

Abstract
During vesicular stomatitis virus (VSV) infection, host protein synthesis is inhibited, while synthesis of viral proteins increases. VSV infection causes inhibition of host transcription and RNA transport. Therefore, most host mRNAs in the cytoplasm of infected cells were synthesized before infection. However, viral mRNAs are synthesized throughout infection and are newer than preexisting host mRNAs. To determine if the timing of appearance of mRNAs in the cytoplasm affected their translation during VSV infection, we transfected reporter mRNAs into cells at various times relative to the time of infection and measured their rate of translation in mock- and VSV-infected cells. We found that translation of mRNAs transfected during infection was not inhibited but that translation of mRNAs transfected prior to infection was inhibited during VSV infection. Based on these data, we conclude that the timing of viral mRNA appearance in the cytoplasm is responsible, at least in part, for the preferential translation of VSV mRNAs. A time course measuring translation efficiencies of viral and host mRNAs showed that the translation efficiencies of viral mRNAs increased between 4 and 8 h postinfection, while translation efficiencies of host mRNAs decreased. The increased translation efficiency of viral mRNAs occurred in cells infected with an M protein mutant virus that is defective in host shutoff, demonstrating that the enhanced translation of viral mRNA is genetically separable from inhibition of translation of host mRNA.
Vesicular stomatitis virus (VSV) is a member of the rhabdovirus family and is widely studied as a model of negative-sense single-stranded RNA viruses. Like many negative-strand viruses, VSV replicates in the cytoplasm of infected cells, and viral mRNAs are transcribed from the viral genome by the viral RNA-dependent RNA polymerase (RDRP). VSV transcription produces five mRNAs that encode the five major viral proteins. These mRNAs are similar in structure to host mRNAs. Their 5′ ends contain 2′-O-methylated adenosine capped by 7-methyl guanosine linked by 5′-5′ triphosphate (22, 30, 31, 39, 40, 44). VSV mRNAs also have a 3′ poly(A) tail that is similar in length to that of cellular mRNAs (16, 19, 20). The synthesis of VSV mRNAs, including the 5′ and 3′ end modifications, is accomplished entirely in the cytoplasm by the viral RDRP (23).
Translation of VSV mRNAs, and of all viral mRNAs, is dependent on the host cell translation machinery. During virus infection, the cellular translation machinery is often modified, leading to a decrease in synthesis of host proteins, while viral protein synthesis increases. Many viruses, such as picornaviruses, influenza viruses, and VSV, are thought to inhibit host protein synthesis in order to suppress cellular antiviral responses (28). Viruses have developed a variety of mechanisms to inhibit host protein synthesis while viral mRNAs are preferentially translated. Understanding the mechanisms behind preferential translation of viral mRNAs is critical for understanding viral replication. Furthermore, cellular mechanisms controlling translation are often elucidated by studying translation during viral infection.
During VSV infection, host gene expression is rapidly inhibited by the matrix (M) protein. The M protein inhibits host gene expression at multiple levels, including transcription (1, 2, 4, 13), transport of mRNA to the cytoplasm (12, 17, 37, 38), and translation (2, 27, 32, 43). Previous experiments have shown that host translation is inhibited at the initiation step (7, 27) and is likely due to modification of the cap-binding eukaryotic initiation factor 4F (eIF4F) (8, 9, 11). However, it seems paradoxical that translation of host mRNAs would be inhibited while translation of viral mRNAs proceeds, since VSV mRNAs are structurally similar to host mRNAs. Yet in cells infected with VSV, as host protein synthesis is inhibited, viral protein synthesis becomes predominant (8, 9, 29, 32, 43, 45). The goal of the experiments presented here was to determine why viral mRNAs are translated during the time that translation of host mRNAs is inhibited.
Several viruses have been shown to allow preferential translation of viral mRNAs through the use of cis-acting sequences. For example, picornavirus mRNAs contain cis-acting internal ribosome entry sites that recruit the translation machinery (21, 34, 35). We showed previously that the preferential translation of VSV mRNAs is independent of cis-acting sequences in viral mRNA (46). The data presented here show that the timing of mRNA synthesis relative to infection is involved in controlling translation in VSV-infected cells. We found that in vitro-transcribed mRNA introduced into VSV-infected cells by transfection after infection was resistant to VSV-induced inhibition of translation, while translation of mRNA transfected into cells before infection was inhibited. This result indicates that the inhibition of host translation reflects the inhibition of translation of preexisting mRNAs together with the lack of production of new host mRNAs due to the inhibition of transcription and transport by M protein. In contrast to that of host mRNAs, the translation efficiency of mRNAs generated by the viral RDRP increased between 4 and 8 h postinfection. This increase in translation efficiency of viral mRNA also occurred in cells infected with an M protein mutant virus that is defective in the inhibition of host translation. This result indicates that inhibition of host translation is not essential for the efficient translation of viral mRNAs.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 7.5% fetal bovine serum (FBS). HeLa cells stably expressing enhanced green fluorescent protein (HeLa-EGFP) were generated by transfection with EGFP-N1 plasmid (Clontech) and selection in medium containing 600 μg/ml G418. Recombinant wild-type VSV (rwt virus), recombinant VSV expressing EGFP as a foreign gene (rwt-EGFP virus), and recombinant M51R-M protein mutant VSV (rM51R-M virus) were described previously (46). Recombinant M51R-M protein mutant VSV expressing EGFP as a foreign gene (rM51R-M-EGFP) was recovered from plasmid DNA, similar to rwt-EGFP virus (46). Virus stocks were prepared as described previously (24). Cells were infected in DMEM with 2% FBS at a multiplicity of infection of 10 PFU/cell to ensure synchronous infection.
Metabolic labeling.
Approximately 7 × 105 cells grown in six-well dishes were washed twice with DMEM without methionine and then incubated in DMEM without methionine for 0.5 h. Cells were pulse labeled for 1 hour in DMEM containing 200 μCi/ml [35S]methionine. Cells were then harvested for immunoprecipitation by adding 500 μl radioimmunoprecipitation assay (RIPA) buffer (0.15 M NaCl, 1% deoxycholic acid, 1% Triton X-100, 10 mM Tris-Cl, pH 7.4, and 0.1% sodium dodecyl sulfate [SDS]) with 1 mg/ml bovine serum albumin (BSA), 10 mM benzamidine, and 10 mM phenylmethylsulfonyl fluoride to each well. Plates containing RIPA buffer were rocked gently until cells were visibly lifted from the dish. Lysates were then centrifuged at 20,000 × g for 15 min at 4°C. For analysis of total protein synthesis, cells were harvested following pulse labeling, using 500 μl RIPA buffer without BSA, and 360 μl of cell extract was added to 40 μl of 10× SDS-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer. For analysis of total protein synthesis, 10 μl of lysate was electrophoresed in a 10 or 12% SDS-PAGE gel. Gels were dried and analyzed by phosphorimaging (Molecular Dynamics). Quantitation was performed using ImageQuant 5.2 (Molecular Dynamics).
Immunoprecipitation.
Immunoprecipitation of EGFP was performed by adding 3.8 μg goat anti-GFP (RDI) to 100 μl of cell lysate. Samples were incubated overnight at 4°C. Twenty microliters of protein G-Sepharose (Sigma) in NETN buffer (20 mM Tris-Cl, pH 8.0, 1 mM EDTA, 150 mM NaCl, 0.5% NP-40, and 4% BSA) was added and incubated for 1 h. Samples were centrifuged at 500 × g at 4°C, and pellets were washed five times with 400 μl of RIPA buffer with high SDS (1% SDS). Five microliters of SDS loading buffer was added to final pellets, and samples were heated to ∼95°C, separated in 10 or 12% SDS-PAGE gels, and analyzed as described above.
Northern blotting.
RNAs were harvested from 6 × 106 HeLa cells by using 3 ml of Trizol (Invitrogen) according to the manufacturer's specifications. Five micrograms of RNA harvested from stably transfected cells or 0.5 μg of RNA harvested from HeLa cells infected with rVSV-EGFP or rM51R-M-EGFP was run in a 1.2% glyoxal agarose gel. The gel was incubated in 800 ml of transfer buffer (0.01 M NaOH, 3 M NaCl) for 20 min and then transferred to a GeneScreen Plus (Perkin-Elmer) hybridization transfer membrane by upward capillary transfer (42). An [α-32P]dCTP-labeled EGFP probe was prepared using a Prime-a-Gene kit (Promega). Membranes were probed using ExpressHyb hybridization solution (BD Biosciences Clontech) according to the manufacturer's specifications. Membranes were analyzed by phosphorimaging as described above.
In vitro translation.
RNAs harvested at 8 h postinfection by using Trizol (Invitrogen) were used to direct translation in rabbit reticulocyte lysates (Promega). Reaction mixtures had a 0.1-ml total volume, with 27 μg total RNA or 1.6 μg of poly(A) RNA added. Poly(A) RNA was isolated from total RNA by using oligo(dT)-cellulose columns (Amersham Biosciences) and two rounds of purification. Translation reactions were carried out in a water bath at 30°C for 2 h and stopped by incubation on ice. Three microliters of each reaction mix was removed for analysis of all products of protein synthesis. Remaining volumes were diluted in 1.2 ml RIPA buffer plus 1 mg/ml BSA, 10 mM benzamidine, and 10 mM phenylmethylsulfonyl fluoride. EGFP was immunoprecipitated as described above.
In vitro transcription.
EGFP mRNA was transcribed in vitro using an mMessage mMachine SP6 kit (Ambion) according to the manufacturer's instructions, using the EGFP template plasmid pSD4.EGFP. Template DNA was linearized by cleavage with SalI restriction endonuclease and ethanol precipitated prior to transcription. The pSD4.EGFP plasmid directed transcription of EGFP mRNA with a 36-nucleotide (nt) 5′ untranslated region (UTR) and a 124-nt 3′ UTR that contained 66 nt of poly(A) sequence. Most of the mRNA produced by this transcription reaction is capped, with half of the 7mG(5′)ppp(5′)G cap structures in the proper orientation (25). The final RNA product was precipitated from the transcription reaction solution by using lithium chloride according to the manufacturer's instructions.
mRNA transfection.
mRNA was transfected into HeLa cells in six-well dishes. Transfections were done when the cell density was between 40 and 70% confluence, using a transit-mRNA transfection kit (Mirus Bio Corporation). Transfection mixtures were prepared with mRNA as described by the manufacturer and were added to cells over 1 ml of fresh growth medium (DMEM with 7.5% FBS). Infections of transfected cells were done by removing the transfection medium and immediately adding virus, suspended in DMEM with 7.5% FBS, to the wells.
RESULTS
During VSV infection, the inhibition of host transcription and mRNA transport prevents new host mRNAs from reaching the cytoplasm. During this time, viral transcription is the primary source of newly synthesized mRNAs. These events also coincide with the inhibition of host translation and the predominance of viral protein synthesis. These observations suggested that the timing of transcription may be involved in controlling translation in VSV-infected cells; specifically, the most recently synthesized mRNAs may be translated preferentially. We tested whether the time of appearance of mRNAs in the cytoplasm relative to the time of infection was involved in controlling translation by transfecting cells with in vitro-transcribed EGFP reporter mRNAs at various times relative to the time of infection.
The purpose of the experiments shown in Fig. 1 was to determine the time required after transfection for mRNA to enter the cytoplasm and direct translation. mRNA was synthesized in vitro, using SP6 RNA polymerase (SP6-EGFP) in the presence of a cap analog, from a vector that directed transcription of an mRNA with an EGFP open reading frame flanked by a short vector-derived 5′ UTR and a 3′ UTR containing a poly(A) tract. HeLa cells were transfected with SP6-EGFP mRNA, and translation rates at various times after transfection were determined by pulse labeling with [35S]methionine. EGFP synthesis was then analyzed by immunoprecipitating and measuring labeled EGFP by SDS-PAGE and phosphorimaging. Figure 1A shows a representative phosphorimage of EGFP synthesized at various times posttransfection. Figure 1B shows results from four experiments normalized to EGFP translation at 8 h posttransfection. At 2 h posttransfection, EGFP synthesis was already detectable. By 4 h posttransfection, EGFP synthesis had almost reached a maximum level, which was maintained throughout the time course. These results indicate that the minimum time required for transfected SP6-EGFP mRNA to be translated optimally is around 4 h.
FIG. 1.
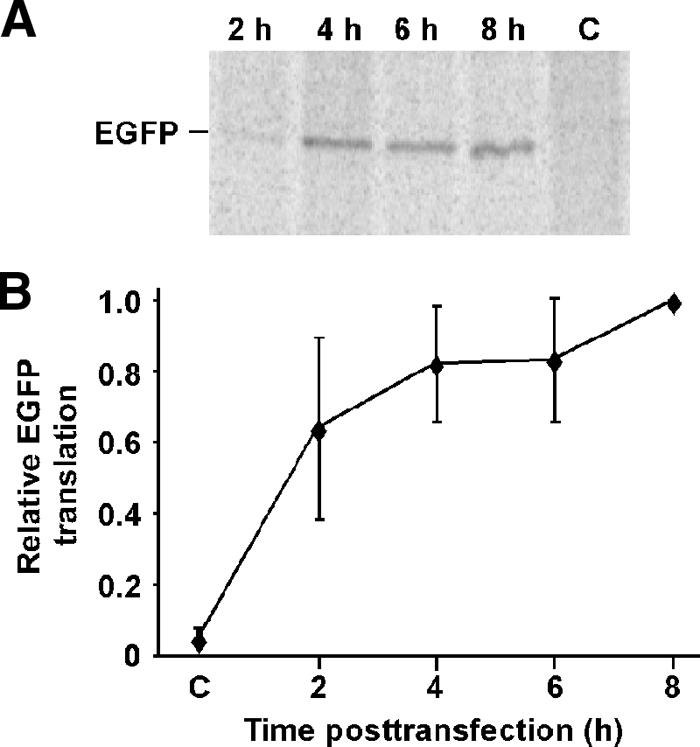
Time course of translation of transfected SP6-EGFP mRNA. (A) Phosphorimage of immunoprecipitated EGFP, analyzed by SDS-PAGE, from HeLa cells that were transfected with in vitro-transcribed EGFP mRNA (SP6-EGFP) and pulse labeled with [35S]methionine at various times after transfection. Control cells (C) were not transfected. (B) Quantification of EGFP labeling at various times after transfection of SP6-EGFP mRNA into HeLa cells. Data are expressed as fractions of the maximum at 8 h posttransfection and are means for four experiments (± standard errors [SE]).
To determine if newly transfected mRNAs were resistant to VSV-induced inhibition of translation, HeLa cells were transfected with SP6-EGFP mRNA at three different times relative to the time of infection. Figure 2A shows a timeline of the experiment. The time of infection or mock infection is the zero time point, and transfection times are shown relative to this time point. Cells were transfected with SP6-EGFP mRNA 22 h before infection, 4 h before infection, or 1 h after infection with a recombinant VSV encoding a wild-type M protein (rwt virus). At 5.5 h postinfection, cells were pulse labeled with [35S]methionine for 1 h. The time of the pulse was short relative to the rate of EGFP turnover, so the incorporation of the label reflected the rate of EGFP synthesis. The three time points for transfection were chosen based on the data in Fig. 1. mRNAs transfected 22 h before infection would have been in the translating polysomes for 14 to 18 h at the time of infection, similar to host mRNAs that are turned over slowly (15). mRNAs transfected 4 h before infection would be entering polysomes from about the time of infection until about 4 h postinfection. mRNAs transfected at 1 h postinfection would be entering polysomes from approximately 5 to 9 h postinfection, when viral protein synthesis is optimal. We did not attempt to transfect cells at later times postinfection due to concern that the viral cytopathic effects beginning around 4 h postinfection would alter the transfection efficiency.
FIG. 2.

EGFP mRNA transfected concurrent with infection is translated preferentially. (A) Experimental design for analyzing EGFP synthesis in HeLa cells transfected with SP6-EGFP mRNA at three different times relative to the time of infection, set as time zero. SP6-EGFP mRNA was transfected 22 h before infection, 4 h before infection, or 1 h after infection with rwt virus. (B) Phosphorimages of immunoprecipitated EGFP from cells transfected 22 h before infection, 4 h before infection, or 1 h after infection. Each time of transfection was performed as a separate experiment. (C) Phosphorimage of total cell lysates of cells that were transfected 22 h before infection or of untransfected cells that were subsequently mock infected or infected with rwt virus. Inhibition of total protein synthesis by rwt virus was similar for all three times of transfection. (D) EGFP synthesis or total host protein synthesis in rwt virus-infected cells relative to that in mock-infected cells that were transfected with SP6-EGFP at different times relative to the time of infection. Data shown are means ± SE for experiments performed on at least three different days for each time of transfection (−22 h, n = 4; −4 h, n = 6; 1 h, n = 5).
Translation rates of transfected SP6-EGFP mRNAs were determined by immunoprecipitating EGFP from cell lysates and analyzing it by SDS-PAGE and phosphorimaging (Fig. 2B). In mock-infected cells, EGFP synthesis was easily detected. However, in rwt virus-infected cells that were transfected 22 or 4 h before infection, EGFP was synthesized at much lower rates. The intense band for rwt virus-infected cell lysates that migrates slightly slower than EGFP is nonspecifically immunoprecipitated VSV M protein. This was shown previously by its precipitation in control samples, using an irrelevant antibody (46).
The key result in Fig. 2 is that when SP6-EGFP mRNA was transfected at 1 h postinfection, EGFP was synthesized to higher levels in rwt virus-infected cells than in mock-infected cells (Fig. 2B). These results show that mRNA transfected during infection is resistant to VSV-induced translation inhibition, while mRNA transfected before infection is translationally inhibited. In controls for the specificity of immunoprecipitation (Fig. 2B), there was a faint band from a nonspecific host protein that migrated similarly to EGFP in the untransfected cells that were mock infected. This band did not appear in the immunoprecipitation product from rwt virus-infected cells. Because of the nonspecifically immunoprecipitated protein, EGFP labeling for transfected cells that were mock infected was likely to be overestimated, while quantitation of the EGFP labeling intensity for rwt virus-infected cells transfected at 1 h postinfection was likely to be more accurate because the nonspecific protein was not labeled in these cells, while EGFP was labeled. Therefore, these results showing that SP6-EGFP mRNA is resistant to VSV-induced inhibition of translation when transfected around the time of infection are a conservative underestimate of this resistance.
As controls for the effect of transfection on the rates of translation of host and viral proteins, total cellular protein synthesis was also analyzed by SDS-PAGE and phosphorimaging (Fig. 2C). Transfection of SP6-EGFP mRNA slightly reduced levels of total protein synthesis compared to those of untransfected controls, independent of infection. Consequently, overall host translation was not reduced as dramatically during rwt virus infection of transfected cells as during infection of untransfected cells (Fig. 2C). Quantitation of host protein synthesis indicated that in transfected cells, rwt virus infection reduced host protein synthesis by around 60%, whereas in untransfected cells, rwt virus infection reduced translation by around 80%. Results for total host protein synthesis were similar for each transfection time. Transfection had little, if any, effect on viral protein synthesis.
Rates of EGFP synthesis from transfected SP6-EGFP mRNA, as well as total host protein synthesis, relative to those in mock-infected cells were determined by quantifying results from multiple experiments similar to the experiments shown in Fig. 2B and C (Fig. 2D). When cells were transfected 22 h before infection, translation of SP6-EGFP mRNA was reduced by rwt virus infection to less than half of the level in mock-infected cells, similar to the reduction of overall translation in these cells (Fig. 2D). When cells were transfected 4 h before infection, translation of SP6-EGFP mRNA was not inhibited as much as it was for mRNA transfected 22 h before infection or as much as inhibition of overall translation in these cells (Fig. 2D). However, when SP6-EGFP mRNA was transfected 1 h after infection, EGFP was translated almost twofold better in rwt virus-infected cells than in mock-infected cells. The translation rate of SP6-EGFP mRNA transfected 1 h after infection into rwt virus-infected cells relative to mock-infected cells was significantly different from that of overall protein synthesis (P = 0.007, using a two-sample t test). These data show that mRNAs introduced into the cytoplasm during infection or close to the time of infection are resistant to VSV-induced inhibition of translation.
Because VSV mRNAs turn over more rapidly than do host mRNAs (15, 36), there may be a continuous supply of newly synthesized viral mRNAs appearing in the cytoplasm of infected cells, which is resistant to the inhibition of translation. Alternatively, translation of VSV mRNAs may become inhibited at later times postinfection as VSV mRNAs become progressively older. To distinguish these possibilities, the translation efficiencies of host- and virus-derived mRNAs were measured over the period when translation of most host mRNAs is increasingly inhibited. Furthermore, we also analyzed cells infected with a recombinant M protein mutant virus (rM51R-M virus) defective in inhibiting host gene expression (2, 8). For these experiments, we used HeLa cells that stably express EGFP from transfected EGFP plasmid DNA (HeLa-EGFP cells) and recombinant viruses that express EGFP as a foreign gene: rwt-EGFP virus has a wild-type M protein, and rM51R-M-EGFP virus has a mutant M protein and is defective in inhibiting host gene expression.
Figure 3A shows the rates of EGFP synthesis in HeLa-EGFP cells infected with rwt or rM51R-M virus and in HeLa cells infected with rwt-EGFP or rM51R-M-EGFP virus expressing EGFP as a foreign gene. EGFP synthesis was measured by pulse labeling with [35S]methionine at 4, 8, or 12 h postinfection, followed by immunoprecipitation, SDS-PAGE, and phosphorimaging. Extracts containing virus-derived EGFP were diluted 1:20 prior to immunoprecipitation. A representative image obtained from duplicate cell cultures at 8 h postinfection is shown. Data from three separate experiments were quantified and are shown relative to the rate for mock-infected HeLa-EGFP control cells. EGFP translation in HeLa-EGFP cells was inhibited to a greater extent following infection with rwt virus than with rM51R-M virus (Fig. 3A, left panel), as expected based on previous studies with these viruses (2, 8).
FIG. 3.

Determination of translation efficiencies of host-derived and virus-derived EGFP mRNAs at different times after infection with virus containing wild-type or mutant M protein. (A) Rate of EGFP synthesis from host versus viral mRNA. HeLa-EGFP cells were mock infected or infected with rwt or M51R-M virus, and HeLa cells were infected with rwt-EGFP or M51R-M-EGFP virus. Cells were radiolabeled at 4, 8, or 12 h postinfection, and EGFP was immunoprecipitated and analyzed by SDS-PAGE and phosphorimaging. Extracts containing virus-derived EGFP were diluted 1:20 prior to immunoprecipitation. Each condition was analyzed in duplicate cell cultures. A representative image obtained at 8 h postinfection is shown. Data from three separate experiments were quantified and are shown relative to the rate for HeLa-EGFP mock-infected control cells (means ± SE) for HeLa-EGFP cells infected with rwt or M51R-M virus (left) and for HeLa cells infected with rwt-EGFP or rM51R-M-EGFP virus (right). (B) EGFP mRNA levels in HeLa-EGFP cells that were mock infected or infected with rwt or M51R-M virus and in HeLa cells infected with rwt-EGFP or M51R-M-EGFP virus. A representative Northern blot obtained from duplicate cell cultures at 8 h postinfection is shown. Extracts containing virus-derived EGFP mRNA were diluted 1:10 prior to Northern analysis. Data from three separate experiments were quantified and are shown relative to the mRNA level in HeLa-EGFP mock-infected control cells (means ± SE) for HeLa-EGFP cells infected with rwt or M51R-M virus (left) and for HeLa cells infected with rwt-EGFP or rM51R-M-EGFP virus (right). (C) EGFP translation efficiencies were calculated as protein synthesis (A) divided by the mRNA level (B) and are given relative to the efficiency in mock-infected HeLa-EGFP control cells.
While translation of host-derived EGFP mRNA was reduced during VSV infection, translation of virus-derived mRNA occurred at high levels (notice the difference in scale for the two graphs in Fig. 3A). In HeLa cells infected with rwt-EGFP virus, EGFP synthesis increased between 4 and 8 h postinfection but decreased by 12 h postinfection (Fig. 3A, right panel). In HeLa cells infected with rM51R-M-EGFP virus, synthesis of EGFP was similar to that in HeLa cells infected with rwt-EGFP virus at 4 and 8 h postinfection. However, at 12 h postinfection, EGFP synthesis remained high in cells infected with the recombinant M protein mutant virus expressing EGFP, in contrast to cells infected with rwt-EGFP virus (Fig. 3A, right panel).
EGFP mRNA levels were determined by Northern blotting. A representative Northern blot obtained from duplicate cultures at 8 h postinfection is shown in Fig. 3B. Extracts containing virus-derived EGFP mRNA were diluted 1:10 prior to Northern analysis. Data from three separate experiments were quantified and are shown relative to the mRNA levels in mock-infected HeLa-EGFP control cells and also expressed as ratios to the levels in mock-infected HeLa-EGFP cells (Fig. 3B). For HeLa-EGFP cells infected with rwt virus, the level of EGFP mRNA was not significantly different from the level observed in mock-infected cells until 12 h postinfection. At this time, the EGFP mRNA level in rwt virus-infected cells was reduced to 0.28 relative to that in mock-infected HeLa-EGFP control cells (Fig. 3B, left panel), likely due to decay of EGFP mRNA following inhibition of its synthesis by wt M protein. In HeLa-EGFP cells infected with rM51R-M virus, EGFP mRNA levels were not significantly different from the control level at any time point tested (Fig. 3B, left panel). In HeLa cells infected with recombinant viruses expressing EGFP as a foreign gene, EGFP mRNA levels were consistent throughout the time course, at levels 10- to 12-fold above the level in mock-infected HeLa-EGFP control cells (Fig. 3B, right panel).
Translation efficiencies were determined by dividing the EGFP translation rates (Fig. 3A) by the EGFP mRNA levels (Fig. 3B) and are shown in Fig. 3C as ratios to the translation efficiency in mock-infected HeLa-EGFP cells. Infection of HeLa-EGFP cells by rwt virus dramatically reduced the translation efficiency of EGFP, to 0.10 relative to the control, by 12 h postinfection. Infection by rM51R-M virus also reduced the translation efficiency of EGFP mRNA in HeLa-EGFP cells, but to a lesser extent, as expected based on previously reported data regarding translation in cells infected with these viruses (8, 9).
In contrast to host-derived EGFP mRNA, translation efficiencies of virus-derived EGFP mRNA increased during infection with both rwt-EGFP and rM51R-EGFP viruses (Fig. 3C). Translation efficiencies of EGFP mRNAs from the recombinant viruses at 4 h postinfection were similar to the translation efficiency of host-derived EGFP mRNA. However, the translation efficiencies of virus-derived EGFP mRNAs increased between 4 and 8 h postinfection, to around 2.0. Twelve hours after infection with rwt-EGFP virus, the translation efficiency of EGFP mRNA declined to 1.2 relative to that of the control (Fig. 3C). The EGFP translation efficiency in HeLa cells infected with rM51R-M-EGFP remained high throughout the time course (Fig. 3C). These results indicate that inhibition of host translation does not affect translation efficiency of viral mRNAs before 8 h postinfection and therefore separate the ability of VSV to inhibit host translation from its ability to promote translation of viral mRNAs.
In vitro translation assays were used to determine whether the increase in translation efficiencies of virus-derived mRNAs from 4 to 8 h postinfection was due to differences in viral mRNAs synthesized at different times postinfection (e.g., changes in cap structure, etc.). Total RNA from HeLa cells infected with EGFP-expressing viruses was added to rabbit reticulocyte lysate translation reaction mixtures so that the levels of EGFP mRNA were similar. Translation reactions were carried out in the presence of [35S]methionine, and EGFP was immunoprecipitated and analyzed by SDS-PAGE and phosphorimaging. A representative gel is shown in Fig. 4A. The EGFP mRNA level in each reaction mix was determined by Northern blotting (Fig. 4B), and relative translation efficiencies of EGFP mRNA in vitro (Fig. 4C) were determined by dividing the relative intensities of EGFP immunoprecipitated from in vitro translation reactions by the relative intensities of EGFP mRNA levels from Northern blotting.
FIG. 4.
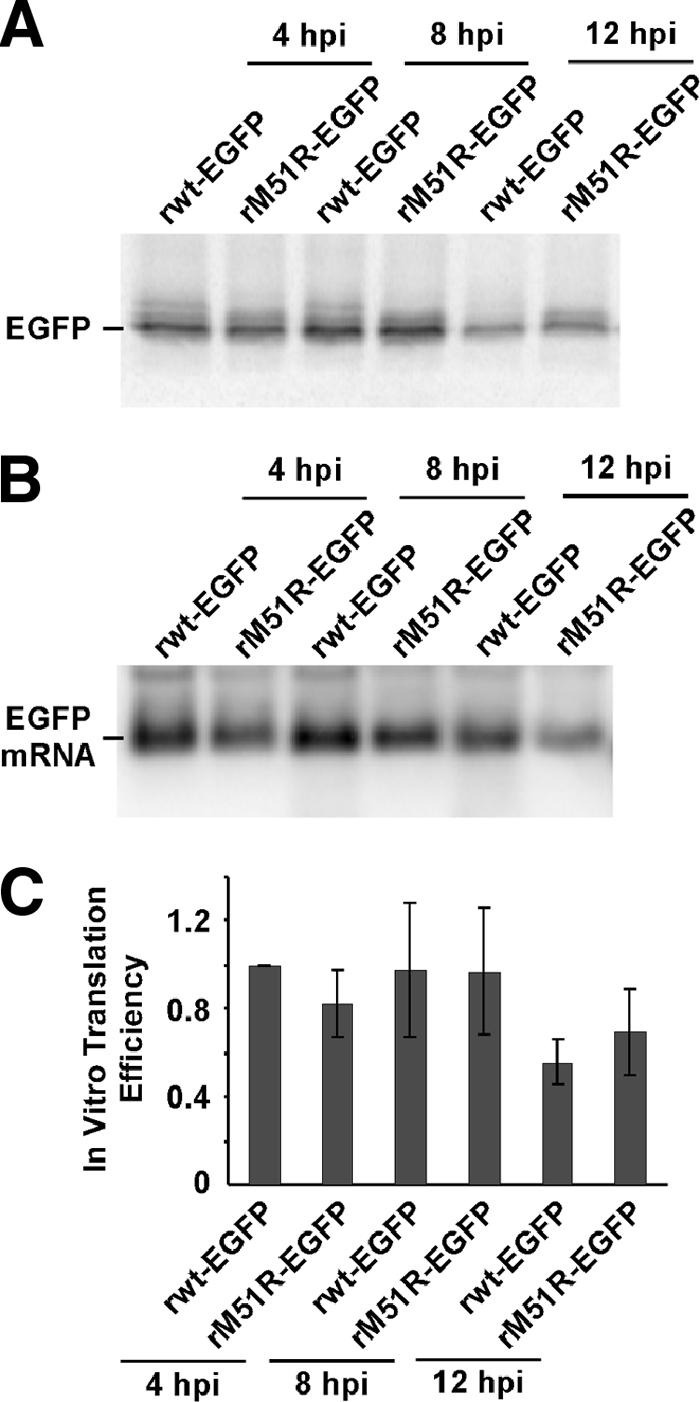
Virus-derived EGFP mRNA harvested at various times postinfection directs translation in vitro with similar efficiencies. (A) Phosphorimage of immunoprecipitated EGFP synthesized in rabbit reticulocytes programmed with total RNA harvested from HeLa cells infected with recombinant rwt-EGFP or M51R-M-EGFP virus at 4, 8, or 12 h postinfection (hpi). Translation reaction mixes also contained total RNA harvested from HeLa cells so that each reaction mix contained the same amount of total RNA. (B) Phosphorimage of Northern blot of EGFP mRNA samples like those used for in vitro translation reactions. (C) In vitro translation efficiencies of EGFP mRNAs relative to the efficiency of translation of EGFP mRNA harvested from HeLa cells infected with rwt-EGFP virus at 4 h postinfection.
EGFP mRNAs harvested from infected cells at various times postinfection were all translated with similar efficiencies in vitro. Based on these data, we concluded that the increase in translation efficiencies of virally derived EGFP mRNAs from 4 to 8 h postinfection was not due to differences in the mRNAs. Instead, these differences were the result of changes in the translation program of infected cells.
DISCUSSION
The predominance of viral protein synthesis in cells where host translation is inhibited is an important feature of infection displayed by many viruses. VSV infections are particularly striking in the extent to which viral protein synthesis predominates over the synthesis of host proteins. Mechanisms that viruses use to achieve preferential translation of viral mRNAs are varied but often rely on cis-acting sequences, such as the internal ribosome entry site sequences used by picornaviruses and the tripartite leader sequences of adenovirus late mRNAs. However, we showed previously that VSV mRNAs are preferentially translated through a mechanism that does not appear to involve cis-acting sequences (46). Instead, as shown here, the translation program of infected cells is altered such that the timing of transcription and appearance in the cytoplasm of VSV mRNAs allows them to be translated preferentially. Furthermore, the data presented here as well as data previously reported (10) demonstrate that the inhibition of host translation is genetically separable from the promotion of translation of viral mRNAs.
The resistance of newly appearing mRNAs to VSV-induced inhibition of translation was shown by transfecting mRNAs into cells at various times relative to the time of infection (Fig. 2). The resistance of SP6-EGFP mRNA transfected after infection to VSV-induced translation inhibition also confirms the sequence independence of efficient translation of VSV mRNAs. Because VSV mRNAs are also new to infected cells, these results imply that viral mRNAs are resistant to VSV-induced inhibition of translation. This resistance can account in part for the efficient translation of viral mRNAs relative to that of host mRNAs. Furthermore, viral mRNAs are turned over much more rapidly than most host mRNAs (half-life of 1.5 to 2 h versus 6 to 12 h) (15, 36) and are continuously replenished by the viral transcriptase throughout the time course of infection. Thus, there is a continuous supply of “new” mRNAs that are resistant to the mechanism of host shutoff. These data also imply that the inhibition of host translation is dependent on the inhibition of host transcription and mRNA transport by the VSV M protein. The ability of VSV to inhibit host mRNA transcription and transport has never been separated genetically from its ability to inhibit host translation. During infection with M protein mutant viruses defective in shutoff of host gene expression, the continued translation of host-derived mRNAs (Fig. 3) may be due to the failure to shut off host transcription and transport.
Figure 5 shows a model by which the translation program may be altered in infected cells, resulting in selective translation of newly introduced mRNAs. Previous data have shown that inhibition of translation of host mRNAs is due at least in part to alterations in the eIF4F cap-binding complex, reflected in the dephosphorylation of the cap-binding subunit eIF4E (8, 9, 11). According to this model, dephosphorylation of eIF4E (Fig. 5A) leads to inhibition of translation of preexisting host mRNAs (Fig. 5B). Reinitiation of translation is prevented by shuttling of these mRNAs into translationally inactive mRNPs (41, 46), where they are stably maintained in infected cells (Fig. 5C). While VSV M protein inhibits the production of newly synthesized host mRNA, viral transcription provides a continuous supply of newly synthesized viral mRNAs, which are translated by the reprogrammed translation apparatus (Fig. 5D). It might be expected that viral mRNAs would be less sensitive than host mRNAs to alterations in the eIF4F cap-binding complex because of their relatively short and unstructured 5′ UTRs. We have shown previously that extending the 5′ UTR of virally derived EGFP mRNA to a length typical of host mRNAs has little, if any, effect on translation (46). Nonetheless, it will be important to determine whether there is a difference between host and viral mRNAs in their dependence on eIF4F.
FIG. 5.

Model for preferential translation of newly appearing mRNAs during VSV infection. (A and B) Modification of translation machinery during VSV infection, indicated by dephosphorylation of eIF4E (A), leads to inhibition of translation of preexisting host mRNAs (B). (C) Reinitiation of translation is prevented by shuttling of these mRNAs into translationally inactive mRNPs. (D) Viral transcription provides a continuous supply of newly synthesized viral mRNAs, which are translated by the reprogrammed translation apparatus. (E) M protein enhances translation of viral mRNAs.
Our results also show that not only are viral mRNAs resistant to inhibition, but there is actually an increase in the translation efficiency of viral mRNAs between 4 and 8 h postinfection, the same time when translation of host mRNAs is inhibited (Fig. 3). Our results also showed an increased translation of SP6-EGFP mRNA transfected 1 h after VSV infection relative to that in mock-infected cells (Fig. 2), at a time when translation of host mRNAs is reduced. For virally derived mRNA, this increase in translation efficiency was seen with a virus containing wt M protein as well as an M protein mutant virus defective in inhibiting host gene expression. These results show that efficient translation of viral mRNAs or other newly appearing mRNAs is not dependent on inhibition of host translation. During infection with rwt-EGFP virus, host translation is inhibited and viral translation proceeds efficiently, at least until 8 h postinfection, but decreases between 8 and 12 h postinfection (Fig. 3C). This decrease in viral translation efficiency may be due to inhibition of the translation initiation factor eIF2α by phosphorylation, which has been shown to occur around this time (8). Alternatively, virally derived mRNAs may be subject to inhibition by shuttling to inactive mRNPs (Fig. 5C), similar to host-derived mRNAs at late times postinfection.
Our laboratory has previously described an M protein mutant virus with a complementary phenotype to that of rM51R-M mutant virus. This recombinant virus contains a replacement of four hydrophobic amino acids (φ) with polar amino acids in a surface-exposed loop in the M protein structure. This mutant virus shuts off host translation as effectively as does virus with a wild-type M protein but is defective in promoting or allowing translation of viral mRNAs (10). This result proves that there is a genetic dissociation of the abilities of M protein to shut off host translation and to promote viral translation (or translation of any newly appearing mRNAs). Furthermore, this result implicates the viral M protein in enhancing the translation of newly synthesized viral mRNAs (Fig. 5E).
The increase in translation efficiency of viral mRNAs between 4 and 8 h postinfection is not due to inherent changes in mRNA translatability over time. This was shown by measuring the in vitro translation efficiencies of viral mRNAs harvested at various times postinfection (Fig. 4). We also measured the translation efficiency of SP6-EGFP mRNA, which we found to be similar to that of virally derived EGFP mRNA (our unpublished data). We previously reported that the translatability in vitro of HeLa-EGFP mRNA from stably transfected cells was not altered as a result of infection with VSV (46), similar to previous results with host mRNAs (27). However, HeLa-EGFP mRNA was translated much less efficiently in vitro than was virally derived EGFP mRNA, by 24-fold, suggesting that there were cis-acting structural differences between host-derived and virally derived EGFP mRNAs (46). Because in vitro translation levels of viral and SP6-EGFP mRNAs were similar, we now think that there is something that interferes with in vitro translation of HeLa-EGFP mRNA. In support of this idea, we have found that HeLa cell-derived actin mRNA is translated similarly in vitro to SP6 in vitro-transcribed actin mRNA (our unpublished data).
Timing of mRNA entry into the cytoplasm may also play a role in regulating translation in cells infected with other viruses in addition to VSV. For example, analysis of translation in influenza virus-infected cells originally suggested that cis-acting viral sequences were important for the expression of reporter mRNAs expressed from plasmids in virus-infected cells (14, 33). However, Cassetti et al. showed that translation of mRNAs introduced into influenza virus-infected cells by transfection was not inhibited, regardless of the presence or absence of viral sequences (5). These results showed that cis-acting sequences are not required for translation during influenza virus infection. Instead, Cassetti et al. suggested that cis-acting sequences are required only for transport of mRNA to the cytoplasm during influenza virus infection. They concluded that the introduction of mRNA into the cytoplasm allowed preferential translation during influenza virus infection. In their study, the timing of introduction of mRNA into the cytoplasm was not addressed specifically. However, the timing of mRNA introduction into influenza virus-infected cells may be critical to its resistance to inhibition of translation.
The differential translation of newly appearing versus preexisting mRNAs also likely plays a role in the poliovirus-induced inhibition of host translation. Expression of the poliovirus 2A protease results in cleavage of the scaffolding subunit of the eIF4F complex (eIF4G) at different rates, depending on the eIF4G isoform present in the complex (eIF4GI > eIF4GII). Cleavage of eIF4GI preferentially inhibits translation of newly synthesized mRNAs, while cleavage of eIF4GII is required to inhibit translation of preexisting mRNAs (6). Likewise, cleavage of poly(A)-binding protein by poliovirus 3C protease in vitro preferentially inhibits the translation of newly initiated mRNAs but has little effect on ribosome recycling of mRNAs whose translation was initiated prior to cleavage (26).
The preference for newly synthesized mRNAs by the translation machinery is likely to be a normal phenomenon that is exploited by VSV and perhaps other viruses. For example, the shuttling of preexisting mRNAs to inactive mRNPs (41, 46) and the preferential translation of newly synthesized mRNAs (shown here) are reminiscent of the changes in mRNA distribution during certain types of stress responses, in which preexisting mRNAs are shuttled into stress granules while newly synthesized mRNAs are preferentially translated (3, 18). Addressing these similarities would help to define the mechanism that leads to preferential translation of newly transcribed mRNAs in VSV-infected cells.
Acknowledgments
We thank Maryam Ahmed and Elizabeth Pettit-Kneller for helpful advice and comments on the manuscript.
This work was supported by NIH grant RO1AI052304.
Footnotes
Published ahead of print on 19 December 2007.
REFERENCES
- 1.Ahmed, M., and D. S. Lyles. 1998. Effect of vesicular stomatitis virus matrix protein on transcription directed by host RNA polymerases I, II, and III. J. Virol. 728413-8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed, M., M. O. McKenzie, S. Puckett, M. Hojnacki, L. Poliquin, and D. S. Lyles. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 774646-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson, P., and N. Kedersha. 2006. RNA granules. J. Cell Biol. 172803-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black, B. L., and D. S. Lyles. 1992. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J. Virol. 664058-4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cassetti, M. C., D. L. Noah, G. T. Montelione, and R. M. Krug. 2001. Efficient translation of mRNAs in influenza A virus-infected cells is independent of the viral 5′ untranslated region. Virology 289180-185. [DOI] [PubMed] [Google Scholar]
- 6.Castello, A., E. Alvarez, and L. Carrasco. 2006. Differential cleavage of eIF4GI and eIF4GII in mammalian cells. Effects on translation. J. Biol. Chem. 28133206-33216. [DOI] [PubMed] [Google Scholar]
- 7.Centrella, M., and J. Lucas-Lenard. 1982. Regulation of protein synthesis in vesicular stomatitis virus-infected mouse L-929 cells by decreased protein synthesis initiation factor 2 activity. J. Virol. 41781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Connor, J. H., and D. S. Lyles. 2005. Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 28013512-13519. [DOI] [PubMed] [Google Scholar]
- 9.Connor, J. H., and D. S. Lyles. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 7610177-10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor, J. H., M. O. McKenzie, and D. S. Lyles. 2006. Role of residues 121 to 124 of vesicular stomatitis virus matrix protein in virus assembly and virus-host interaction. J. Virol. 803701-3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dratewka-Kos, E., I. Kiss, J. Lucas-Lenard, H. B. Mehta, C. L. Woodley, and A. J. Wahba. 1984. Catalytic utilization of eIF-2 and mRNA binding proteins are limiting in lysates from vesicular stomatitis virus infected L cells. Biochemistry 236184-6190. [DOI] [PubMed] [Google Scholar]
- 12.Faria, P. A., P. Chakraborty, A. Levay, G. N. Barber, H. J. Ezelle, J. Enninga, C. Arana, J. van Deursen, and B. M. Fontoura. 2005. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell 1793-102. [DOI] [PubMed] [Google Scholar]
- 13.Ferran, M. C., and J. M. Lucas-Lenard. 1997. The vesicular stomatitis virus matrix protein inhibits transcription from the human beta interferon promoter. J. Virol. 71371-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garfinkel, M. S., and M. G. Katze. 1993. Translational control by influenza virus. Selective translation is mediated by sequences within the viral mRNA 5′-untranslated region. J. Biol. Chem. 26822223-22226. [PubMed] [Google Scholar]
- 15.Goldstein, E. S., and S. Penman. 1973. Regulation of protein synthesis in mammalian cells. V. Further studies on the effect of actinomycin D on translation control in HeLa cells. J. Mol. Biol. 80243-254. [DOI] [PubMed] [Google Scholar]
- 16.Gupta, A. K., M. Mathur, and A. K. Banerjee. 2002. Unique capping activity of the recombinant RNA polymerase (L) of vesicular stomatitis virus: association of cellular capping enzyme with the L protein. Biochem. Biophys. Res. Commun. 293264-268. [DOI] [PubMed] [Google Scholar]
- 17.Her, L. S., E. Lund, and J. E. Dahlberg. 1997. Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science 2761845-1848. [DOI] [PubMed] [Google Scholar]
- 18.Holcik, M., and N. Sonenberg. 2005. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6318-327. [DOI] [PubMed] [Google Scholar]
- 19.Hunt, D. M. 1983. Vesicular stomatitis virus mutant with altered polyadenylic acid polymerase activity in vitro. J. Virol. 46788-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunt, D. M., E. F. Smith, and D. W. Buckley. 1984. Aberrant polyadenylation by a vesicular stomatitis virus mutant is due to an altered L protein. J. Virol. 52515-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang, S. K., H. G. Krausslich, M. J. Nicklin, G. M. Duke, A. C. Palmenberg, and E. Wimmer. 1988. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 622636-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keene, J. D., and R. A. Lazzarini. 1976. A comparison of the extents of methylation of vesicular stomatitis virus messenger RNA. Virology 69364-367. [DOI] [PubMed] [Google Scholar]
- 23.Knipe, D. M., P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.). 2007. Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 24.Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 7512169-12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krieg, P. A., and D. A. Melton. 1987. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzymol. 155397-415. [DOI] [PubMed] [Google Scholar]
- 26.Kuyumcu-Martinez, N. M., M. E. Van Eden, P. Younan, and R. E. Lloyd. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 241779-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodish, H. F., and M. Porter. 1980. Translational control of protein synthesis after infection by vesicular stomatitis virus. J. Virol. 36719-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyles, D. S. 2000. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev. 64709-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAllister, P. E., and R. R. Wagner. 1976. Differential inhibition of host protein synthesis in L cells infected with RNA-temperature-sensitive mutants of vesicular stomatitis virus. J. Virol. 18550-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moyer, S. A., G. Abraham, R. Adler, and A. K. Banerjee. 1975. Methylated and blocked 5′ termini in vesicular stomatitis virus in vivo mRNAs. Cell 559-67. [DOI] [PubMed] [Google Scholar]
- 31.Moyer, S. A., and A. K. Banerjee. 1976. In vivo methylation of vesicular stomatitis virus and its host-cell messenger RNA species. Virology 70339-351. [DOI] [PubMed] [Google Scholar]
- 32.Mudd, J. A., and D. F. Summers. 1970. Protein synthesis in vesicular stomatitis virus-infected HeLa cells. Virology 42328-340. [DOI] [PubMed] [Google Scholar]
- 33.Park, Y. W., and M. G. Katze. 1995. Translational control by influenza virus. Identification of cis-acting sequences and trans-acting factors which may regulate selective viral mRNA translation. J. Biol. Chem. 27028433-28439. [DOI] [PubMed] [Google Scholar]
- 34.Pelletier, J., G. Kaplan, V. R. Racaniello, and N. Sonenberg. 1988. Cap-independent translation of poliovirus mRNA is conferred by sequence elements within the 5′ noncoding region. Mol. Cell. Biol. 81103-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334320-325. [DOI] [PubMed] [Google Scholar]
- 36.Pennica, D., P. S. Cohen, and H. L. Ennis. 1981. Mechanism of vesicular stomatitis virus mRNA decay. Arch. Biochem. Biophys. 208403-408. [DOI] [PubMed] [Google Scholar]
- 37.Petersen, J. M., L. S. Her, and J. E. Dahlberg. 2001. Multiple vesiculoviral matrix proteins inhibit both nuclear export and import. Proc. Natl. Acad. Sci. USA 988590-8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen, J. M., L. S. Her, V. Varvel, E. Lund, and J. E. Dahlberg. 2000. The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol. Cell. Biol. 208590-8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rhodes, D. P., and A. K. Banerjee. 1975. 5′-Terminal sequence of vesicular stomatitis virus mRNAs synthesized in vitro. J. Virol. 1733-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhodes, D. P., S. A. Moyer, and A. K. Banerjee. 1974. In vitro synthesis of methylated messenger RNA by the virion-associated RNA polymerase of vesicular stomatitis virus. Cell 3327-333. [DOI] [PubMed] [Google Scholar]
- 41.Rosen, C. A., H. L. Ennis, and P. S. Cohen. 1982. Translational control of vesicular stomatitis virus protein synthesis: isolation of an mRNA-sequestering particle. J. Virol. 44932-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 43.Stanners, C. P., A. M. Francoeur, and T. Lam. 1977. Analysis of VSV mutant with attenuated cytopathogenicity: mutation in viral function, P, for inhibition of protein synthesis. Cell 11273-281. [DOI] [PubMed] [Google Scholar]
- 44.Toneguzzo, F., and H. P. Ghosh. 1976. Characterization and translation of methylated and unmethylated vesicular stomatitis virus mRNA synthesized in vitro by ribonucleoprotein particles from vesicular stomatitis virus-infected L cells. J. Virol. 17477-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wertz, G. W., and J. S. Youngner. 1972. Inhibition of protein synthesis in L cells infected with vesicular stomatitis virus. J. Virol. 985-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitlow, Z. W., J. H. Connor, and D. S. Lyles. 2006. Preferential translation of vesicular stomatitis virus mRNAs is conferred by transcription from the viral genome. J. Virol. 8011733-11742. [DOI] [PMC free article] [PubMed] [Google Scholar]