

Abstract
Herpes simplex virus type 1 (HSV-1) mutants that fail to express the viral immediate-early protein ICP0 have a pronounced defect in viral gene expression and plaque formation in limited-passage human fibroblasts. ICP0 is a RING finger E3 ubiquitin ligase that induces the degradation of several cellular proteins. PML, the organizer of cellular nuclear substructures known as PML nuclear bodies or ND10, is one of the most notable proteins that is targeted by ICP0. Depletion of PML from human fibroblasts increases ICP0-null mutant HSV-1 gene expression, but not to wild-type levels. In this study, we report that depletion of Sp100, another major ND10 protein, results in a similar increase in ICP0-null mutant gene expression and that simultaneous depletion of both proteins complements the mutant virus to a greater degree. Although chromatin assembly and modification undoubtedly play major roles in the regulation of HSV-1 infection, we found that inhibition of histone deacetylase activity with trichostatin A was unable to complement the defect of ICP0-null mutant HSV-1 in either normal or PML-depleted human fibroblasts. These data lend further weight to the hypothesis that ND10 play an important role in the regulation of HSV-1 gene expression.
Herpes simplex virus type 1 (HSV-1) is a common human pathogen that causes recurrent infections through its ability to establish a latent state in sensory ganglia after primary epithelial infections (for a general review, see reference 58). Lytic HSV-1 infection is characterized by abundant transcription from the entire viral genome in a temporal cascade of immediate-early (IE), early, and late gene products. The IE gene products regulate the expression of later classes of viral genes. In contrast, lytic cycle genes are repressed during latency, and only the latency-associated transcripts (LATs; derived from a single locus that lies countersense to the IE gene encoding ICP0) are expressed in readily detectable amounts (8, 55, 69). The IE protein ICP0 is a RING finger E3 ubiquitin ligase (4) that is required for efficient entry into the lytic cycle and which can induce reactivation of latent or quiescent genomes (reviewed in references 12, 14, 15, 29-31, and 55). ICP0 influences many cellular pathways, and one of its most prominent activities is its ability to localize to and disrupt nuclear substructures known as PML nuclear bodies (also known as ND10) (reviewed in references 10, 14, 16, and 43). This disruption occurs through ICP0-induced degradation of PML (17), the key component of ND10 which is required for assembly of these structures (34, 76). HSV-1 mutants that fail to express ICP0 or that express mutant ICP0 proteins that lack RING finger activity are unable to disrupt ND10 or to degrade PML (4, 11, 17, 44, 45). Such mutants have a profound defect in HSV-1 gene expression after infection of limited-passage human fibroblasts (9, 21, 62, 63).
The strong correlation between the effects of ICP0 on ND10 and its requirement for lytic virus infection prompted the hypothesis that ND10 might have a repressive effect on HSV-1 gene expression and thereby constitute an intrinsic antiviral defense. Although high-level expression of PML isoforms III, IV, and VI (6, 22, 42) did not impede HSV-1 infection, recent work established that depletion of PML from human fibroblasts increased the infectivity of ICP0-null mutant HSV-1 (20) and that of both wild-type (wt) human cytomegalovirus (HCMV) and a mutant HCMV deficient in IE72, which has functional similarities to ICP0 (65, 66). Furthermore, hDaxx, another major ND10 protein and an interaction partner of PML, has been found to be involved in repression of HCMV IE gene expression. The repressive effects of hDaxx are relieved by the HCMV tegument protein pp71 (5, 57, 59, 72). Sp100, yet another major ND10 component, has been implicated in repression of HSV-1 gene expression (48) and in the regulation of Epstein-Barr virus transcription (40). The accumulating evidence that several ND10 proteins are involved in the repression or regulation of viral gene expression lends support to the hypothesis outlined above. However, depletion of PML by no means completely eliminates the defect of ICP0-null mutant HSV-1 (20), suggesting that other factors must also be involved in the ICP0-null mutant phenotype in human fibroblasts. These could be connected with ND10 or could involve other cellular pathways, such as ICP0 interactions with histone deacetylase (HDAC) enzymes and their associated proteins, as suggested in a number of recent studies (25, 41, 53, 75).
This study set out to determine whether depletion of Sp100 either by itself or in combination with PML could influence the gene expression and plaque formation efficiencies of ICP0-null mutant HSV-1. We found that depletion of Sp100, like that of PML, improved ICP0-null mutant HSV-1 replication and that simultaneous depletion of both proteins further decreased repression of the mutant virus. Although simultaneous depletion of PML and Sp100 substantially increased the infectivity of ICP0-null mutant HSV-1, the mutant virus was not complemented to wt levels. Therefore, PML and Sp100 are both involved in the regulation of HSV-1 gene expression, but additional factors must be invoked to explain the full extent of the defect of ICP0-null mutant HSV-1 in human fibroblasts.
MATERIALS AND METHODS
Viruses and cells.
HSV-1 strain 17+ was the wt strain used, and the ICP0 null mutant dl1403 was derived from this strain. Viruses in1863 and dl1403/CMVlacZ are derivatives of the above that contain the lacZ gene under the control of the HCMV promoter/enhancer inserted into the tk gene (kindly provided by Chris Preston). HSV-1 mutant tsK includes a temperature-sensitive lesion in ICP4 (54), and mutant virus in1374 contains the tsK lesion, a deletion of the ICP0 gene, and a mutation within VP16 that inactivates its ability to stimulate IE gene expression (56). All viruses were grown in baby hamster kidney (BHK) cells and titrated in U2OS cells, in which ICP0 is not required for efficient replication of HSV-1. Viruses tsK and in1374 were propagated at the permissive temperature of 31°C, and the latter was grown in the presence of 2.5 mM hexamethylene bisacetamide (56). All viruses were used at multiplicities of infection (MOIs) based on their PFU titers in U2OS cells, regardless of the cell type used (15). U2OS cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. BHK cells were grown in Glasgow modified Eagle's medium supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. Primary human fibroblasts isolated from human foreskin tissue (HF cells; Department of Urology, University of Erlangen) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. HepaRG hepatocyte cells (23) were grown in William's medium E supplemented with 10% FBS Gold (PAA Laboratories Ltd.), 2 mM glutamine, 5 μg/ml insulin, and 0.5 μM hydrocortisone. All cell growth media were supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin. Lentivirus-transduced cells were maintained with continuous antibiotic selection, as appropriate.
Lentiviruses, transduction, and shRNA sequences.
HF cells were transduced with lentivirus vectors expressing either a control anti-luciferase short hairpin RNA (shRNA), anti-PML shRNA (19), or anti-Sp100 shRNA to produce the HF-shLuci, HF-shPML1, and HF-shSp100-2 cell lines, respectively. The anti-luciferase and anti-PML shRNA sequences were the same as those utilized to prepare the siLuci and siPML2 cells described previously (20, 65). Note that the siPML2 sequence of the 2006 studies is the same as that named shPML1 in this paper. The sense-strand DNA sequence of the anti-Sp100 shRNA was 5′-GTGAGCCTGTGATCAATAA, which corresponds to a sequence common to all Sp100 isoforms. The sequence starts within codon 350 and is preceded by the sequence AA. Processing of the transcribed shRNA results in production of a 21-bp small interfering RNA that is complementary to the above sequence and which has additional complementary U residues at its 3′ end, resulting from Pol II terminating within a poly(T) tract following the hairpin sequence. The shRNA sequence was built into double-stranded oligonucleotides (using a BD Biosciences design tool) for cloning into a lentivirus plasmid vector based on pLKO.1puro, from which lentivirus stocks were derived as described previously (19, 20). Transduced cells were selected with puromycin (initially used at 1 μg/ml and then reduced to 0.5 μg/ml during subsequent passages) and maintained in medium containing puromycin. Derivatives of the above plasmid vectors that expressed neomycin resistance were also constructed. Cells transduced with lentiviruses made from these plasmids were selected with G418 (initially used at 1 mg/ml and then reduced to 0.4 mg/ml during subsequent passages).
Plaque assays with transduced cell lines.
Cells were seeded into 24-well dishes at 1 × 105 cells per well and then infected the following day with appropriate sequential threefold dilutions of in1863 or dl1403/CMVlacZ. After virus adsorption, the cells were overlaid with medium containing 1% human serum, and then the cells were stained for β-galactosidase-positive plaques 24 h later, as described previously (35). Relative probabilities of plaque formation were calculated by comparing the numbers of plaques on the different cell lines at each separate dilution of virus. This approach overcomes the problem of the highly nonlinear nature (with respect to virus dilution) of plaque formation by ICP0-null mutant viruses in human fibroblasts (15, 62, 63).
Infections and Western blot analysis.
Cells were seeded into 24-well dishes at 1 × 105 cells per well and then infected the following day with wt HSV-1 or mutant dl1403, as stated in the relevant figure legend. Cell monolayers were washed twice with phosphate-buffered saline before being harvested in sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer. Proteins were resolved in 7.5% sodium dodecyl sulfate gels and then transferred to nitrocellulose membranes by Western blotting. ICP0, ICP4, UL42, and actin were detected using anti-ICP0 mouse monoclonal antibody (MAb) 11060, anti-ICP4 MAb 58S, anti-UL42 MAb Z1F11, and anti-actin MAb AC-40 (Sigma-Aldrich), as previously described (13). PML was detected with MAb 5E10 (64), and Sp100 was detected with rabbit serum SpGH (28).
Immunofluorescence and confocal microscopy.
Cells on 13-mm glass coverslips were infected with either wt or ICP0-null mutant HSV-1 at the chosen multiplicity and harvested at the times detailed in the figure legends. The cells were fixed and prepared for immunofluorescence. ICP4 was detected with MAb 58S, PML was detected with rabbit serum r8 or MAb 5E10, Sp100 was detected with rabbit serum SpGH or rat serum r26 (28), and hDaxx was detected with rabbit serum r1866 (52). The secondary antibodies used were fluorescein isothiocyanate-conjugated sheep anti-mouse immunoglobulin G (IgG; Sigma) or Cy3-conjugated goat anti-rabbit or goat anti-rat IgG (Amersham). The samples were examined using a Zeiss LSM 510 confocal microscope with 488-nm and 543-nm laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap. The images were exported as TIFF files and then processed using Photoshop.
RESULTS
ICP0-null mutant HSV-1 exhibits enhanced plaque formation and gene expression in Sp100-depleted human fibroblasts.
Previous work has established that depletion of PML from human fibroblasts increases the probability of plaque formation and enhances gene expression of ICP0-null mutant HSV-1 (20). These observations and parallel studies using HCMV add weight to the hypothesis that PML and ND10 contribute to an intrinsic cellular defense that represses herpesvirus gene expression (reviewed in references 13, 14, and 16). However, it is clear that PML cannot be the sole cellular factor involved in HSV-1 genome repression because the enhancement of ICP0-null mutant replication in PML-depleted cells is modest compared to that expected if repression were completely lifted (20). Accordingly, we set out to test whether other components of ND10 are also involved in HSV-1 genome repression in the absence of ICP0.
Sp100 is a major ND10 component that, like PML, is extensively modified by SUMO-1 (60, 61), is expressed from an interferon-inducible gene (24, 27), and is expressed as a series of multiple isoforms as a result of alternative splicing (28). The SUMO modification status of Sp100 is dependent on PML (20). Sp100-A is the most predominant isoform, while the longer isoforms (Sp100-B, -C, and -HMG) include a SAND domain (and an HMG domain in the case of Sp100-HMG) that is likely to be involved in DNA binding (3). Sp100-B is a repressor of gene expression in transfection reporter assays (71), and Sp100 isoforms B, C, and HMG (but not A) were found to repress HSV-1 IE gene expression and to be involved in interferon-mediated repression of HSV-1 gene expression through a mechanism that requires an intact SAND domain (48). To investigate further the role of Sp100 in HSV-1 infection of human fibroblasts, we constructed lentivirus vectors that express shRNAs that target sequences common to all alternatively spliced Sp100 transcripts. Preliminary experiments established two shRNA sequences that result in depleted Sp100 expression in transfected HeLa cells and showed that such depletion allows increased ICP0-null mutant gene expression in this cell type (data not shown). The more active anti-Sp100 shRNA sequence was inserted into a lentivirus vector to enable efficient transduction of limited-passage human fibroblasts and establishment of a cell line expressing this shRNA (HF-shSp100-2 cells). Control cells expressing an shRNA sequence corresponding to the luciferase gene were established in parallel (HF-shLuci cells). HF-shSp100-2 cells exhibit substantially reduced levels of all Sp100 isoforms, as analyzed by Western blotting (Fig. 1A) and by immunofluorescence (Fig. 2, row 2). Up to 90% of the cells exhibited levels of fluorescence staining of Sp100 that were not distinguishable from nonspecific background fluorescence.
FIG. 1.

Depletion of Sp100 from human fibroblasts allows increased plaque formation and gene expression by ICP0-null mutant HSV-1. (A) HF cells were transduced with lentiviruses expressing control and anti-Sp100 shRNAs to give cell lines HF-shLuci and HF-shSp100-2, and then cell extracts were analyzed for Sp100 expression by Western blotting using anti-Sp100 rabbit serum SpGH. The positions of the molecular weight markers and the likely identities of the major Sp100 isoforms are marked. (B) HF-shLuci and HF-shSp100-2 cells were infected with wt and ICP0-null mutant HSV-1 at low MOIs suitable for plaque assays (the starting MOI was approximately 0.01 for the wt and 1.0 for the mutant, and then five further sequential threefold dilutions were used), and then the relative probability of plaque formation at a given dilution of each virus in HF-shSp100-2 cells was calculated with respect to that in HF-shLuci cells. The data represent the means for six independent repeat experiments using two different isolates of the cell lines. The error bars show the standard errors of the means (SEM). (C) HF-shLuci and HF-shSp100-2 cells were infected with wt or ICP0-null mutant HSV-1 at an MOI of 2 PFU per cell, and then samples were harvested 4, 6, and 8 h after infection for analysis of expression of ICP4 and UL42 by Western blotting. The blots were also probed for actin as a loading control.
FIG. 2.
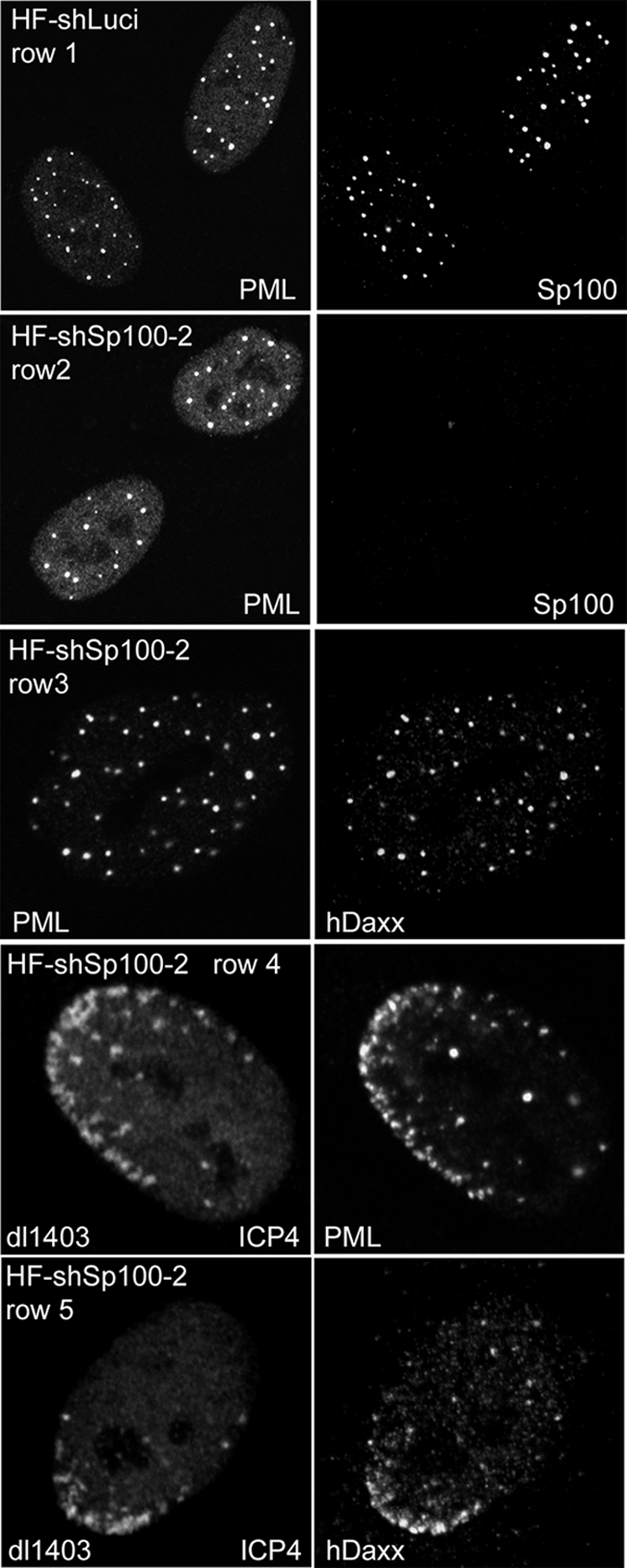
Depletion of Sp100 does not affect the localization of PML or hDaxx within ND10 or their relocalization to sites associated with HSV-1 genomes. Each row shows the two channels of the same field of view stained as indicated. Row 1, HF cells transduced with a control lentivirus stained for PML (left) and Sp100 (right); row 2, HF-Sp100-2 cells were analyzed in parallel and stained for PML (left) and Sp100 (right) (the images in both panels were captured under identical conditions); row 3, HF-Sp100-2 cells stained for PML (left) and hDaxx (right); rows 4 and 5, HF-Sp100-2 cells infected at a low MOI with ICP0-null mutant virus dl1403 and then stained the following day for ICP4 (left) and either PML (row 4) or hDaxx (row 5). Images of cells at the edges of plaques showing typical asymmetric foci of ICP4 in early replication compartments associated with either PML or hDaxx are illustrated.
HF-shLuci and HF-shSp100-2 cells were then used to determine the probability of plaque formation of wt and ICP0-null mutant HSV-1 strains containing a lacZ reporter gene. Subsequent staining of cells for β-galactosidase activity facilitated the identification of plaques (35). Because the plaque-forming efficiency of ICP0-null mutant HSV-1 is highly nonlinear with respect to virus dilution in human fibroblasts, threefold dilutions of virus were used, and the numbers of plaques at each dilution were compared between the two cell lines. This method of analysis gives relative probabilities of plaque formation at a given virus dose and therefore provides a more accurate comparison than does estimation of apparent virus titer, since the latter varies considerably with virus dilution. The experiment was repeated several times with independently derived pairs of cell lines. The results indicate a clear increase in the probability of ICP0-null mutant HSV-1 plaque formation, on the order of fivefold, in HF-shSp100-2 cells, whereas wt HSV-1 plaque formation is very similar in the two cell types (Fig. 1B).
HSV-1 gene expression was analyzed in the two cell types by infecting them with wt and ICP0-null mutant viruses at an MOI of 2 and then harvesting samples 4, 6, and 8 h after virus adsorption. The levels of ICP4 and UL42 expression (typical representatives of the IE and E classes of HSV-1 proteins) were similar after wt virus infection of the two cell lines, whereas their expression from the ICP0-null mutant virus was enhanced in the Sp100-depleted cells (Fig. 1C). These data are consistent with previous work (48) and indicate that the presence of endogenous Sp100 isoforms decreases the efficiency of ICP0-null mutant HSV-1 gene expression in cells in which the ICP0-null mutant phenotype is most pronounced.
Depletion of Sp100 does not affect the localization of other major ND10 components or their relocalization to sites associated with HSV-1 genomes.
It has been established that components of ND10, such as PML, Sp100, and hDaxx, relocate to establish novel ND10-like structures that are in close association with HSV-1 genomes soon after they enter the nucleus. In a wt virus infection, this process is very short-lived because expression of ICP0 leads to degradation of PML, a loss of SUMO-modified Sp100 isoforms, and dispersal of the other ND10 proteins. However, this recruitment is far more pronounced in the absence of ICP0 because PML remains stable (18, 21). Surprisingly, despite PML being required for normal ND10 assembly in uninfected cells, the ND10 proteins Sp100 and hDaxx are recruited efficiently into ND10-like foci associated with ICP0-null mutant HSV-1 genomes during infection of PML-depleted cells (20). We found that Sp100 is not required for the localization of hDaxx into ND10 in uninfected cells (Fig. 2, compare rows 1 and 3). Using the technique of detecting the locations of HSV-1 genomes and early replication compartments by staining for the viral transcriptional regulator ICP4, we observed that Sp100 was not required for the recruitment of either PML or hDaxx into the ND10-like foci that form in association with ICP4-defined viral nucleoprotein complexes (Fig. 2, rows 4 and 5).
Simultaneous depletion of both PML and Sp100 from human fibroblasts significantly enhances the replication efficiency of ICP0-null HSV-1.
Depletion of either PML (20) or Sp100 (Fig. 1) independently increases the fitness of ICP0-null mutant HSV-1, but in both cases the increases are modest compared to the full extent of the defects of such viruses (increases on the order of 5-fold in both cases, compared to a total defect on the order of 1,000-fold in HF cells). Therefore, we investigated whether simultaneous depletion of both proteins further increases the probability of plaque formation by the mutant virus. Cells were first depleted of Sp100, and then these cells were transduced a second time with lentiviruses expressing a different selectable marker and either shLuci or shPML-1 shRNA to create HF-shS/L and HF-shS/P cells. In parallel, the control HF-shLuci cells were transduced with a lentivirus expressing the same control and anti-PML shRNAs (HF-shL/L and HF-shL/P cells). It proved to be difficult to establish cell lines in which both PML and Sp100 were depleted to the extents possible in the single-transduction cell lines. Because depletion of PML causes redistribution of Sp100 into a more diffuse localization pattern of reduced fluorescence intensity, it was also difficult to determine precisely the proportion of cells extensively depleted of both proteins. Several independent batches of shS/P cells were produced, with estimated proportions of doubly depleted cells varying from 25% to 70%.
HF-shL/L, HF-shS/L, HF-shL/P, and HF-shS/P cells were infected with wt and ICP0-null mutant HSV-1 to determine the relative probabilities of plaque formation. While plaque formation by the wt virus was equally efficient in the four cell lines, there was an enhancement on the order of 15-fold in the probability of plaque formation by the ICP0-null mutant at a given virus input in shS/P cells (Fig. 3). This increase was a further three- to fivefold greater than that in parallel cells depleted individually of PML or Sp100. Despite this improved probability of plaque formation, simultaneous depletion of both PML and Sp100 does not eliminate the ICP0-null mutant defect, since this is on the order of 1,000-fold in parental HF cells.
FIG. 3.

HF cells depleted of both PML and Sp100 allow a greater probability of plaque formation by ICP0-null mutant HSV-1 than do cells depleted of either protein alone. HF cells doubly transduced with lentiviruses expressing anti-luciferase control shRNAs (shL/L) or combinations of control and anti-Sp100 or anti-PML shRNAs (shL/S and shL/P) or both anti-Sp100 and anti-PML shRNAs (shS/P) were isolated using lentiviruses with either neomycin or puromycin selection cassettes. The cells were used to determine the probability of plaque formation by either wt (in1863) or ICP0-null mutant (dl1403/CMVlacZ) virus by staining for β-galactosidase activity 24 h after low-MOI infection. The experimental details of the MOIs and dilutions used were similar to those outlined in the legend to Fig. 1. The error bars represent SEM.
Depletion of either PML or Sp100 increases ICP0-null mutant HSV-1 replication efficiency in a human hepatocyte cell line.
Although we found that human fibroblasts depleted of both PML and Sp100 were routinely more permissive for ICP0-null mutant HSV-1 infection than cells depleted of either protein alone, the extent of the increase varied between independent sets of isolated cells. This may have been related to the difficultly of establishing cell lines highly depleted of both proteins. Therefore, we sought a different cell system with which to test the conclusions arising from the work with HF cells. We found that HepaRG cells were ideally suited for this purpose. HepaRG cells have an epithelial phenotype and were derived from the liver of a patient suffering from hepatocarcinoma (23). They have few chromosomal abnormalities and retain the ability to differentiate into cells with most of the markers of normal hepatocytes and are therefore similar in many respects to primary human hepatocytes (23). Initially, HepaRG cells were transfected with plasmids expressing the shLuci, shPML-1, and shSp100-2 shRNAs, and individual clones were isolated after puromycin selection. Two independent (each) PML- and Sp100-depleted clones (HAP1, HAP2, HAS9, and HAS10) were analyzed, along with the parental HepaRG cells and a control shLuci cell line (HAL7). Western blotting confirmed that the cells were depleted of either PML or Sp100 as expected (Fig. 4A). As in the case of human fibroblasts (20), depletion of PML caused the loss of the likely SUMO-modified form Sp100-A (Fig. 4A).
FIG. 4.

Effects of simultaneous or individual depletion of PML and Sp100 on HSV-1 infection in nontransformed human hepatocytes. Individual clones of HepaRG cells expressing shRNAs targeting luciferase (HAL7), PML (HAP1 and HAP2), or Sp100 (HAS9 and HAS10) were isolated. Cells depleted of both PML and Sp100 (HAP1/shS2 and HAS10/shP1) were derived by lentivirus transduction of HAP1 and HAS10 cells, respectively. (A) Western blot analysis of PML, Sp100, and an actin loading control in HAL7, HAP1, HAS10, and HAP1/shS2 cells. The various isoforms of PML and Sp100 are marked. The filter was probed sequentially for the two proteins. (B to D) The relative probability of plaque formation by either wt (in1863) or ICP0-null mutant (dl1403/CMVlacZ) virus in the complete set of cell lines was tested by staining for β-galactosidase activity 24 h after low-MOI infection. Panel C shows the plaque formation probabilities of the ICP0-null mutant virus in the cell lines depleted individually of PML or Sp100. The data in panel C are repeated in panel D, which also includes the data from plaque assays with cells depleted of both PML and Sp100. HepA, naïve HepaRG cells. The experimental details of MOIs and dilutions used were similar to those outlined in the legend to Fig. 1. The error bars represent SEM.
Plaque assays indicated that the probability of plaque formation of the ICP0-null mutant HSV-1 was increased by depletion of either PML or Sp100 from HepaRG cells (Fig. 4C), while that of the wt virus was unaffected (Fig. 4B). The two independent clones of the PML- and Sp100-depleted cells gave similar results. Western blot analysis of infected HepaRG, HAL7, HALP1, and HALS10 cells confirmed the expected increase in ICP0-null mutant but not wt HSV-1 gene expression (Fig. 5).
FIG. 5.

Western blot analysis of wt and ICP0-null mutant HSV-1 infection of HepaRG, HAL7, HAP1, and HAS10 cells. Cells were infected with wt HSV-1 strain 17 (top) or ICP0-null mutant dl1403 (bottom) at an MOI of 2 PFU per cell in both cases. Samples harvested at the indicated times were analyzed by Western blotting for ICP4, UL42, actin, and in the case of the wt virus, ICP0. m, control mock-infected lanes; hpi, hours post-virus adsorption.
Simultaneous depletion of both PML and Sp100 from human hepatocytes significantly enhances the probability of plaque formation by ICP0-null HSV-1.
HAP1 and HAS10 cells were transduced with lentiviruses expressing a different selectable marker and the shSp100-2 and shPML-1 shRNAs, respectively, to create cells depleted of both proteins (HAP1/shS2 and HAS10/shP1 cells). In contrast to the case for the experiments using human fibroblasts, populations of cells highly depleted of both proteins could be isolated quite easily (Fig. 4A). Immunofluorescence analysis indicated that >90% of the cells were highly depleted of both proteins, in that the fluorescence staining intensity was diffuse and no higher than the background (data not shown).
Plaque assays with these cells revealed a further increase in the probability of plaque formation of the ICP0-null mutant over that seen in the singly depleted cells (Fig. 4D), while having no effect on wt HSV-1 plaque formation (Fig. 4B). The extent of this increase was such that it was frequently not possible to compare the numbers of plaques on the control and doubly depleted cells at a given virus dilution because those giving a reliable number of plaques on the control cells resulted in far too many plaques to count on the depleted cells. Therefore, the data in Fig. 4D were calculated on the basis of the further increases in plaque numbers in the doubly depleted cells over their singly depleted parents.
To eliminate the possibility that the results were influenced by the use of clonal cell lines, the experiments were repeated by making mixed cell populations depleted of either PML, Sp100, or both by using lentivirus transduction at both stages. In this case, all singly transduced cells (HALL, HALP1, and HALS2 cells [cells transduced with lentiviruses expressing shLuci, shPML-1, and shSp100-2 shRNAs, respectively]) were transduced a second time with lentiviruses expressing a different selectable marker and each of the shRNAs. These second-generation cells were named HALL/L, HALP1/L, HALS2/L, HALP1/S2, and HALS2/P1 cells. Western blot analysis confirmed the expected high levels of depletion of either PML, Sp100, or both, depending on the presence of the shPML-1 and/or shSp100-2 shRNA (Fig. 6A). Plaque assays indicated that while the probabilities of plaque formation by the wt virus were similar for all cell lines (Fig. 6B), those of the ICP0-null mutant were enhanced in the singly depleted cells, to similar extents to those in the clonal cell lines used previously (Fig. 6C). Again, there was a further enhancement of plaque formation probability in the doubly depleted cells (Fig. 6C). Western blot experiments demonstrated that wt HSV-1 gene expression was equivalent in HepaRG, HALL, HALP1, and HALP1/S2 cells, while ICP0-null mutant HSV-1 exhibited enhanced UL42 expression in HALP1 cells compared to that in the two control cell lines, and this was greater still in HALP1/S2 cells (Fig. 7).
FIG. 6.

Effects of simultaneous or individual depletion of PML and Sp100 on HSV-1 infection in nontransformed human hepatocytes. HepaRG cells transduced with lentiviruses expressing shRNAs targeting luciferase (HALL), PML (HALP1), or Sp100 (HALS2) were isolated. These cell lines were then transduced a second time with lentiviruses with a different selectable marker expressing the same shRNAs to give cell lines HALL/L, HALP1/L, HALS2/L, HALP1/S2, and HALS2/P1 (L, S2, and P1 indicate shRNAs Luci, shSp100-2, and shPML1, respectively). (A) Western blot analysis of PML, Sp100, and an actin loading control in HepaRG, HALL, HALP1, HALS2, HALP1/S2, and HALS2/P1 cells. The various isoforms of PML and Sp100 are marked. The filter was probed sequentially for the two proteins. (B and C) The relative probability of plaque formation by either wt (in1863) (B) or ICP0-null mutant (dl1403/ CMVlacZ) (C) virus in the complete set of cell lines was tested by staining for β-galactosidase activity 24 h after low-MOI infection. HepA, naïve HepaRG cells. The experimental details of MOIs and dilutions used were similar to those outlined in the legend to Fig. 1. The error bars represent SEM.
FIG. 7.

Western blot analysis of wt and ICP0-null mutant HSV-1 infection of HepaRG, HALL, HALP1, and HALP1/S2 cells. Cells were infected with wt HSV-1 strain 17 (top) or ICP0-null mutant dl1403 (bottom) at an MOI of 5 PFU per cell in both cases. Samples harvested at the indicated times were analyzed by Western blotting for ICP4, ICP0, and UL42. 0, control mock-infected lanes; hpi, hours post-virus adsorption.
Repression of the genome of mutant HSV-1 that fails to express active VP16, ICP0, and ICP4 is less efficient in hepatocytes depleted of both PML and Sp100.
HSV-1 mutants that lack the IE gene transactivation function of VP16, express a severely truncated form of ICP0, and include a temperature-sensitive (ts) allele of ICP4 are highly susceptible to cell-mediated repression following infection of human fibroblasts (56). Such viruses, exemplified by mutant in1374, can be used to infect cells at a relatively high MOI (3 PFU per cell), such that a large proportion of cells receive one or more viral genomes but only a negligible proportion exhibit detectable levels of viral gene expression by 24 h after infection (56). Such expression is most conveniently detected through the HCMV-driven lacZ marker gene in in1374. We recently demonstrated that compared to control cells, a higher proportion of PML-depleted fibroblasts failed to repress in1374 expression 24 h after infection, although repression still occurred in the majority of PML-depleted cells (19). We investigated whether in1374 is similarly repressed in hepatocytes and whether depletion of PML, Sp100, or both alters the efficiency of repression of the defective viral genome.
The HAL series of cells was infected with in1374 at an MOI of 3, in either the presence or absence of virus tsK (MOI of 2), and then the cells were incubated at 38.5°C for 24 h before being stained for β-galactosidase activity. Virus tsK expresses wt ICP0 and the same ts allele of ICP4 as that in in1374, thereby disabling cell-mediated repression of viral gene expression and allowing expression of the marker lacZ gene. Therefore, the coinfection experiment indicates the total number of cells that have been infected with in1374 (56). We found that, as in human fibroblasts, repression of in1374 was very efficient in both parental and control hepatocytes, with only a small number of β-galactosidase-positive cells (0.2%) (Fig. 8), whereas coinfection with tsK resulted in over 90% of the cells being positive (data not shown). The number of cells in which the viral genome escaped repression was increased for PML-depleted cells (consistent with the situation in fibroblasts) (19) and, to a lesser extent, Sp100-depleted cells (Fig. 8). The number of β-galactosidase-positive cells was further increased by depletion of both PML and Sp100 (Fig. 8). However, it was clear that depletion of both PML and Sp100 did not render in1374 gene expression independent of ICP0, VP16, and ICP4 in the majority of cells.
FIG. 8.

The proportion of cells in which the highly defective HSV-1 mutant in1374 escapes initial repression is increased by depletion of either PML or Sp100 individually and, to a greater extent, by depletion of both proteins simultaneously. HepaRG, HALL, HALP1, HALS2, and HALP1/S2 cells were infected with in1374 (MOI of 3 PFU per cell), with or without tsK (MOI of 2 PFU per cell). All virus adsorptions were performed at 37°C, and then the cells were incubated at 38.5°C overnight. The cells were stained for β-galactosidase activity the following day. For HepaRG and HALL cells, the total number of blue cells was counted to determine the proportion of positive cells in the population. There were too many blue cells to count by this method for the other cell types. Therefore, the number of positive cells in five random fields of view, using a ×25 objective lens, was counted for all cell types. The percentages of positive cells for HALP1, HALS2, and HALP1/S2 cells were determined by multiplying the total percentage of positive cells in the controls by the relative increases in cell numbers in the random fields of view for the other cell types.
hDaxx can be recruited to the sites of HSV-1 genomes in cells depleted of both PML and Sp100.
Although there was a substantial increase in the probability of plaque formation by the ICP0-null mutant in HepaRG cells depleted of both PML and Sp100, the mutant was not complemented to wt virus levels. By comparing the absolute plaque-forming titers of stocks of wt and ICP0-null mutant HSV-1 in HepaRG, HALP1/S2, and U2OS cells (in which ICP0 is not required for HSV-1 infection) (74), we estimate that the effect of depleting both PML and Sp100 from HepaRG cells is to increase the plaque-forming efficiency to approximately 20% of that of the wt virus in hepatocytes. This observation raises the question of which cellular factors are responsible for the remaining repression of the ICP0-null mutant HSV-1 genome. These factors may or may not be related to the components of ND10.
If the cellular response that leads to the accumulation of ND10 proteins in close association with parental HSV-1 genomes is indeed relevant to the repression of those genomes that occurs in the absence of ICP0, the relatively modest increases in ICP0-null mutant replication in the absence of either PML or Sp100 individually could be related to the continued recruitment of other ND10 proteins (20) (Fig. 2). Because the ICP0-null mutant is still subject to repression, albeit less efficiently, in HepaRG cells depleted of both PML and Sp100, we questioned whether recruitment of hDaxx to the sites associated with parental viral genomes and early replication compartments also occurs in these cells. We used the approach of examining the locations of ND10 proteins in cells asymmetrically infected at the edges of developing plaques of ICP0-null mutant HSV-1 in relation to the sites of viral genomes identified by binding of the HSV-1 transcriptional activator ICP4, as described above and in previous studies (18, 21). We found that even in the apparent absence of both PML and Sp100, hDaxx was still recruited to the sites of parental HSV-1 genomes and to early replication compartments in the absence of ICP0 (Fig. 9). Recruitment of hDaxx to ICP4-defined HSV-1 nucleoprotein complexes also occurred in HALP1 and HALS2 cells, as efficiently as that in the parental HepaRG cells (data not shown). Whether recruitment of hDaxx or other ND10 proteins contributes to repression of HSV-1 gene expression in the absence of ICP0 is currently under study.
FIG. 9.
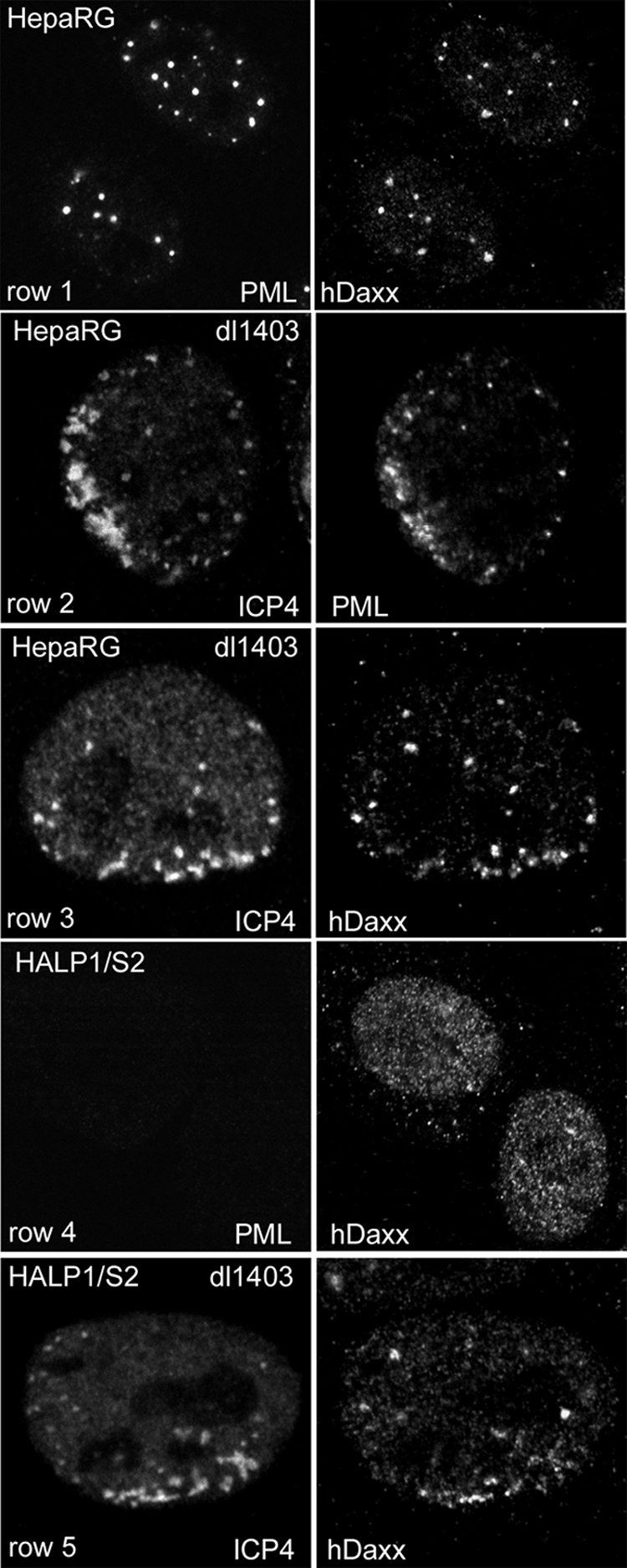
The major ND10 component hDaxx is recruited to the sites of parental HSV-1 genomes in the absence of PML and Sp100. Each row shows the two channels of the same field of view stained as indicated. Row 1, hDaxx was present in ND10 structures in HepaRG cells; rows 2 and 3, HepaRG cells were infected with ICP0-null mutant virus dl1403 at a low MOI and then stained the following day for ICP4 (left) and either PML (row 2) or hDaxx (row 3). Images of cells at the edges of plaques showing typical asymmetric foci of ICP4 in early replication compartments associated with either PML or hDaxx are illustrated. Row 4, HALP1/S2 cells were stained for PML (left) and hDaxx (right); row 5, HALP1/S2 cells were infected with ICP0-null mutant virus dl1403 at a low MOI and then stained the following day for ICP4 (left) and hDaxx (right). An image of a cell at the edge of a plaque showing typical asymmetric foci of ICP4 in early replication compartments associated with hDaxx is illustrated.
The repression of HSV-1 infection that occurs in both human fibroblasts and hepatocytes in the absence of ICP0 is independent of histone deacetylation.
Several previous studies have implicated cellular HDACs in the mechanism by which HSV-1 genomes become repressed. It has been established that repressed and latent genomes have epigenetic markers consistent with deacetylated histones and heterochromatin, while active viral genes are preferentially associated with an open chromatin structure characterized by acetylated histones (32, 36-38). ICP0 and its bovine herpesvirus orthologue have been found to interact with certain HDACs (41, 75) or proteins that interact with HDACs and regulate their activity (25, 26, 53). Sodium butyrate, which inhibits HDAC activity, partially relieves the defect of ICP0-null mutant HSV-1 in SK-N-SH cells (53), while trichostatin A (TSA) (another HDAC inhibitor) reverses CoREST-mediated repression of HSV-1 IE promoters in transfected cells (51). However, the role of HDACs in the phenotype of simple ICP0-null mutant HSV-1 in human fibroblasts has not been reported. We considered the possibility that the repression of HSV-1 gene expression attributed to ND10 components could be mediated through regulation of HDACs or that the repression of ICP0-null mutant genomes that continues in the absence of PML and/or Sp100 could be the result of HDAC activity. Therefore, we investigated whether TSA or sodium butyrate could increase ICP0-null mutant HSV-1 gene expression or plaque formation in human fibroblasts or HepaRG cells and their PML- and/or Sp100-depleted derivatives.
Surprisingly, TSA treatment did not stimulate ICP0-null mutant HSV-1 plaque formation in either human fibroblasts or HepaRG cells, although it did increase plaque numbers in Vero cells (Fig. 10B). In fact, TSA had a slight inhibitory effect on plaque formation in fibroblasts when it was present in the medium throughout the assay. Furthermore, TSA treatment did not increase ICP0-null mutant HSV-1 replication in any of the HepaRG-derived cell lines depleted of PML and/or Sp100 (Fig. 10C). Similarly, there was no evidence that ICP0-null mutant HSV-1 could be complemented by inhibition of HDAC activity using either TSA or sodium butyrate in either normal or PML-depleted human fibroblasts (data not shown). In all of the above experiments, the cells were pretreated with TSA for 30 min before the addition of the virus, and then the drug was maintained in the medium throughout the assay. However, in the case of IE1-deficient HCMV, although the presence of TSA in the medium during the actual infection could be inhibitory to virus replication (M. Nevels and C. Paulus, personal communication), pretreatment of cells with TSA or other HDAC inhibitors was found to overcome the IE1 defect (49). Accordingly, we tested the effect of pretreatment of cells for times of between 2 and 24 h with TSA at concentrations from 50 to 500 nM on plaque formation by ICP0-null mutant HSV-1. The drug was removed immediately prior to virus infection. Preliminary experiments indicated that the effects seen were not increased further by using TSA concentrations of >200 nM or pretreatments of longer than 2 h. Using this approach, the inhibitory effect of TSA did not occur, and instead, there was a slight increase, on the order of twofold, in ICP0-null mutant HSV-1 in human fibroblasts (Fig. 10D). This increase did not occur in either PML-depleted fibroblasts or HepaRG cells (Fig. 10D). Pretreatment with sodium butyrate for 2 h prior to virus infection, followed by removal of the drug, caused a similar slight increase in ICP0-null mutant HSV-1 plaque formation in HF cells while not affecting that of the wt (data not shown).
FIG. 10.

Inhibition of HDACs does not increase plaque formation by ICP0-null mutant HSV-1 in human fibroblasts and hepatocytes or in hepatocytes depleted of PML, Sp100, or both proteins simultaneously. (A) HF, HepaRG, U2OS, and Vero cells were either pretreated or not treated with TSA (50 nM) for 30 min and then infected at a low MOI with wt HSV-1 for plaque assays. TSA was present in the treated samples throughout virus adsorption and subsequent incubation. Plaques were counted 48 h after infection. The results show relative plaque counts in the presence and absence of TSA. (B) The experiment described for panel A was conducted in parallel with ICP0-null mutant dl1403. (C) A similar experiment was conducted with ICP0-null mutant dl1403 in HALL, HALP1, HALS2, and HALP1/S2 cells, and plaques were counted 38 h after infection. (D) The indicated cell types were pretreated with 200 nM TSA for 2 h prior to plaque assays of ICP0-null mutant HSV-1. The drug was washed out at the time of infection. The error bars represent standard deviations.
These data are consistent with the previous conclusion that TSA does not complement HSV-1 mutant in1814 (50), which contains a mutation in VP16 such that it is unable to stimulate IE gene expression and thus has a phenotype similar to that of ICP0-defective mutants (1). Indeed, as in our experiments with dl1403, TSA had a slight inhibitory effect on in1814 plaque formation in human fibroblasts (50). We conclude that the repression of ICP0-null mutant HSV-1 gene expression and plaque formation that occurs in human fibroblasts and hepatocytes cannot be alleviated simply by inhibiting HDAC activity. Therefore, other factors, which have yet to be determined, must be required for the remaining repression of ICP0-null mutant HSV-1 genomes that occurs in cells depleted of both PML and Sp100.
DISCUSSION
This study demonstrates that independent depletion of either PML or Sp100 increases the efficiencies of gene expression and plaque formation by ICP0-null mutant HSV-1 and that cells from which both proteins have been depleted exhibit further enhancement of replication of the mutant virus. These conclusions were confirmed using two different cell types. PML has been implicated in an intrinsic defense against several DNA viruses (reviewed in reference 16), but the mechanism by which PML and ND10 contribute to HSV-1 genome repression remains unknown. Since PML is required for the assembly of ND10 in uninfected cells, it has the potential to affect the localization and/or biochemical properties of all other ND10 component proteins. For example, depletion of PML has a pronounced effect on the SUMO modification status of Sp100-A (20; this study). Therefore, it was formally possible that the improved replication of ICP0-null mutant HSV-1 in PML-depleted cells could be explained by the downstream effect on Sp100, particularly as the effects of PML depletion and HSV-1 infection on Sp100 expression are apparently identical (20). The fact that independent depletion of either PML or Sp100 results in similar increases in ICP0-null mutant HSV-1 replication lends initial support for this hypothesis. However, since depletion of both proteins results in an increased effect, it is unlikely that the effect of PML depletion is mediated solely through Sp100.
Simultaneous depletion of both PML and Sp100 does not restore ICP0-null mutant HSV-1 plaque formation to wt virus levels in either human fibroblasts or hepatocytes. The remaining defect of the ICP0-null mutant in such cells could be due to factors either related to or independent of other ND10 proteins. An obvious candidate cellular protein that could be involved in HSV-1 gene expression is hDaxx, which binds to PML (34) and is involved in repression of gene expression (39, 46, 68) and chromatin modification (33, 73). Furthermore, high-level expression of hDaxx inhibits the HCMV major IE promoter, while depletion of hDaxx increases HCMV IE gene expression (72). The repressive effect of hDaxx is targeted by the HCMV tegument protein pp71, and the defect in IE gene expression of pp71-deficient HCMV can be overcome by depletion of hDaxx (5, 57, 59). However, our preliminary data indicate that depletion of hDaxx has only a small effect on ICP0-null mutant HSV-1 in human fibroblasts. It would be interesting to test whether depletion of hDaxx in combination with other ND10 proteins, such as PML and Sp100, or depletion of other ND10 proteins involved in chromatin metabolism also impacts the phenotype of ICP0-null mutant HSV-1.
The mechanism by which ND10 proteins accumulate at sites associated with HSV-1 genomes is unknown. It is not dependent on either PML or Sp100, yet it occurs rapidly in an infected cell. The speed at which this process takes place and the fact that it is counteracted by ICP0 are highly suggestive of its biological significance. Elucidation of the proteins and mechanisms that are required for these events could be extremely important. Whatever the cellular factors involved in this recruitment, we know that neither de novo viral protein synthesis nor transcription is required (19). These observations point to the conclusion that it is entry into the nucleus of the viral genome itself that triggers the process. Although the viral genome may be coated with the small polyamines that are present within the viral capsid, it will otherwise be naked until chromatin assembly begins to take place. It is likely that chromatin assembly begins rapidly once the viral genome is in the nucleus, and the recruitment of ND10 proteins may be one aspect of this process. Indeed, several ND10 proteins are known to be involved directly in chromatin metabolism (13).
Given the obvious importance of chromatin assembly on viral genomes and the highly likely scenario that repression of the genomes reflects their assembly into a repressed chromatin structure, it is surprising that HDAC inhibitors do not complement the ICP0-null mutant defect, at least in the human cell types used here (Fig. 10). This is in apparent contrast to observations that TSA stimulates reactivation of latent or quiescent wt and ICP0-null mutant HSV-1 or enhances ICP0-null mutant viral gene expression in a variety of neuronal cell types (2, 7, 47, 53, 67). A failure of TSA to significantly stimulate HSV-1 gene expression in human fibroblasts infected with ICP0-null mutants was also noted in a recent study, although this work clearly demonstrated an effect of the drug on a subset of murine neurons, particularly within 24 h of infection (67). The authors proposed a very interesting model in which the degree of repression of the HSV-1 genome in latently or quiescently infected neurons varies considerably, such that in some cells reactivation can be achieved by TSA treatment but, with time, an increasing proportion of cells become refractory to TSA and can be reactivated only by ICP0 (67). They proposed that the mechanisms of reactivation induced by TSA and ICP0 may differ or, at least, that histone deacetylation is but one step in the process of reactivation (67).
We note that there is a distinction between the events of reactivation from latency, during which a repressed chromatin structure including hypoacetylated histones (37, 38, 70) must be reversed, and the formation of a viral chromatin structure during the early stages of infection. It is not necessary to invoke a direct role for HDACs in the assembly of a repressed chromatin state. The histones that are initially loaded onto viral chromatin may be unmodified, so it is feasible that they are subsequently modified with the epigenetic markers of repressed chromatin without the need for deacetylation. The demonstrated presence of acetylated histones on transcriptionally active viral genomes (32, 36) could be explained as easily by inhibition of repressive histone modifications by viral activators as by inhibition of histone deacetylation. An important future goal is to determine whether the response of ND10 proteins to the entry of HSV-1 genomes into the nucleus is related to regulation through chromatin assembly and, if so, how.
Acknowledgments
The work in Glasgow was supported by the Medical Research Council and, in part, by the European Commission Framework 6 SME-STREP project TargetHerpes. The Heinrich-Pette-Institut is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit und Soziale Sicherung. This work was supported by grants from the DFG and by Stiftung für neurovirale Erkrankungen to H.S.
We thank Roel van Driel (Amsterdam) and Hans Will (Hamburg) for generous provision of anti-PML and anti-Sp100 antibodies, respectively. Thomas Stamminger (Erlangen) developed the retroviral anti-PML shRNA technology on which the lentiviral system used here is based. Chris Preston supplied viruses in1863, dl1403/CMVlacZ, and in1374. Susann Cordes and Karine Pradeau contributed to the early stages of this work. All members of the R.D.E. laboratory (Chris Boutell, Jill Murray, Amanda Sykes, and Vera Lukashchuk) also contributed during the development of this work.
Footnotes
Published ahead of print on 26 December 2007.
REFERENCES
- 1.Ace, C. I., T. A. McKee, J. M. Ryan, J. M. Cameron, and C. M. Preston. 1989. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J. Virol. 632260-2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arthur, J. L., C. G. Scarpini, V. Connor, R. H. Lachmann, A. M. Tolkovsky, and S. Efstathiou. 2001. Herpes simplex virus type 1 promoter activity during latency establishment, maintenance, and reactivation in primary dorsal root neurons in vitro. J. Virol. 753885-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottomley, M. J., M. W. Collard, J. I. Huggenvik, Z. Liu, T. J. Gibson, and M. Sattler. 2001. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 8626-633. [DOI] [PubMed] [Google Scholar]
- 4.Boutell, C., S. Sadis, and R. D. Everett. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantrell, S. R., and W. A. Bresnahan. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 806188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chelbi-Alix, M. K., and H. de The. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18935-941. [DOI] [PubMed] [Google Scholar]
- 7.Danaher, R. J., R. J. Jacob, M. R. Steiner, W. R. Allen, J. M. Hill, and C. S. Miller. 2005. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 11306-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Efstathiou, S., and C. M. Preston. 2005. Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res. 111108-119. [DOI] [PubMed] [Google Scholar]
- 9.Everett, R. D. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 701185-1202. [DOI] [PubMed] [Google Scholar]
- 10.Everett, R. D. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 207266-7273. [DOI] [PubMed] [Google Scholar]
- 11.Everett, R. D. 2000. ICP0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 749994-10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everett, R. D. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22761-770. [DOI] [PubMed] [Google Scholar]
- 13.Everett, R. D. 2006. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol. 8365-374. [DOI] [PubMed] [Google Scholar]
- 14.Everett, R. D. 2006. The roles of ICP0 during HSV-1 infection, p. 39-64. In R. M. Sandri-Goldin (ed.), Alpha herpesviruses. Molecular and cellular biology. Caister Academic Press, Wymondham, United Kingdom.
- 15.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 781763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett, R. D., and M. K. Chelbi-Alix. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89819-830. [DOI] [PubMed] [Google Scholar]
- 17.Everett, R. D., P. Freemont, H. Saitoh, M. Dasso, A. Orr, M. Kathoria, and J. Parkinson. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 726581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everett, R. D., and J. Murray. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 795078-5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everett, R. D., J. Murray, A. Orr, and C. M. Preston. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 8110991-11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett, R. D., S. Rechter, P. Papior, N. Tavalai, T. Stamminger, and A. Orr. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 807995-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Everett, R. D., G. Sourvinos, C. Leiper, J. B. Clements, and A. Orr. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J. Virol. 781903-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everett, R. D., and A. Zafiropoulos. 2004. Visualization by live-cell microscopy of disruption of ND10 during herpes simplex virus type 1 infection. J. Virol. 7811411-11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gripon, P., S. Rumin, S. Urban, J. Le Seyec, D. Glaise, I. Cannie, C. Guyomard, J. Lucas, C. Trepo, and C. Guguen-Guillouzo. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 9915655-15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grotzinger, T., T. Sternsdorf, K. Jensen, and H. Will. 1996. Interferon-modulated expression of genes encoding the nuclear-dot-associated proteins Sp100 and promyelocytic leukemia protein (PML). Eur. J. Biochem. 238554-560. [DOI] [PubMed] [Google Scholar]
- 25.Gu, H., Y. Liang, G. Mandel, and B. Roizman. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 1027571-7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu, H., and B. Roizman. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. USA 10417134-17139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guldner, H. H., C. Szostecki, T. Grotzinger, and H. Will. 1992. IFN enhance expression of Sp100, an autoantigen in primary biliary cirrhosis. J. Immunol. 1494067-4073. [PubMed] [Google Scholar]
- 28.Guldner, H. H., C. Szostecki, P. Schroder, U. Matschl, K. Jensen, C. Luders, H. Will, and T. Sternsdorf. 1999. Splice variants of the nuclear dot-associated Sp100 protein contain homologies to HMG-1 and a human nuclear phosphoprotein-box motif. J. Cell Sci. 112733-747. [DOI] [PubMed] [Google Scholar]
- 29.Hagglund, R., and B. Roizman. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 782169-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halford, W. P., C. D. Kemp, J. A. Isler, D. J. Davido, and P. A. Schaffer. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 756143-6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halford, W. P., and P. A. Schaffer. 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 753240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrera, F. J., and S. J. Triezenberg. 2004. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 789689-9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hollenbach, A. D., C. J. McPherson, E. J. Mientjes, R. Iyengar, and G. Grosveld. 2002. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 1153319-3330. [DOI] [PubMed] [Google Scholar]
- 34.Ishov, A. M., A. G. Sotnikov, D. Negorev, O. V. Vladimirova, N. Neff, T. Kamitani, E. T. Yeh, J. F. Strauss III, and G. G. Maul. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147221-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jamieson, D. R., L. H. Robinson, J. I. Daksis, M. J. Nicholl, and C. M. Preston. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J. Gen. Virol. 761417-1431. [DOI] [PubMed] [Google Scholar]
- 36.Kent, J. R., P. Y. Zeng, D. Atanasiu, J. Gardner, N. W. Fraser, and S. L. Berger. 2004. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 7810178-10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kubat, N. J., A. L. Amelio, N. V. Giordani, and D. C. Bloom. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J. Virol. 7812508-12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kubat, N. J., R. K. Tran, P. McAnany, and D. C. Bloom. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 781139-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li, H., C. Leo, J. Zhu, X. Wu, J. O'Neil, E. J. Park, and J. D. Chen. 2000. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell. Biol. 201784-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ling, P. D., R. S. Peng, A. Nakajima, J. H. Yu, J. Tan, S. M. Moses, W. H. Yang, B. Zhao, E. Kieff, K. D. Bloch, and D. B. Bloch. 2005. Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J. 243565-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lomonte, P., J. Thomas, P. Texier, C. Caron, S. Khochbin, and A. L. Epstein. 2004. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 786744-6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopez, P., R. J. Jacob, and B. Roizman. 2002. Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J. Virol. 769355-9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maul, G. G. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20660-667. [DOI] [PubMed] [Google Scholar]
- 44.Maul, G. G., and R. D. Everett. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 751223-1233. [DOI] [PubMed] [Google Scholar]
- 45.Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 742679-2690. [DOI] [PubMed] [Google Scholar]
- 46.Michaelson, J. S., and P. Leder. 2003. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 116345-352. [DOI] [PubMed] [Google Scholar]
- 47.Miller, C. S., R. J. Danaher, and R. J. Jacob. 2006. ICP0 is not required for efficient stress-induced reactivation of herpes simplex virus type 1 from cultured quiescently infected neuronal cells. J. Virol. 803360-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Negorev, D. G., O. V. Vladimirova, A. Ivanov, F. Rauscher III, and G. G. Maul. 2006. Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J. Virol. 808019-8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nevels, M., C. Paulus, and T. Shenk. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 10117234-17239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicholl, M. J., and C. M. Preston. 1996. Inhibition of herpes simplex virus type 1 immediate-early gene expression by alpha interferon is not VP16 specific. J. Virol. 706336-6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pinnoji, R. C., G. R. Bedadala, B. George, T. C. Holland, J. M. Hill, and S. C. Hsia. 2007. Repressor element-1 silencing transcription factor/neuronal restrictive silencer factor (REST/NRSF) can regulate HSV-1 immediate-early transcription via histone modification. Virol. J. 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pluta, A. F., W. C. Earnshaw, and I. G. Goldberg. 1998. Interphase-specific association of intrinsic centromere protein CENP-C with HDaxx, a death domain-binding protein implicated in Fas-mediated cell death. J. Cell Sci. 1112029-2041. [DOI] [PubMed] [Google Scholar]
- 53.Poon, A. P., H. Gu, and B. Roizman. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. USA 1039993-9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Preston, C. M. 1979. Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature-sensitive mutant tsK. J. Virol. 29275-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Preston, C. M. 2000. Repression of viral transcription during herpes simplex virus latency. J. Gen. Virol. 811-19. [DOI] [PubMed] [Google Scholar]
- 56.Preston, C. M., and M. J. Nicholl. 1997. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 717807-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Preston, C. M., and M. J. Nicholl. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 871113-1121. [DOI] [PubMed] [Google Scholar]
- 58.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 59.Saffert, R. T., and R. F. Kalejta. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 803863-3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sternsdorf, T., K. Jensen, B. Reich, and H. Will. 1999. The nuclear dot protein sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J. Biol. Chem. 27412555-12566. [DOI] [PubMed] [Google Scholar]
- 61.Sternsdorf, T., K. Jensen, and H. Will. 1997. Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 1391621-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stow, E. C., and N. D. Stow. 1989. Complementation of a herpes simplex virus type 1 Vmw110 deletion mutant by human cytomegalovirus. J. Gen. Virol. 70695-704. [DOI] [PubMed] [Google Scholar]
- 63.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 672571-2585. [DOI] [PubMed] [Google Scholar]
- 64.Stuurman, N., A. de Graaf, A. Floore, A. Josso, B. Humbel, L. de Jong, and R. van Driel. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 101773-784. [DOI] [PubMed] [Google Scholar]
- 65.Tavalai, N., P. Papior, S. Rechter, M. Leis, and T. Stamminger. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 808006-8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tavalai, N., P. Papior, S. Rechter, and T. Stamminger. 2007. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 82126-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Terry-Allison, T., C. A. Smith, and N. A. DeLuca. 2007. Relaxed repression of herpes simplex virus type 1 genomes in murine trigeminal neurons. J. Virol. 8112394-12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Torii, S., D. A. Egan, R. A. Evans, and J. C. Reed. 1999. Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J. 186037-6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagner, E. K., and D. C. Bloom. 1997. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 10419-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang, Q. Y., C. Zhou, K. E. Johnson, R. C. Colgrove, D. M. Coen, and D. M. Knipe. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 10216055-16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilcox, K. W., S. Sheriff, A. Isaac, and J. L. Taylor. 2005. SP100B is a repressor of gene expression. J. Cell Biochem. 95352-365. [DOI] [PubMed] [Google Scholar]
- 72.Woodhall, D. L., I. J. Groves, M. B. Reeves, G. Wilkinson, and J. H. Sinclair. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 28137652-37660. [DOI] [PubMed] [Google Scholar]
- 73.Xue, Y., R. Gibbons, Z. Yan, D. Yang, T. L. McDowell, S. Sechi, J. Qin, S. Zhou, D. Higgs, and W. Wang. 2003. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. USA 10010635-10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yao, F., and P. A. Schaffer. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 696249-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang, Y., Y. Jiang, V. Geiser, J. Zhou, and C. Jones. 2006. Bovine herpesvirus 1 immediate-early protein (bICP0) interacts with the histone acetyltransferase p300, which stimulates productive infection and gC promoter activity. J. Gen. Virol. 871843-1851. [DOI] [PubMed] [Google Scholar]
- 76.Zhong, S., S. Muller, S. Ronchetti, P. S. Freemont, A. Dejean, and P. P. Pandolfi. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 952748-2752. [PubMed] [Google Scholar]