

Abstract
Reaction of the Schiff-base complex [Co(acetylacetonate-ethylenediimine)(NH3)2]+ with metmyoglobin at pH 6.5 yields a partially folded protein containing six Co(III) complexes. Although half of its α-helical secondary structure is retained, absorption and CD spectra indicate that the tertiary structure in both B-F and AGH domains is disrupted in the partially folded protein. In analogy to proton-induced unfolding, it is likely that the loss of tertiary structure is triggered by metal-ion binding to histidines. Cobalt(III)-induced unfolding of myoglobin is unique in its selectivity (other proteins are unaffected) and in allowing the isolation of the partially folded macromolecule (the protein does not refold or aggregate upon removal of free denaturant).
Keywords: protein unfolding, cobalt complexes
Partially folded proteins are polypeptides with substantial secondary structure in a largely disordered tertiary structure (1–4). They are often referred to as “molten globules” (1, 4, 5). Because of increased flexibility, hydrophobic amino acid residues may become exposed in a molten globule, leading to a higher membrane affinity (as compared with a fully folded protein), an attribute that could be of importance in biological processes (1, 6). Aggregates formed in vivo from partially folded proteins contribute to a number of important human disease states, such as Alzheimer’s disease, other amyloidoses, and the prion diseases (7–9). So far, partially folded proteins have been obtained from their more stable folded precursors either under equilibrium conditions in nonphysiological denaturing media or as short-lived kinetic intermediates in rapid folding experiments (1), but, with the exception of protein aggregates (7–9), they have not been isolated.¶
The reaction between a cobalt(III) Schiff base complex, [Co(acetylacetonate-ethylenediimine)(NH3)2]† (1) (10, 11, 42), and metmyoglobin (metMb) yields a partially folded protein isolated in a biologically relevant medium.¶ Uniquely, most other proteins are not affected by 1 under the same conditions, demonstrating that this novel unfolding method can be highly selective. Mechanistic studies indicate that the irreversibility and selectivity of unfolding by 1 originate in the strong bond formed preferentially between cobalt and an imidazole nitrogen of a histidine. Our findings open the way for applications based on the unique properties of molten globules as toxin-like prodrugs (6). 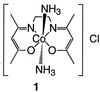
MATERIALS AND METHODS
Materials.
[Co(acacen)L2]Cl (acacen = acetylacetonate-ethylene-diimine; L = NH3, 1; L = imidazole, 2; L = 4-Me-imidazole, 3; L = N-Me-imidazole, 4; L = 2-Me-imidazole, 5; L = MeNH2, 6) complexes were prepared by literature methods (10). Horse skeletal muscle metMb and apoMb (Sigma) were of the highest purity available and were used as received.
Spectroscopy.
UV/vis absorption spectra were acquired on a Hewlett Packard HP 8452 diode array spectrophotometer. CD spectra were measured on an Aviv 62DS spectropolarimeter at 25°C (1-mm cell for far-UV CD; 10-mm cell for near-UV CD). Measurements were made on the incubation mixtures, without additional treatment, before dissociation of weakly bound Co(III) complexes. Note, however, that removal of free 1 by dialysis did not change the spectrum of the incubation product of metMb with 11 equivalents of 1. The 200-MHz 1H NMR spectrum (hemin region) of metMb (1 mM) with excess 2 ([2]0 = 12 mM) in 0.05M sodium phosphate buffer (pH 7.0) at 25°C was measured on a Bruker (Billerica, MA) AM200.
Unfolding Experiments.
Different amounts from a stock solution of the relevant cobalt complex in 0.01 M sodium phosphate buffer solution at pH 6.5 were added to a metMb or apoMb solution in the same medium. Volume corrections with pure buffer solution (so that a final reaction volume of 20 ml would be reached) were done before addition of the cobalt solution. The final protein concentration was 1⋅10− 5 M (except for determining the concentration dependence of the 222-nm CD signal on the protein concentration). Incubation was for 96 h at 22°C. The protein products were analyzed after dialysis of the free cobalt, unless otherwise mentioned. Dialysis at 4°C was either from bags (3,000-D cutoff) for 12 h against pure buffer solutions or from Centricon concentrators (3,000 D, Amicon).
Determination of the Cobalt-to-Protein Binding Ratio.
Final protein concentrations were determined by the Folin–Ciocalteu method (12). [1] after dialysis was determined by graphite furnace atomic absorption of cobalt [duplicate 20-μl samples were examined, by using argon as the inert gas; the absorbance was measured at 240.7 nm (0.2-nm slit, 1 s) on a Varian SpectrAA-20, equipped with PSC-56 and GTA-96]. Binding ratios are rounded to the nearest whole number.
Attempts to Reverse the Unfolding.
These experiments were carried out on 10−5-M solutions of the unfolded protein from which the free unfolding reagent was removed by dialysis. The reaction with excess dithionite was run for 3 h under argon before the UV/vis spectrum of the solution was recorded. The product was exposed to air, and the spectrum was remeasured. The reactions with excess imidazole (25, 200, and 105) were followed periodically by UV/vis absorption. The reaction with 25-fold excess hemin (solubilized in a minimum volume of a 0.01-M NaOH solution) was allowed to proceed at room temperature for 24 h. The solution was passed through a Sephadex G-25 column (1.5⋅20 cm) to separate the free hemin, and the absorption spectrum was acquired.
Ligand Exchange.
Reactions of complexes 4 and 5 with free imidazole occur in water. UV/vis absorptions of these reaction mixtures were monitored at three or four different wavelengths (per experiment) in which the absorbance difference between the starting compound and the expected product (prepared separately) was maximized. The Co(III) complex concentration was 10 μM, and the free ligand was in 25- and 200-fold excess. NMR spectral analysis of the initial and final solution established the identity of the products.
RESULTS AND DISCUSSION
Incubation of metMb with 1 leads to broadening, a strong decrease in the molar extinction coefficient and a blue shift of the Soret band in the absorption spectrum from 409 (ɛ = 1.7⋅105) to 392 nm (ɛ ≈ 5.9⋅104 M−1⋅cm−1) (Fig. 1). The intraligand π–π* band of 1 shifts from 334 to 340 nm, with no apparent change in extinction coefficient (ɛ334 = 7100 M−1⋅cm−1). A minimum Co(III)-to-protein ratio of 11 to 1 is required for the full spectroscopic transformation (Fig. 2A). The far UV CD shows a 50% decrease in the α-helical secondary structure of metMb (Fig. 2B)‖ that parallels the decrease in the Soret band (Fig. 2A). The loss of the near-UV CD (Fig. 2D) indicates diminished packing around the aromatic amino acids. At an initial 1:metMb ratio of 11:1, virtually no near-UV CD signal is observed.
Figure 1.

UV/vis absorption spectral changes during the incubation of metMb and 1 in 0.01 M sodium phosphate, at pH 6.5 and 25°C, with 15-min intervals between measurements. [metMb]0 = 10 μM, [1]0 = 0.18 mM.
Figure 2.

(A) Soret absorbance as a function of the initial concentration ratios of metMb and 1. (B and C) CD at 222 nm after incubating metMb with (B) 1 or (C) 2. (D) Near-UV CD after incubating metMb with 11 equivalents of 1. All spectra were taken after 96 h at 22°C in 0.01 M sodium phosphate at pH 6.5. [metMb]0 = 10 μM.
To verify that the loss of CD signal intensity loss is not related to protein aggregation in experiments conducted with 10 μM metMb or less, the dependence of the CD signal at 222 nm on the metMb concentration was determined (keeping [metMb]:[1] = 10 and other conditions as reported in Fig. 2). A linear correlation was obtained between 1 and 18 μM metMb (data not shown).
The reaction is irreversible in the sense that extensive dialysis leaves an average of six cobalt complexes associated with the protein and that the product retains 1 for prolonged periods in solution even in the absence of free 1. External reagents reverse the reaction. Excess dithionite reduces the hemin to ferroheme and causes cobalt dissociation from the protein [probably by reducing Co(III) to labile Co(II)]. Exposure to air regenerates metMb. A large excess of imidazole leads to slow, incomplete recovery of the Soret band. Excess hemin does not affect the product UV/vis spectrum.
ApoMb reacts with 1 much faster than metMb, and a smaller 1-to-apoMb ratio completes the transformation [≈6:1 1:apoMb (Fig. 3A)]. The product is identical with the one obtained from metMb and excess 1 [overlapping far-UV CD (Fig. 3B), with 6 Co(III) complexes bound to the protein]. The observation that these products are the same indicates that 1 causes hemin dissociation from the active site of metMb.** The similarity of the electronic spectrum of metMb after reaction with 1 with that of free hemin in a polar medium (15) also supports this conclusion.
Figure 3.

(A) CD at 222 nm after incubating apoMb (7.5 μM) with 1 and 2. (B) Far-UV CD after incubating metMb and apoMb with 11 equivalents of 1. Conditions as given in the Fig. 2 legend.
The far-UV CD and UV/vis spectral changes (Fig. 2 A and B) are similar to those obtained upon treatment of metMb with acid or low concentration guanidine⋅HCl (16–18), where it is well established that there is disruption of the tertiary structure in the B-F folding domain (19–21). Evidence for unfolding by 1 in this region is based on the fact that the active site is contained within this domain and includes our observation of hemin dissociation and the correlation between that dissociation (monitored by the Soret absorption) and the changes in the protein secondary structure (as indicated by [Θ]222) (Fig. 2 A and B).
Because the near-UV CD bands at λ > 268 nm are due to electronic transitions in tryptophans and tyrosines (22, 23), they probe the tertiary structure of horse myoglobin only in the AGH domain, where such residues are found (24). The disappearance of the near-UV CD features suggests that this domain loses its tertiary fold upon reaction with 1. In contrast, near-UV CD (25–27) and other studies (19–21) indicate that the AGH domain remains largely intact upon myoglobin unfolding by acid or low concentration guanidine⋅HCl.
The partially unfolded Co(III)-myoglobin can be described as a molten globule (1, 4, 5) because it retains much secondary structure but very little tertiary structure (Fig. 2D). Because all free denaturant can be removed, this is in every sense an isolated, kinetically stable, partially folded protein.
[Co(acacen)(imidazole)2]+ (2) reacts differently than 1 with metMb. Only one molecule of 2 binds each protein, causing no change in the Soret absorption and only a small change in the far-UV CD (Fig. 2C). The 4-methyl- and N-methyl-imidazole derivatives of 1 (complexes 3 and 4) behave as 2 toward metMb (only 1 cobalt binds), whereas the 2-methyl-imidazole and methylamine derivatives (complexes 5 and 6) react like 1 (6 cobalts bind to the protein). Nevertheless, 2 unfolds apoMb. The CD unfolding profile at 222 nm is similar to that of 1 (Fig. 3A), although only three cobalt complexes are bound to the isolated product.
Some of the imidazole liberated by the reaction of metMb with 2 binds at the active site, generating low spin imidazole-bound hemin in equilibrium with high spin water-bound hemin. This conclusion is based on the paramagnetic 1H-NMR spectrum of metMb with excess 2, which is a superposition of the spectra of native metMb and imidazole-bound metMb (data not shown). Removal of the imidazole by dialysis regenerates a pure high spin species.‡‡ When this product is reacted with excess 2, no low spin product is observed by 1H NMR because the cobalt binding site remains occupied after removal of unreacted 2. This evidence strongly suggests that the reaction of metMb with 2 involves cleavage of the cobalt-imidazole bond and replacement of this ligand by a protein side chain with ligating capabilities.
The small changes in wavelength (6-nm shift) and extinction coefficient of the π-π* absorption band of 1 (Fig. 1) indicate that it binds to a nitrogen donor of the protein. Although these values are similar for ammonia, alkylamines, pyridine, and substituted imidazoles as axial ligands (10, 11), they do change upon replacement of a nitrogen donor by an oxygen donor (28). Moreover, attempts to prepare [Co(acacen)(amino acid)2]+ complexes in which amino acids bind η1 through a carboxylate oxygen have failed, resulting in binding only to the amino group (29). Ligand exchange experiments demonstrate that 1 and 2 prefer histidines over lysines. Indeed, two imidazole equivalents replace both ammines of 1 in water at 25°C, whereas no reaction is observed between 2 and two equivalents of n-butylamine. Thus, we conclude that the protein ligand in the 1-Mb derivative is a histidine imidazole.
The kinetic lability of the Co(III) axial ligand is important in determining the reaction product with metMb. This is apparent upon comparing complexes 2, 3, 4, and 5. Ligand exchange of 5 with excess imidazole is much faster than the analogous reaction with 4 and is attributed to steric repulsion between the methyl group at the imidazole 2-position and the cis disposed Schiff base. Because 2, 3, and 4 bind only to one (or two, if bridging, see below) of the exposed histidines of metMb,†† it would appear that ligand exchange on the cobalt center of 2, 3, or 4 is assisted by interactions with the protein.§§ The fact that, during unfolding, apoMb binds six molecules of 1, but only three of 2, supports this suggestion.
Why do 1 and 5 unfold metMb, whereas other histidine modifiers {2, [Ru(NH3)5(H2O)]2+ (31) and [Ru(en)2(H2O)2]2+ (en = 1,2-ethylenediamine) (32)} do not? An intriguing explanation is that 1 and 5, which could dissociate sequentially both axial ligands, bind to two trans-disposed histidines.¶¶ Possible support for this suggestion is the observation that six (on average) molecules of 1 bind to unfolded myoglobin. Horse myoglobin has 11 histidines, therefore this number may reflect five pairs of cobalt-bridged histidines, plus a histidine that is bound predominantly to a nonbridging cobalt (interprotein linking by cobalt is also a possibility).
The interaction of Co(III) with one or more surface histidines is a likely initial step in the unfolding of metMb. The relative rates of unfolding by 1 are metMb ≪ apoMb, indicating that hemin dissociation is rate-determining. Hence, the parallel changes of the hemin absorption at 408 nm (Fig. 2A) and the backbone far-UV CD (Fig. 2B) imply that hemin dissociation precedes unfolding, as suggested for metMb unfolding under equilibrium conditions** (33). Barrick and Baldwin have proposed that two protons per protein unfold apoMb (34) by disrupting the structurally important His24 to His119 hydrogen bond (35). Like Co(III), Zn2+ and Cu2+ associate with the histidines of metMb, probably triggering partial unfolding (15, 36). It is reasonable, therefore, to propose that metal cations play a role similar to that of protons in their interactions with apoMb.
The unfolding of metMb by 1 is highly selective. Under the same reaction conditions, neither the absorption nor the far-UV CD spectra of cytochrome c, azurin, thrombin, and thermolysin (37) are affected by treatment with 1; and the catalytic activity of carbonic anhydrase is not diminished (37). Unlike metMb, these proteins do not possess enough accessible metal-ion binding sites, and they probably do not have histidines that are involved in structurally critical hydrogen bonds (unlike apoMb), thus explaining their markedly different behavior toward Co(III). By using suitably structured metal complexes, it should be possible to unfold proteins rich in other hydrogen bond-forming amino acids, in addition to those that possess a single hydrogen bond that is critical for tertiary structure stabilization.
Molten globules are superior to folded proteins in their ability to translocate across or insert into membranes (6) because they have increased affinity for hydrophobic surfaces (1). Therefore, an isolatable partially folded protein can be a powerful new type of pro-drug, functioning in a manner similar to that suggested for some bacterial toxins (6). Metal-ion-induced unfolding also may be used for selective protein precipitation, thereby aiding the formation of solid inclusion bodies, which could lead to improvements in large scale protein biosynthesis (8).
Acknowledgments
We thank Drs. Bassil Dahiyat and Michel E. Goldberg for discussions. O.B. acknowledges Rothchild and Fulbright postdoctoral fellowships. This work was supported by the National Science Foundation, the Arnold and Mabel Beckman Foundation, and the Redox Pharmaceutical Corporation.
ABBREVIATIONS
- metMb
oxidized form of the horse skeletal muscle myoglobin
- apoMb
horse skeletal muscle myoglobin without the heme group
- acacen
acetylacetonate-ethylene-diimine
- Me
methyl
Footnotes
Some proteins in their natural state may be partially folded (42). The Co(III)–myoglobin complex is a rare isolatable example of a partially folded species obtained from a naturally folded precursor.
Myoglobin unfolded by urea and guanidinium chloride retains virtually no helicity (13).
The dissociated hemin is not separated from the cobalt–myoglobin product by dialysis, as confirmed by iron atomic absorption measurements. It probably binds nonspecifically to the unfolded protein, as observed by Hargrove and Olson (14).
The position and intensity of the band at 340 nm confirm that the acacen ligand remains bound to cobalt. The π-π* transition of the uncomplexed ligand is at 323 nm (11).
As many as six surface histidines on metMb can be modified (30). Three [Ru(NH3)5]2+ complexes can be bound to metMb without perturbing the protein structure (31).
This assistance probably depends on neighboring side chain interactions with the cobalt complex. Protonation of the axial ligand by an acidic residue is a likely possibility.
Cobalt(III) Schiff-base complexes differ from other d6 complexes in their higher axial ligand labilities (29).
References
- 1.Ptitsyn O B. Adv Prot Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 2.Roder H, Colon W. Curr Opin Struct Biol. 1997;7:15–28. doi: 10.1016/s0959-440x(97)80004-8. [DOI] [PubMed] [Google Scholar]
- 3.Privalov P L. J Mol Biol. 1996;258:707–725. doi: 10.1006/jmbi.1996.0280. [DOI] [PubMed] [Google Scholar]
- 4.Ptitsyn O B. Nat Struct Biol. 1996;3:488–490. doi: 10.1038/nsb0696-488. [DOI] [PubMed] [Google Scholar]
- 5.Ewbank J J, Creighton T E, Hayer-Hartl M K, Hartl F U. Nat Struct Biol. 1995;2:10–11. doi: 10.1038/nsb0195-10a. [DOI] [PubMed] [Google Scholar]
- 6.van der Goot F G, Lakey J H, Pattus F. Trends Cell Biol. 1992;2:343–348. [PubMed] [Google Scholar]
- 7.Jaenicke R, Seckler R. Adv Prot Chem. 1997;50:1–59. doi: 10.1016/s0065-3233(08)60318-6. [DOI] [PubMed] [Google Scholar]
- 8.Betts S, Haase-Pettingell C, King J. Adv Prot Chem. 1997;50:243–264. doi: 10.1016/s0065-3233(08)60323-x. [DOI] [PubMed] [Google Scholar]
- 9.Fink A L. Folding Design. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 10.Böttcher A, Takeuchi T, Hardcastle K I, Meade T J, Gray H B, Cwikel D, Kapon M, Dori Z. Inorg Chem. 1997;36:2498–2504. [Google Scholar]
- 11.Costa G, Mestroni G, Tauzher G, Stefani L. J Organomet Chem. 1966;6:181–187. [Google Scholar]
- 12.Layne E. Methods Enzymol. 1975;3:447–454. [Google Scholar]
- 13.Nishii I, Kataoka M, Goto Y. J Mol Biol. 1995;250:223–228. doi: 10.1006/jmbi.1995.0373. [DOI] [PubMed] [Google Scholar]
- 14.Hargrove M S, Olson J S. Biochemistry. 1996;35:11310–11318. doi: 10.1021/bi9603736. [DOI] [PubMed] [Google Scholar]
- 15.Cann J R. Biochemistry. 1964;3:714–722. doi: 10.1021/bi00893a020. [DOI] [PubMed] [Google Scholar]
- 16.Puett D. J Biol Chem. 1973;248:4623–4634. [PubMed] [Google Scholar]
- 17.Bismuto E, Colona G, Irace G. Biochemistry. 1983;22:4165–4170. doi: 10.1021/bi00287a001. [DOI] [PubMed] [Google Scholar]
- 18.Palaniappan V, Bocian D F. Biochemistry. 1994;33:14264–14274. doi: 10.1021/bi00251a039. [DOI] [PubMed] [Google Scholar]
- 19.Hughson F M, Wright P E, Baldwin R L. Science. 1990;249:1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- 20.Jennings P A, Wright P E. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 21.Eliezer D, Yao J, Dyson H J, Wright P E. Nat Struct Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 22.Strickland E H. CRC Crit Rev Biochem. 1974;2:113–175. doi: 10.3109/10409237409105445. [DOI] [PubMed] [Google Scholar]
- 23.Sirangelo I, Bismuto E, Tavassi S, Irace G. Eur Biophys J. 1998;27:27–31. doi: 10.1007/s002490050107. [DOI] [PubMed] [Google Scholar]
- 24.Evans S V, Brayer G D. J Biol Chem. 1988;263:4263–4268. [PubMed] [Google Scholar]
- 25.Gast K, Damaschun H, Misselwitz R, Müller-Frohne M, Zirwer D, Damaschun G. Eur Biophys J. 1994;23:297–305. doi: 10.1007/BF00213579. [DOI] [PubMed] [Google Scholar]
- 26.Irace G, Bismuto E, Savy F, Colona G. Arch Biochem Biophys. 1986;244:459–469. doi: 10.1016/0003-9861(86)90614-4. [DOI] [PubMed] [Google Scholar]
- 27.Fink A L, Oberg K A, Seshadri S. Folding Design. 1998;3:19–25. doi: 10.1016/S1359-0278(98)00005-4. [DOI] [PubMed] [Google Scholar]
- 28.Sima J. Polish J Chem. 1994;68:1689–1697. [Google Scholar]
- 29.Fujii Y. Bull Chem Soc Jpn. 1972;45:3084–3092. [Google Scholar]
- 30.Konopka K, Waskell L. Biochim Biophys Acta. 1988;954:189–200. doi: 10.1016/0167-4838(88)90071-4. [DOI] [PubMed] [Google Scholar]
- 31.Toi H, La Mar G N, Margalit R, Che C-M, Gray H B. J Am Chem Soc. 1984;106:6213–6217. [Google Scholar]
- 32.Che C-M, Margalit R, Chiang H-J, Gray H B. Inorg Chim Acta. 1987;135:33–35. [Google Scholar]
- 33.Konermann L, Rosell F I, Mauk A G, Douglas D J. Biochemistry. 1997;36:6448–6454. doi: 10.1021/bi970353j. [DOI] [PubMed] [Google Scholar]
- 38.Barrick D, Baldwin R L. Biochemistry. 1993;32:3790–3796. doi: 10.1021/bi00065a035. [DOI] [PubMed] [Google Scholar]
- 39.Barrick D, Hughson F M, Baldwin R L. J Mol Biol. 1994;237:588–601. doi: 10.1006/jmbi.1994.1257. [DOI] [PubMed] [Google Scholar]
- 40.Hartzell C R, Hardman K D, Gillespie J M, Gurd F R N. J Biol Chem. 1967;242:47–53. [PubMed] [Google Scholar]
- 41.Takeuchi T. Ph.D. thesis. Pasadena, CA: California Institute of Technology; 1996. [Google Scholar]
- 42.Baskakov I, Bolen D W. J Biol Chem. 1998;273:4831–4834. doi: 10.1074/jbc.273.9.4831. [DOI] [PubMed] [Google Scholar]