exposure to LPS and TNF-α showed that a relatively short exposure to either agonist was sufficient to sensitize the cells to Stx2 and that both agonists stimulated intracellular signaling pathways within a short time. Cell viability assays indicated that the p38 mitogen-activated protein kinase (MAPK) inhibitors SB202190 and SB203580 and the general protein synthesis inhibitor cycloheximide inhibited both the LPS and TNF-α sensitization of HUVEC to Stx2, while all other inhibitors tested did not inhibit this sensitization. Additionally, SB202190 reduced the cellular globotriaosylceramide content under LPS- and TNF-α-induced conditions. In conclusion, our results show that LPS and TNF-α induction of Stx2 sensitivity in HUVEC is mediated through a pathway that includes p38 MAPK. These results indicate that inhibition of p38 MAPK in endothelial cells may protect a host from the deleterious effects of Stx2.
Escherichia coli O157:H7 is a food-borne pathogen mainly associated with undercooked contaminated beef products (1). Most cases of E. coli O157:H7 infection are sporadic; however, numerous outbreaks have been reported in temperate areas of the world (5, 6, 67). Once ingested, the bacteria form attaching and effacing lesions on colonic intestinal epithelial cells (46). It is here that the bacteria release many different agents, including Shiga toxin 1 (Stx1) and Stx2 (3). Stx1 and Stx2 are the causative agents of hemolytic-uremic syndrome (HUS) (70), which is the most frequent cause of acute renal failure in young children. HUS involves a combination of symptoms, including hemolytic anemia, thrombocytopenia, and acute renal failure, appearing most frequently in children less than 4 years of age (9).
The Stxs consist of an enzymatically active A subunit protein bound noncovalently to a pentamer of B-subunit proteins that are responsible for binding to cells (13). Stx1 and Stx2 bind to the glycolipid receptor Galα1-4Galβ1-4GlcCeramide, also known as globotriaosylceramide (Gb3), CD77, Pk antigen, or GL-3 (50), and enter the target cell via receptor-mediated endocytosis. Once the toxin is within the endosomes, various mechanisms have been proposed for retrograde transport through the Golgi network and endoplasmic reticulum and into the cytosol (49). Once the toxin is in the cytosol, the Stx A subunit removes a specific adenine from rRNA and inhibits protein synthesis (16).
Gb3 is a neutral glycosphingolipid that is either expressed constitutively or induced under inflammatory conditions by types of cells within the kidney. These cells include human glomerular endothelial cells, glomerular visceral epithelial cells (podocytes), proximal tubule cells, and mesangial cells (22, 65). Gb3 levels are increased on brain microvascular endothelial cells (15) after treatment with tumor necrosis factor alpha (TNF-α), providing a rationale for Stx-induced brain injury (51). Finally, Gb3 can be induced on human umbilical vein endothelial cells (HUVEC) with lipopolysaccharide (LPS), TNF-α, or interleukin-1β (IL-1β) and has been widely used as an in vitro model for endothelial damage by Stx (27, 29, 37-39, 60, 68).
These in vitro agonists have a potential role in HUS. Increased concentrations of TNF-α in serum have been associated with HUS patients (36) along with increased concentrations of the soluble TNF receptors p55 and p75 (61). In addition, the urinary TNF-α level was elevated in patients in the acute phase of HUS compared to controls (26), suggesting a renal origin for this cytokine. Finally, when Stx was presented systematically to a transgenic mouse, TNF-α promoter activity was observed exclusively within the kidney (21). LPS has also been implicated in the pathogenesis of HUS. Anti-LPS antibodies belonging to the O157:H7 serotype have been found in the serum of HUS patients (2) along with clinical evidence of endotoxemia (31). It has also been shown that in a primate model LPS is required in combination with Stx1 to induce the clinical hallmarks of HUS (52). Finally, a complete murine model of HUS requires LPS in addition to Stx2 (28).
The goal of the present study was to determine the intracellular pathways that LPS and TNF-α utilize in HUVEC to induce Gb3 and subsequent Stx2 sensitivity. Different pharmacological inhibitors, along with RNA interference, were employed to test known pathways used by these two agents in HUVEC to determine which of these pathways are involved in induction of Stx2 sensitivity. The results indicate that both stimuli use a p38 mitogen-activated protein kinase (MAPK)-dependent pathway to stimulate Stx2 sensitivity.
MATERIALS AND METHODS
Materials.
Recombinant human TNF-α (∼600 pg/U) was a gift from Dainippon Pharmaceutical Co., Ltd. (Osaka, Japan). Stx2 was immunoaffinity purified from E. coli DH5α(pJES120) lysates obtained from A. D. O'Brien as described previously (41). Neutral red dye, cycloheximide, and E. coli O55:B5 LPS which was purified by gel filtration chromatography and gamma irradiated were purchased from Sigma Aldrich (St. Louis, MO). Wortmannin, Bay11-7082, SN50, SN50M, SB202190, SB203580, 2-phenyl-1,2-benzisoselenazol-3(2H)-one (Ebselen), SP600125, Jak inhibitor 1, U0126, and myristoylated protein kinase Cζ (PKCζ) pseudosubstrate were purchased from EMD Biosciences (San Diego, CA). A CCK-8 cell viability kit was purchased from Dojindo Molecular Technologies (Gaithersburg, MD). The RNAiFect transfection reagent was purchased from Qiagen (Valencia, CA). SMARTpool small interfering RNA (siRNA) duplexes for PKCι were purchased from Dharmacon (Lafayette, CO). Anti-PKCι, anti-p38, and anti-phospho-p38 antibodies were purchased from BD Biosciences (San Jose, CA). PD98059, anti-c-Jun, anti-phospho-c-Jun, anti-IRF-1, anti-Akt, anti-phospho-Akt, anti-phospho-CREB, and anti-I-κBα antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-β-actin antibody was purchased from Abcam (Cambridge, MA). A DuoSet human IL-6 enzyme-linked immunosorbent assay (ELISA) kit and anti-ICAM-1 antibody were purchased from R&D Systems, Inc. (Minneapolis, MN). Anti-mouse immunoglobulin G and horseradish peroxidase (HRP)-tagged antibody were purchased from Amersham (Piscataway, NJ). Anti-rabbit HRP-tagged antibody was purchased from Zymed (San Francisco, CA). Anti-CREB and anti-sheep HRP-tagged antibodies were purchased from Upstate (Lake Placid, NY). All inhibitor targets are listed in Table 1.
TABLE 1.
Effects of various signal transduction inhibitors on TNF-α− or LPS-induced Stx2 cytotoxicitya
| Inhibitor | Source | Target | LPS | TNF-α | Positive control | Reference(s) |
|---|---|---|---|---|---|---|
| Wortmannin | EMD Biosciences | Phosphatidylinositol 3-kinase | − | − | pAkt | 33 |
| PKCζ pseudosubstrate | Biosource | PKCζ | 50% | − | ELISA, IL-6 | 14, 44 |
| PKCι siRNA | Dharmacon | PKCι | − | − | PKCι | 25 |
| Bay11-7082 | EMD Biosciences | IκB kinase | − | − | Cell-based ELISA, ICAM-1 | 43 |
| SN50 | EMD Biosciences | NF-κB | − | − | Cell-based ELISA, ICAM-1 | 35 |
| SB202190 | EMD Biosciences | p38 | + | + | NA | 19 |
| SB203580 | EMD Biosciences | p38 | + | + | NA | 19 |
| Ebselen | EMD Biosciences | Traf2/ASK1 | ND | − | Toxic | 66 |
| SP600125 | EMD Biosciences | JNK | − | − | p-c-Jun | 30 |
| Jak inhibitor 1 (pyridone 6) | EMD Biosciences | JAK | − | − | IRF-1 | 34, 57 |
| PD98059 | Cell Signaling Technology | MEK1/2 | − | − | pERK1/2 | 42 |
| U0126 | EMD Biosciences | MEK1/2 | − | − | pERK1/2 | 17 |
| Cycloheximide | Sigma Aldrich | Protein synthesis | + | + | NA | 60 |
HUVEC were incubated with the indicated inhibitors for up to 1 h (see Materials and Methods), treated with 200 U/ml TNF-α or 10 μg/ml LPS with the inhibitor for 24 h, and then treated with 1 nM Stx2 for 24 h. CCK-8 or neutral red cell viability assays were performed to determine if an inhibitor protected against TNF-α- or LPS-induced Stx2 cytotoxicity. +, protection; −, inhibitor had no effect; 50%, LPS-induced Stx2 cytotoxicity was reduced by one-half; ND, not determined. The data are representative of at least three independent experiments. Positive controls indicate cellular responses that were modified by identical concentrations of the inhibitors in HUVEC. Supporting data are shown in Fig. S1 in the supplemental material. NA, not applicable.
Cell culture.
Primary HUVEC were purchased from VEC Technologies (Rensselaer, NY) and grown in MCDB 131 medium (Mediatech/CellGro) supplemented with 1 μg/ml hydrocortisone (Sigma Aldrich), 10 ng/ml epidermal growth factor (Collaborative Biomedical Products), 100 μg/ml endothelial cell mitogen (Biomedical Technologies Inc.), 2 mM l-glutamine (Gibco), 10% fetal bovine serum (HyClone), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Mediatech/CellGro). Cells were incubated at 37°C with 5% CO2 and 90% humidity in Falcon 75-cm2 flasks (BD Biosciences). All experiments were performed using HUVEC at passages 2 to 5.
Immunoblotting.
HUVEC were seeded at a concentration of 400,000 cells/well, allowed to attach and grow overnight to confluence in six-well plates, and treated as indicated below. Following treatment, wells were aspirated, and 400 μl Laemmli sample buffer (Bio-Rad) with 5% 2-mercaptoethanol was added for lysis. To generate lysates for use in immunoblots, cells were harvested by scraping, DNA was sheared by passing cells through a 28-gauge needle more than six times, and proteins were denatured at 95°C for 4 min. Equal volumes of lysate were separated using sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), transferred to a polyvinylidene difluoride membrane, and probed with primary and HRP-conjugated secondary antibodies. Bound HRP was detected by enhanced chemiluminescence according to the manufacturer's instructions (Western Lightning, Perkin Elmer). To assess loading controls, membranes were stripped by washing them thoroughly with phosphate-buffered saline (PBS)-Tween, incubating them in 62.5 mM Tris-HCl (pH 6.8)-2% SDS-100 mM 2-mercaptoethanol at 50°C for 30 min, and rewashing them prior to reprobing.
Cell viability assays.
HUVEC were seeded at a concentration of 24,000 cells/well and allowed to attach and grow overnight to confluence in 96-well plates, and then they were treated as indicated below. Following treatment, either neutral red (37) or CCK-8 cell viability assays were performed according to the manufacturer's instructions (Dojindo Molecular Technologies).
Pharmacological inhibition of different pathways.
HUVEC were seeded as described above for the cell viability assays. At different times prior to stimulation with either LPS or TNF-α, the following inhibitors were diluted in media and added to the cells: at 1 h prior to stimulation, wortmannin (1, 10, or 100 nM), PKCζ pseudosubstrate (50, 60, or 80 μM), SN50 (100 μM), SP600125 (500 or 10 μM), Jak inhibitor 1 (100 or 500 nM), and PD98059 (10, 25, or 100 μM); at 45 min prior to stimulation, Bay11-7082 (20 μM) and U0126 (40, 60, 100, or 500 nM or 1 μM); at 40 min prior to stimulation, SB202190 (1, 10, 20, 50, or 100 μM) and SB203580 (20 μM); at 30 min prior to stimulation, Ebselen (1, 10, or 100 μM); and at the time of stimulation, cycloheximide (0.5, 2, or 5 μg/ml). When appropriate, dimethyl sulfoxide diluent controls were included. LPS and TNF-α were then added to the cells in the presence of the inhibitors for 24 h. The medium was then replaced with or without 1 nM Stx2 for 24 h, and cell viability assays were performed as described above. In separate experiments, HUVEC were incubated with the maximum tested dose of an inhibitor and treated with TNF-α for 20 min or with LPS for 4 h; then they were analyzed by immunoblotting as described above or fixed in 96-well plates for a cell-based ELISA, or supernatants were analyzed for secreted IL-6 by an ELISA performed according to the manufacturer's instructions (R&D Systems).
Cell-based ELISA.
Cells were treated with Bay11-7082 and SN50 as described above, treated with LPS or TNF-α, and incubated for 4 h at 37°C. Following incubation, cells were fixed with a 1% glutaraldehyde solution for 20 min. Cells were then washed with PBS and blocked with PBS containing 1% bovine serum albumin for 1 h. After blocking, cells were incubated with a 1:1,000 dilution of anti-ICAM-1 antibody (overnight) and anti-sheep HRP (2 h). The signal was visualized with a 3,3′,5,5′-tetramethylbenzidine substrate solution and 2 N H2SO4 and quantified by determining the optical density at 450 nm with a reference wavelength of 570 nm. Following collection of ELISA data, cells were stained with crystal violet and values were normalized to the relative cell number. This protocol was adapted from the protocol described in the general FACE kit manual from Active Motif (Carlsbad, CA).
RNA interference knockdown of PKCι.
Transfections were performed using the RNAiFECT handbook guidelines, with the following modifications. HUVEC were seeded at a concentration of 12,000 cells/well in 96-well plates and allowed to attach overnight. The 25-μl (total volume) transfection mixture consisted of 0.125 μg siRNA, 0.75 μl RNAiFECT reagent, and 23.75 μl medium (control cells received PBS in place of the siRNA suspension). Transfection was carried out for 4 h, and then the transfection mixture was replaced with 200 μl medium and the cells were incubated at 37°C for 24 h. Then the assay was performed as described above for the cell viability assay.
TLC of Gb3.
Equal amounts of HUVEC were seeded in Falcon 75-cm2 flasks, and the cells were allowed to grow to confluence. Cells were then treated as described in the figure legends, trypsinized, and counted. Neutral glycolipids were then isolated from the cells and analyzed by the thin-layer chromatography (TLC)-Stx1 subunit B overlay method as described previously (47). TLC plates were analyzed by densitometry, and the amounts were normalized to the cell number and Gb3 amount interpolated by comparison with authentic glycolipid standards (Matreya LLC) on each plate.
Statistics.
Cell viability data were expressed as means ± standard deviations. Student's two-sample t tests were used to compare cell viability values where indicated. A P value of <0.01 was considered significant.
RESULTS
LPS and TNF-α induce Stx2 sensitivity in HUVEC.
HUVEC are relatively insensitive to the cytotoxic effects of Stx2 compared to other subtypes of endothelial cells; however, their sensitivity can be increased by preincubating the cells with LPS or TNF-α (27, 37-39, 48, 59, 60). We were able to confirm these findings by preincubating HUVEC with 0.01 to 100 μg/ml LPS or 0.2 to 2,000 U/ml TNF-α for 24 h prior to treatment of the cells with 1 nM Stx2. As shown in Fig. 1A and B, both LPS and TNF-α had a dose-dependent effect on Stx2 sensitivity. We further characterized the system by adding either 10 μg/ml LPS or 200 U/ml TNF-α to HUVEC for various times prior to 24 h of Stx2 challenge. Figure 1C shows both the LPS- and TNF-α-induced Stx2 sensitivities after the first hour of incubation and that induction remained relatively constant throughout the 48-h time course. TNF-α was slightly more potent than LPS at the 8-, 12-, 16-, and 24-h time points at these doses. In all additional experiments described here 10 μg/ml LPS or 200 U/ml TNF-α was used for 24 h prior to Stx2 addition.
FIG. 1.
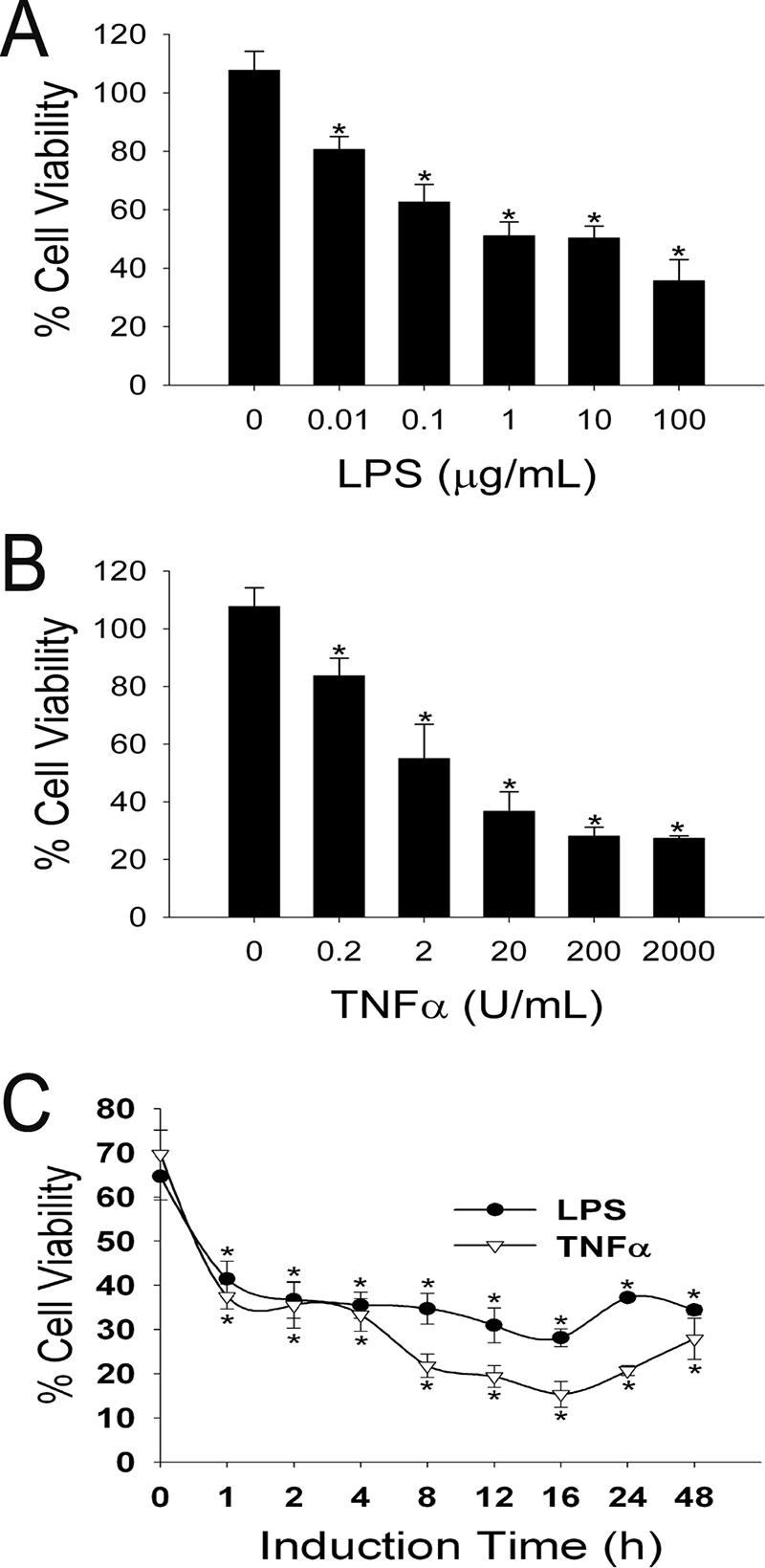
TNF-α and LPS induce Stx2 cytotoxicity in a dose- and time-dependent manner. (A and B) Confluent HUVEC were incubated with the indicated concentrations of (A) LPS or (B) TNF-α for 24 h before treatment with 1 nM Stx2 for 24 h. (C) Confluent HUVEC were incubated with either 10 μg/ml LPS or 200 U/ml TNF-α for the indicated times before treatment with 1 nM Stx2 for 24 h. Cell viability assays were performed, and the results were expressed as the percent viability for identical treatments for each concentration and time point without Stx2; the error bars indicate standard deviations (n = 4). An asterisk indicates that the value is statistically significantly (P < 0.01) different from the value for samples without LPS or TNF-α.
LPS and TNF-α induce similar signal transduction pathways at different rates.
LPS and TNF-α stimulate a wide range of downstream effects through many different pathways (for reviews, see references 11, 40, and 64). To characterize a sample of these events in HUVEC, confluent monolayers of HUVEC were treated with either LPS or TNF-α for up to 1 h, cells were lysed, and proteins were analyzed by immunoblotting. We found that TNF-α (Fig. 2A) induced the NF-κB and p38 MAPK pathways within 5 min and with between 10 and 20 min of stimulation, respectively. I-κBα degradation was observed in a similar time frame, with the levels dropping below the detectable limits by 20 min poststimulation. In comparison, LPS showed a much slower induction profile with these pathways. LPS-induced phosphorylation of p38 MAPK reached maximal levels at 1 h poststimulation and did not change throughout the remaining times tested (Fig. 2B). Additional experiments showed that there were negligible changes in these proteins prior to 1 h poststimulation (data not shown). LPS-induced degradation of I-κBα showed a similar temporal response to p38 MAPK. In all cases, phosphorylation of p38 coincided with phosphorylation of the downstream transcription targets CREB and ATF-1 (Fig. 2A and B).
FIG. 2.

TNF-α and LPS induce intracellular pathway activation at different rates. Confluent HUVEC were incubated with (A) TNF-α and (B) LPS for the indicated times and lysed. Lysates were processed by SDS-PAGE and analyzed by immunoblotting.
Differential inhibition of LPS- or TNF-α-induced Stx2 sensitivity.
Since LPS and TNF-α stimulate many different pathways, pharmacological inhibitors and siRNAs were used to determine which of these pathways was involved in LPS- or TNF-α-induced sensitization of HUVEC to Stx2 (Table 1; see Fig. S1 in the supplemental material). Of all the signal transduction inhibitors tested, only the p38 MAPK inhibitors SB202190 and SB203580 fully inhibited both LPS- and TNF-α-induced Stx2 sensitivity. However, the PKCζ pseudosubstrate added prior to LPS or TNF-α induction inhibited LPS-induced sensitivity by 50%. This inhibition was the same when the pseudosubstrate concentration was increased to 80 μM. All pseudosubstrate concentrations above 80 μM were cytotoxic. As expected, cycloheximide was also effective at inhibiting both LPS and TNF-α induction of Stx2 sensitivity, which is consistent with previous results (60).
p38 MAPK is required for Stx2 sensitization.
p38 MAPK has been shown to be involved in Stx-mediated cell death in other cell types (7, 24, 54). To determine which phase (i.e., induction or Stx2 challenge) of the experiment the p38 inhibitors targeted, cells were treated with inhibitor before and/or after treatment with the inducer (Fig. 3A). As shown in Fig. 3B, both LPS and TNF-α enhanced HUVEC sensitivity to Stx2. The induction of sensitivity was reversed when a p38 inhibitor was given prior to treatment with LPS or TNF-α. Notably, the noninduced cells were also more resistant to Stx2 challenge (P < 0.01). However, when the p38 inhibitor was given after induction and before Stx2 challenge, no significant changes were observed. The cells that were exposed to the inhibitor throughout the experiment were indistinguishable from the cells that were incubated with the inhibitor only prior to and concurrently with LPS or TNF-α.
FIG. 3.

Inhibition of p38 reduces TNF-α and LPS induction of Stx2 cytotoxicity. (B) HUVEC were treated with SB202190 (a p38 MAPK inhibitor) before or after induction and then challenged with Stx2. Details are as follows (A). For preinduction, one of the following was added to the cells for 40 min: 20 μM SB202190 or control medium. For induction one of the following was added to the cells for 24 h: SB202190, TNF-α, LPS, TNF-α plus SB202190, LPS plus SB202190, or control medium. For postinduction one of the following was added to the cells for 40 min: SB202190 or control medium. For challenge one of the following was added to the cells for 24 h: SB202190, 1 nM Stx2, SB202190 plus Stx2, or control medium. For the cytotoxicity assay after 24 h of incubation with Stx2, CCK-8 cell viability assays were performed, and the results were expressed as percentages of the viability of an identical treatment without Stx2; the error bars indicate standard deviations (n = 4). An asterisk indicates that the value is statistically significantly (P < 0.01) different from the value for samples not treated with SB202190. The data are representative of three independent experiments. The data obtained with a separate p38 MAPK inhibitor, SB203580, were similar.
p38 is required for LPS or TNF-α induction of Gb3.
It has been shown that Gb3 is the cellular receptor for Stx2 (63) and that Gb3 levels correlate with Stx sensitivity (48, 59, 60, 62). Therefore, to determine the effects of p38 inhibition on levels of Gb3 on HUVEC, cells were treated with LPS or TNF-α alone and after pretreatment with SB202190. Figure 4 shows that pretreatment with the p38 inhibitor SB202190 reduced LPS and TNF-α induction of relative Gb3 levels, a trend that was observed consistently. Note that although the fold change values are different for the individual replicates, in each case the p38 inhibitor reduced LPS- and TNF-α-induced Gb3 levels.
FIG. 4.

p38 inhibition reduces TNF-α and LPS induction of Gb3 in HUVEC. (A) Confluent HUVEC were treated with 20 μM SB202190 40 min prior to and concurrently with 200 U/ml TNF-α or 10 μg/ml LPS for 24 h. Cells were trypsinized and counted, and neutral glycolipids were extracted. Glycolipids were separated by TLC and visualized by using Stx1 subunit B overlay. TLC plates were analyzed by densitometry, and amounts were interpolated by comparison with authentic standards on each plate. (B) Results for TLC-Stx1 subunit B overlay from replicate 2.
Specificity of SB202190.
SB202190 has been shown to be a selective inhibitor of p38 MAPK (12). To verify this specificity under our conditions, HUVEC were treated with 10 and 20 μM SB202190, followed by 200 U/ml TNF-α (Fig. 5A) or 10 μg/ml LPS (Fig. 5B) for 20 min and 1 h, respectively, and the cells were lysed and analyzed by immunoblotting. SB202190 inhibited both TNF-α and LPS induction of CREB and ATF-1 phosphorylation but not degradation of I-κBα. This result is consistent with published data showing that CREB and ATF-1 are exclusively downstream of p38 (20).
FIG. 5.

Specificity of SB202190. Confluent HUVEC were treated with the indicated concentrations of SB202190 for 40 min prior to treatment with (A) TNF-α for 20 min or (B) LPS for 1 h and lysed. Lysates were processed by SDS-PAGE and analyzed by immunoblotting.
DISCUSSION
We show here for the first time that LPS and TNF-α use a p38 MAPK-dependent pathway to sensitize endothelial cells to Stx2-induced cell death. p38 MAPK has been identified as a signaling factor in both Stx-induced cell death (54) and TNF-α induction of Gb3 (55) in other cell types, so we examined if one or both of these roles were evident in HUVEC. Since LPS and TNF-α have been shown to increase Gb3 levels over a 24-h time period (48), p38 inhibitors were used to treat HUVEC before, during, and/or after 24 h of induction using LPS or TNF-α. We found that p38 MAPK inhibitors were effective at reducing LPS- or TNF-α-induced Stx2-mediated cell death only when given prior to the inducer, indicating that in HUVEC, p38 is principally involved in the induction of the Stx2 receptor and not in toxin-mediated cell death. This conclusion is supported by the TLC data which showed that one of the p38 inhibitors, SB202190, reduced LPS- and TNF-α-induced Gb3 levels.
We show here that both LPS and TNF-α induce Stx2-mediated cell death in a dose-dependent manner. These inflammatory mediators have been shown to increase Gb3 and/or Stx sensitivity in kidney cell types such as mesangium (53), tubular epithelial cells (10), and glomerular microvascular endothelial cells even though TNF-α does not affect the retrograde transport of the Stx B subunit in these cells (65). We also show that these cells required minimal times of exposure to LPS or TNF-α in order to have increased sensitivity to Stx2.
Signal transduction of HUVEC in response to LPS and TNF-α is rapid, with TNF-α causing effects in as little as 5 min and LPS causing effects after 1 h of stimulation. This explains why LPS and TNF-α can sensitize HUVEC to the cytotoxic effects of Stx2 after only 1 h of stimulation. Our results are in concert with previous findings that showed that I-κBα degradation occurs more rapidly in the presence of TNF-α than in the presence of LPS (69).
LPS and TNF-α are known to stimulate multiple pathways in HUVEC (11, 40, 64), so we examined each pathway individually using various inhibition methods in order to either include or exclude a pathway's role in Stx2 sensitization. The pharmacological inhibitors that almost completely inhibited LPS and TNF-α induction of Stx2 sensitivity were SB202190 and SB203580, which are closely related inhibitors of p38 MAPK, and cycloheximide, a general inhibitor of protein synthesis. Cycloheximide has been shown to reduce the amount of TNF-α-induced Stx1 binding to human femoral vein endothelial cells and HUVEC (60), theoretically by inhibiting translation of one or more of the biosynthetic enzymes required to make the functional Stx receptor, Gb3. Our hypothesis included the possibility that multiple pathways could be utilized to increase Gb3, and thereby Stx2, sensitization. If multiple pathways were involved, then inhibition of any one of the necessary pathways should partially block LPS- or TNF-α-induced sensitivity to Stx2. Indeed, a PKCζ pseudosubstrate reduced LPS-induced Stx2 sensitivity by 50% in HUVEC. No other inhibitor tested partially reduced LPS-induced Stx2 sensitivity; however, it was shown previously that myristolated PKCζ pseudosubstrate nonspecifically caused the phosphorylation of p38 MAPK, ERK1/2, Akt, and endothelial nitric oxide synthase in porcine pulmonary artery endothelial cells (32), making interpretation of these data difficult. Inhibition of p38 MAPK did not completely reverse LPS- or TNF-α-induced Stx2 sensitivity; however, the negative results obtained with all other signal transduction inhibitors tested imply that if an additional pathway is involved, it is not one of the pathways identified in endothelial cells.
p38 MAPK has been implicated at multiple stages in Stx-mediated HUS. Upon release from the bacterium, Stx interacts with the intestinal epithelium, where p38 MAPK mediates Stx-induced release of IL-8 (58), eIF4E phosphorylation (8), caspase 3 cleavage, and cell death (54). After crossing the intestinal epithelium (23) and entering the vasculature, Stx stimulates circulating monocytes to secrete TNF-α in a p38-dependent manner (4, 18). Once in the vasculature, the main targets of Stx are the brain and kidneys (56). Stricklett et al. showed that p38 MAPK is required for TNF-α induction of Gb3 and Stx1 sensitivity in human brain endothelial cells (55); moreover, Morigi et al. implicated p38 in Stx2-mediated expression of endothelin-1, which is involved in glomerular hemodynamics (45). It is noteworthy that in all cases, inhibiting p38 MAPK would theoretically benefit the host. Thus, p38 MAPK may represent a therapeutic target for reducing the harmful host response to Stx2 if these in vitro results can be verified in an animal model.
In conclusion, p38, a common pathway intermediate, was shown to be involved in sensitization of HUVEC to Stx2 by both LPS and TNF-α. p38 MAPK is required by both agonists to increase cellular Gb3 levels and subsequent Stx2 sensitivity but not for the Stx2-induced cytotoxicity that follows. These findings support the hypothesis that p38 MAPK is an effective target in patients exposed to Stx2 for minimizing symptoms associated with HUS.
Supplementary Material
Acknowledgments
We acknowledge Regina Seaner for expert technical assistance.
Funding for this work was provided by USPHS award AI024431 (T.G.O.).
Editor: B. A. McCormick
Footnotes
Published ahead of print on 17 December 2007.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Alexandre, M., and V. Prado. 2003. Detection of Shiga toxin-producing Escherichia coli in food. Expert Rev. Mol. Diagn. 3105-115. [DOI] [PubMed] [Google Scholar]
- 2.Bitzan, M., E. Moebius, K. Ludwig, D. E. Muller-Wiefel, J. Heesemann, and H. Karch. 1991. High incidence of serum antibodies to Escherichia coli O157 lipopolysaccharide in children with hemolytic-uremic syndrome. J. Pediatr. 119380-385. [DOI] [PubMed] [Google Scholar]
- 3.Boerlin, P. 1999. Evolution of virulence factors in Shiga-toxin-producing Escherichia coli. Cell. Mol. Life Sci. 56735-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cameron, P., S. J. Smith, M. A. Giembycz, D. Rotondo, and R. Plevin. 2003. Verotoxin activates mitogen-activated protein kinase in human peripheral blood monocytes: role in apoptosis and proinflammatory cytokine release. Br. J. Pharmacol. 1401320-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charatan, F. 2006. FDA warns US consumers not to eat spinach after E. coli outbreak. BMJ 333673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, J., and M. W. Griffiths. 1999. Cloning and sequencing of the gene encoding universal stress protein from Escherichia coli O157:H7 isolated from Jack-in-a-Box outbreak. Lett. Appl. Microbiol. 29103-107. [DOI] [PubMed] [Google Scholar]
- 7.Cherla, R. P., S. Y. Lee, P. L. Mees, and V. L. Tesh. 2006. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 79397-407. [DOI] [PubMed] [Google Scholar]
- 8.Colpoys, W. E., B. H. Cochran, T. M. Carducci, and C. M. Thorpe. 2005. Shiga toxins activate translational regulation pathways in intestinal epithelial cells. Cell. Signal. 17891-899. [DOI] [PubMed] [Google Scholar]
- 9.Corrigan, J. J., Jr., and F. G. Boineau. 2001. Hemolytic-uremic syndrome. Pediatr. Rev. 22365-369. [PubMed] [Google Scholar]
- 10.Creydt, V. P., C. Silberstein, E. Zotta, and C. Ibarra. 2006. Cytotoxic effect of Shiga toxin-2 holotoxin and its B subunit on human renal tubular epithelial cells. Microbes Infect. 8410-419. [DOI] [PubMed] [Google Scholar]
- 11.Dauphinee, S. M., and A. Karsan. 2006. Lipopolysaccharide signaling in endothelial cells. Lab Investig. 869-22. [DOI] [PubMed] [Google Scholar]
- 12.Davies, S. P., H. Reddy, M. Caivano, and P. Cohen. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 35195-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donohue-Rolfe, A., G. T. Keusch, C. Edson, D. Thorley-Lawson, and M. Jacewicz. 1984. Pathogenesis of Shigella diarrhea. IX. Simplified high yield purification of Shigella toxin and characterization of subunit composition and function by the use of subunit-specific monoclonal and polyclonal antibodies. J. Exp. Med. 1601767-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duran, A., M. T. Diaz-Meco, and J. Moscat. 2003. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 223910-3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisenhauer, P. B., P. Chaturvedi, R. E. Fine, A. J. Ritchie, J. S. Pober, T. G. Cleary, and D. S. Newburg. 2001. Tumor necrosis factor alpha increases human cerebral endothelial cell Gb3 and sensitivity to Shiga toxin. Infect. Immun. 691889-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endo, Y., K. Tsurugi, T. Yutsudo, Y. Takeda, T. Ogasawara, and K. Igarashi. 1988. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 17145-50. [DOI] [PubMed] [Google Scholar]
- 17.Favata, M. F., K. Y. Horiuchi, E. J. Manos, A. J. Daulerio, D. A. Stradley, W. S. Feeser, D. E. Van Dyk, W. J. Pitts, R. A. Earl, F. Hobbs, R. A. Copeland, R. L. Magolda, P. A. Scherle, and J. M. Trzaskos. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 27318623-18632. [DOI] [PubMed] [Google Scholar]
- 18.Foster, G. H., and V. L. Tesh. 2002. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-alpha. J. Leukoc. Biol. 71107-114. [PubMed] [Google Scholar]
- 19.Goebeler, M., K. Kilian, R. Gillitzer, M. Kunz, T. Yoshimura, E.-B. Brocker, U. R. Rapp, and S. Ludwig. 1999. The MKK6/p38 stress kinase cascade is critical for tumor necrosis factor-alpha-induced expression of monocyte-chemoattractant protein-1 in endothelial cells. Blood 93857-865. [PubMed] [Google Scholar]
- 20.Gustin, J. A., R. Pincheira, L. D. Mayo, O. N. Ozes, K. M. Kessler, M. R. Baerwald, C. K. Korgaonkar, and D. B. Donner. 2004. Tumor necrosis factor activates CRE-binding protein through a p38 MAPK/MSK1 signaling pathway in endothelial cells. Am. J. Physiol. 286C547-C555. [DOI] [PubMed] [Google Scholar]
- 21.Harel, Y., M. Silva, B. Giroir, A. Weinberg, T. B. Cleary, and B. Beutler. 1993. A reporter transgene indicates renal-specific induction of tumor necrosis factor (TNF) by shiga-like toxin. Possible involvement of TNF in hemolytic uremic syndrome. J. Clin. Investig. 922110-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hughes, A. K., D. I. Schmid, and D. E. Kohan. 2002. Sex steroids do not affect shiga toxin cytotoxicity on human renal tubular or glomerular cells. BMC Nephrol. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hurley, B. P., M. Jacewicz, C. M. Thorpe, L. L. Lincicome, A. J. King, G. T. Keusch, and D. W. Acheson. 1999. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect. Immun. 676670-6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda, M., Y. Gunji, S. Yamasaki, and Y. Takeda. 2000. Shiga toxin activates p38 MAP kinase through cellular Ca2+ increase in Vero cells. FEBS Lett. 48594-98. [DOI] [PubMed] [Google Scholar]
- 25.Kanayasu-Toyoda, T., T. Suzuki, T. Oshizawa, E. Uchida, T. Hayakawa, and T. Yamaguchi. 2007. Granulocyte colony-stimulating factor promotes the translocation of protein kinase Cι in neutrophilic differentiation cells. J. Cell. Physiol. 211189-196. [DOI] [PubMed] [Google Scholar]
- 26.Karpman, D., A. Andreasson, H. Thysell, B. S. Kaplan, and C. Svanborg. 1995. Cytokines in childhood hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Pediatr. Nephrol. 9694-699. [DOI] [PubMed] [Google Scholar]
- 27.Kaye, S. A., C. B. Louise, B. Boyd, C. A. Lingwood, and T. G. Obrig. 1993. Shiga toxin-associated hemolytic-uremic syndrome: interleukin-1 beta enhancement of Shiga toxin cytotoxicity toward human vascular endothelial cells in vitro. Infect. Immun. 613886-3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keepers, T. R., M. A. Psotka, L. K. Gross, and T. G. Obrig. 2006. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J. Am. Soc Nephrol. 173404-3414. [DOI] [PubMed] [Google Scholar]
- 29.Keusch, G. T., D. W. Acheson, L. Aaldering, J. Erban, and M. S. Jacewicz. 1996. Comparison of the effects of Shiga-like toxin 1 on cytokine- and butyrate-treated human umbilical and saphenous vein endothelial cells. J. Infect. Dis. 1731164-1170. [DOI] [PubMed] [Google Scholar]
- 30.Kiemer, A. K., N. Bildner, N. C. Weber, and A. M. Vollmar. 2003. Characterization of heme oxygenase 1 (heat shock protein 32) induction by atrial natriuretic peptide in human endothelial cells. Endocrinology 144802-812. [DOI] [PubMed] [Google Scholar]
- 31.Koster, F., J. Levin, L. Walker, K. Tung, R. Gilman, M. Rahaman, M. Majid, S. Islam, and R. Williams. 1978. Hemolytic-uremic syndrome after shigellosis. Relation to endotoxemia and circulating immune complexes. N. Engl. J. Med. 298927-933. [DOI] [PubMed] [Google Scholar]
- 32.Krotova, K., H. Hu, S.-L. Xia, L. Belayev, J. M. Patel, E. R. Block, and S. Zharikov. 2006. Peptides modified by myristoylation activate eNOS in endothelial cells through Akt phosphorylation. Br. J. Pharmacol. 148732-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lal, B. K., S. Varma, P. J. Pappas, I. Hobson, W. Robert, and W. N. Duran. 2001. VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc. Res. 62252-262. [DOI] [PubMed] [Google Scholar]
- 34.Liu, L., A. Paul, C. J. MacKenzie, C. Bryant, A. Graham, and R. Plevin. 2001. Nuclear factor kappa B is involved in lipopolysaccharide-stimulated induction of interferon regulatory factor-1 and GAS/GAF DNA-binding in human umbilical vein endothelial cells. Br. J. Pharmacol. 1341629-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu, X., and Z. Spolarics. 2003. Methemoglobin is a potent activator of endothelial cells by stimulating IL-6 and IL-8 production and E-selectin membrane expression. Am. J. Physiol. 285C1036-C1046. [DOI] [PubMed] [Google Scholar]
- 36.Lopez, E. L., M. M. Contrini, S. Devoto, M. F. de Rosa, M. G. Grana, M. H. Genero, C. Canepa, H. F. Gomez, and T. G. Cleary. 1995. Tumor necrosis factor concentrations in hemolytic uremic syndrome patients and children with bloody diarrhea in Argentina. Pediatr. Infect. Dis. J. 14594-598. [DOI] [PubMed] [Google Scholar]
- 37.Louise, C. B., and T. G. Obrig. 1992. Shiga toxin-associated hemolytic-uremic syndrome: combined cytotoxic effects of Shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect. Immun. 601536-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louise, C. B., and T. G. Obrig. 1991. Shiga toxin-associated hemolytic-uremic syndrome: combined cytotoxic effects of Shiga toxin, interleukin-1beta, and tumor necrosis factor alpha on human vascular endothelial cells in vitro. Infect. Immun. 594173-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Louise, C. B., M. C. Tran, and T. G. Obrig. 1997. Sensitization of human umbilical vein endothelial cells to Shiga toxin: involvement of protein kinase C and NF-kappaB. Infect. Immun. 653337-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Madge, L. A., and J. S. Pober. 2001. TNF signaling in vascular endothelial cells. Exp. Mol. Pathol. 70317-325. [DOI] [PubMed] [Google Scholar]
- 41.Melton-Celsa, A. R., and A. D. O'Brien. 2000. Shiga toxins of Shigella dysenteriae and Escherichia coli, p. 385-406. In M. Aepfelbacher, K. Aktories, and I. Just (ed.), Bacterial protein toxins. Springer, Berlin, Germany.
- 42.Merhi-Soussi, F., Z. Dominguez, O. Macovschi, M. Dubois, G. Nemoz, M. Lagarde, and A.-F. Prigent. 2003. Mechanisms involved in the stimulation of prostacyclin synthesis by human lymphocytes in human umbilical vein endothelial cells. Br. J. Pharmacol. 139321-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miho, N., T. Ishida, N. Kuwaba, M. Ishida, K. Shimote-Abe, K. Tabuchi, T. Oshima, M. Yoshizumi, and K. Chayama. 2005. Role of the JNK pathway in thrombin-induced ICAM-1 expression in endothelial cells. Cardiovasc. Res. 68289-298. [DOI] [PubMed] [Google Scholar]
- 44.Montiel, M., E. P. de la Blanca, and E. Jimenez. 2006. P2Y receptors activate MAPK/ERK through a pathway involving PI3K/PDK1/PKC-zeta in human vein endothelial cells. Cell. Physiol. Biochem. 18123-134. [DOI] [PubMed] [Google Scholar]
- 45.Morigi, M., S. Buelli, C. Zanchi, L. Longaretti, D. Macconi, A. Benigni, D. Moioli, G. Remuzzi, and C. Zoja. 2006. Shiga btoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am. J. Pathol. 1691965-1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nataro, J. P., and J. B. Kaper. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11142-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nutikka, A., B. Binnington-Boyd, and C. A. Lingwood. 2003. Methods for the identification of host receptors for Shiga toxin. Methods Mol. Med. 73197-208. [DOI] [PubMed] [Google Scholar]
- 48.Obrig, T. G., C. B. Louise, C. A. Lingwood, B. Boyd, L. Barley-Maloney, and T. O. Daniel. 1993. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 26815484-15488. [PubMed] [Google Scholar]
- 49.Sandvig, K., and B. van Deurs. 2005. Delivery into cells: lessons learned from plant and bacterial toxins. Gene Ther. 12865-872. [DOI] [PubMed] [Google Scholar]
- 50.Sandvig, K., and B. van Deurs. 1996. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 76949-966. [DOI] [PubMed] [Google Scholar]
- 51.Siegler, R. L. 1995. Hemolytic uremic syndrome in children. Curr. Opin. Pediatr. 7159-163. [DOI] [PubMed] [Google Scholar]
- 52.Siegler, R. L., T. J. Pysher, R. Lou, V. L. Tesh, and F. B. Taylor, Jr. 2001. Response to Shiga toxin-1, with and without lipopolysaccharide, in a primate model of hemolytic uremic syndrome. Am. J. Nephrol 21420-425. [DOI] [PubMed] [Google Scholar]
- 53.Simon, M., T. G. Cleary, J. D. Hernandez, and H. E. Abboud. 1998. Shiga toxin 1 elicits diverse biologic responses in mesangial cells. Kidney Int. 541117-1127. [DOI] [PubMed] [Google Scholar]
- 54.Smith, W. E., A. V. Kane, S. T. Campbell, D. W. Acheson, B. H. Cochran, and C. M. Thorpe. 2003. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 711497-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stricklett, P. K., A. K. Hughes, and D. E. Kohan. 2005. Inhibition of p38 mitogen-activated protein kinase ameliorates cytokine up-regulated shiga toxin-1 toxicity in human brain microvascular endothelial cells. J. Infect. Dis. 191461-471. [DOI] [PubMed] [Google Scholar]
- 56.Taylor, F. B., Jr., V. L. Tesh, L. DeBault, A. Li, A. C. Chang, S. D. Kosanke, T. J. Pysher, and R. L. Siegler. 1999. Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 1541285-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thompson, J. E., R. M. Cubbon, R. T. Cummings, L. S. Wicker, R. Frankshun, B. R. Cunningham, P. M. Cameron, P. T. Meinke, N. Liverton, Y. Weng, and J. A. DeMartino. 2002. Photochemical preparation of a pyridone containing tetracycle: a jak protein kinase inhibitor. Bioorg. Med. Chem. Lett. 121219-1223. [DOI] [PubMed] [Google Scholar]
- 58.Thorpe, C. M., B. P. Hurley, L. L. Lincicome, M. S. Jacewicz, G. T. Keusch, and D. W. Acheson. 1999. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 675985-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van de Kar, N. C., T. Kooistra, M. Vermeer, W. Lesslauer, L. A. Monnens, and V. W. van Hinsbergh. 1995. Tumor necrosis factor alpha induces endothelial galactosyl transferase activity and verocytotoxin receptors. Role of specific tumor necrosis factor receptors and protein kinase C. Blood 85734-743. [PubMed] [Google Scholar]
- 60.van de Kar, N. C., L. A. Monnens, M. A. Karmali, and V. W. van Hinsbergh. 1992. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood 802755-2764. [PubMed] [Google Scholar]
- 61.van de Kar, N. C., R. W. Sauerwein, P. N. Demacker, G. E. Grau, V. W. van Hinsbergh, and L. A. Monnens. 1995. Plasma cytokine levels in hemolytic uremic syndrome. Nephron 71309-313. [DOI] [PubMed] [Google Scholar]
- 62.van Setten, P. A., V. W. van Hinsbergh, T. J. van der Velden, N. C. van de Kar, M. Vermeer, J. D. Mahan, K. J. Assmann, L. P. van den Heuvel, and L. A. Monnens. 1997. Effects of TNF alpha on verocytotoxin cytotoxicity in purified human glomerular microvascular endothelial cells. Kidney Int. 511245-1256. [DOI] [PubMed] [Google Scholar]
- 63.Waddell, T., S. Head, M. Petric, A. Cohen, and C. Lingwood. 1988. Globotriosyl ceramide is specifically recognized by the Escherichia coli verocytotoxin 2. Biochem. Biophys. Res. Commun. 152674-679. [DOI] [PubMed] [Google Scholar]
- 64.Wajant, H., K. Pfizenmaier, and P. Scheurich. 2003. Tumor necrosis factor signaling. Cell Death Differ. 1045-65. [DOI] [PubMed] [Google Scholar]
- 65.Warnier, M., W. Romer, J. Geelen, J. Lesieur, M. Amessou, L. van den Heuvel, L. Monnens, and L. Johannes. 2006. Trafficking of Shiga toxin/Shiga-like toxin-1 in human glomerular microvascular endothelial cells and human mesangial cells. Kidney Int. 702085-2092. [DOI] [PubMed] [Google Scholar]
- 66.Yoshizumi, M., Y. Fujita, Y. Izawa, Y. Suzaki, M. Kyaw, N. Ali, K. Tsuchiya, S. Kagami, S. Yano, S. Sone, and T. Tamaki. 2004. Ebselen inhibits tumor necrosis factor-[alpha]-induced c-Jun N-terminal kinase activation and adhesion molecule expression in endothelial cells. Exp. Cell Res. 2921-10. [DOI] [PubMed] [Google Scholar]
- 67.Yukioka, H., and S. Kurita. 1997. Escherichia coli O157 infection disaster in Japan, 1996. Eur. J. Emerg Med. 4165. [DOI] [PubMed] [Google Scholar]
- 68.Zemunik, T., A. Markotic, and A. Marusic. 2004. Expression of neutral glycosphingolipids in cytokine-stimulated human endothelial cells. Biochemistry (Moscow) 69513-519. [DOI] [PubMed] [Google Scholar]
- 69.Zen, K., A. Karsan, T. Eunson, E. Yee, and J. M. Harlan. 1998. Lipopolysaccharide-induced NF-kappaB activation in human endothelial cells involves degradation of IkappaBalpha but not IkappaBbeta. Exp. Cell Res. 243425-433. [DOI] [PubMed] [Google Scholar]
- 70.Zoja, C., M. Morigi, and G. Remuzzi. 2001. The role of the endothelium in hemolytic uremic syndrome. J. Nephrol. 14(Suppl. 4)S58-S62. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.