

Abstract
Here we report the crystal structure of hemagglutinin (HA) from influenza B/Hong Kong/8/73 (B/HK) virus determined to 2.8 Å. At a sequence identity of ∼25% to influenza A virus HAs, B/HK HA shares a similar overall structure and domain organization. More than two dozen amino acid substitutions on influenza B virus HAs have been identified to cause antigenicity alteration in site-specific mutants, monoclonal antibody escape mutants, or field isolates. Mapping these substitutions on the structure of B/HK HA reveals four major epitopes, the 120 loop, the 150 loop, the 160 loop, and the 190 helix, that are located close in space to form a large, continuous antigenic site. Moreover, a systematic comparison of known HA structures across the entire influenza virus family reveals evolutionarily conserved ionizable residues at all regions along the chain and subunit interfaces. These ionizable residues are likely the structural basis for the pH dependence and sensitivity to ionic strength of influenza HA and hemagglutinin-esterase fusion proteins.
Influenza virus is a negative-stranded RNA virus belonging to the Orthomyxoviridae family. Three types of influenza viruses have been detected: A, B, and C. They all have a major surface glycoprotein, hemagglutinin (HA) for influenza A and B viruses and hemagglutinin-esterase fusion (HEF) protein for influenza C virus (2, 20, 85, 98). Infections caused by influenza A and B viruses remain a major source of human morbidity and mortality worldwide (13, 90).
Influenza B virus circulates exclusively in humans and seals (66), with no subtypes. The first isolated influenza B virus strain was B/Lee/40 (44). Currently, there are two major phylogenetic lineages found in circulation: B/Victoria/2/87 (B/VI) like and B/Yamagata/16/88 (B/YM) like (41, 77, 83). Despite the lack of subtypes, influenza B virus undergoes antigenic variation through genetic reassortment among cocirculating strains of different lineages and antigenic drift from cumulative mutations (17, 48, 49, 56, 70, 83, 101). Influenza B virus HAs have a mutational rate about five times slower than that observed for influenza A virus HAs (3, 10, 15, 19, 34, 41, 44, 45, 81, 94, 101). The antigenic structures of influenza B virus HAs have been studied by sequence analysis of both naturally occurring variants and antibody-selected escape variants (5, 6, 10, 35, 44, 45, 51, 76, 77, 94, 96), using the structure of influenza A H3 HA as a reference (100). However, given the rather low sequence identity between influenza A and B virus HAs, ∼20% for HA1, which is the primary target for antigenic variation, it is hard to define accurately the antigenic sites in this fashion.
We have recently reported two crystal structures of influenza B/Hong Kong/8/73 virus HA (referred to as B/HK HA herein) in complex with human and avian receptor analogs (95). Comparison of these structures with those of influenza A virus HAs provided a structural basis for the receptor-binding properties of influenza A and B virus HAs and suggested the role of residue 222 (H3 numbering) as a key and likely universal determinant for the different binding modes of human receptor analogs by different HAs. Here we report a new crystal structure of unliganded B/HK HA that is determined to 2.8 Å. This structure provides a framework for a detailed understanding of antigenic variation of influenza B virus HAs. Moreover, the B/HK HA structure reveals extensive ionizable residues along the chain and subunit interfaces, which are also found in the structures of influenza A virus HAs (36, 37, 43, 71) and influenza C virus HEF (75, 102). The roles of these ionizable residues in the functions of HA/HEF are discussed.
MATERIALS AND METHODS
Purification of B/HK HA.
The protein of B/HK HA was produced using a vaccinia expression system (9, 86). The homogenized cell suspension was applied to the top of 70% (wt/vol) sucrose in 10 mM Tris, 150 mM NaCl, pH 8.0, and spun at 25,000 rpm for 2 h (Beckman SW28 rotor). The membrane fractions separated by flotation were pelleted by centrifugation at 25,000 rpm for 1 h. Membranes were then resuspended in the same buffer and digested by bromelain (at a protein:bromelain ratio of 10:1 at 28°C with 10 mM 2-mercaptoethanol). After the digestion, the mixture was spun at 55,000 rpm for 15 min (Beckman MLA80 rotor) and the supernatant purified on gradients (5 to 20% sucrose in 10 mM Tris-HCl, pH 8.0, at 38,000 rpm for 16 h; Beckman SW41 rotor). The gradients were harvested into 1-ml fractions and checked for the optical density at 280 nm. The HA-containing fractions were pooled and purified by ion-exchange chromatography. HA was eluted from the column with 150 mM NaCl and then concentrated to 1 to 2 mg/ml.
Protein crystallization and data collection.
Purified protein at 15 mg/ml was used for crystallization trials. Crystals of diamond shape were grown in 100 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], pH 6.5, and 2.5 M ammonium sulfate at 18°C within a week to 100 by 100 by 50 μm. The crystals were flash frozen in liquid nitrogen. The diffraction data were collected at Advanced Photon Source (APS) beamline 8BM. Seven datasets of heavy-atom derivatives were collected at APS beamline BM14C. All datasets were processed using an HKL2000 package (67).
Structure determination, model building, and refinement.
The diffraction pattern of B/HK HA was anisotropic at a high-resolution range, with strong diffractions up to 2.6 Å along one axis but barely beyond 3.0 Å along the other axes. As a result, the completeness of the native data at the highest-resolution shell is only 50.2% (Table 1). Three types of heavy-atom derivatives, K2PtCl4, KAuBr4, and KAu(CN)2, were used for phasing. The first three heavy-atom datasets (datasets 2 to 4) apparently suffered from large errors at resolutions higher than 4 Å. By using SOLVE (93) and MLPHARE in CCP4 (18), a total of 11 sites were located, and the figure of merit was 0.46 using data between 24 and 4 Å. Density modification and phase extension (to 3.2 Å) were carried out using DM in CCP4 (18) and resulted in a map that allowed tracing of the majority of the protein main chains (phase extension to higher than 3.2 Å yielded worse maps). However, there was ambiguity in certain difficult regions in the membrane-distal domain. Four more datasets on heavy-atom derivatives were later collected to 3.0 Å (sets 5 to 8). All seven sets of heavy-atom derivatives together gave a figure of merit of 0.66 up to 3.0 Å. The phase was improved further by solvent flipping and phase extension to 2.8 Å using DM in CCP4 (18). The resulting map was of much higher quality and allowed unambiguous tracing of the entire protein chains of HA1 and HA2. Model building was carried out using the program O (39) and structural refinement using CNS (11) and REFMAC5 in CCP4 (18). Ten-percent reflections of the native data set were set aside for calculating the Rfree factor (Rfactor=∑||F0|−|FC||/∑|F0|). The quality of the model was analyzed using PROCHECK in CCP4 (18). The final model contains 342 residues in HA1, 169 residues in HA2, 11 sugar residues, one sulfate ion, and one water molecule with Rfree and Rcryst factors of 30.9% and 28.0%, respectively (Table 2). The overall temperature B factor (8π2× , where
, where 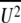 is the mean square displacement of the atomic vibration) is 93.5 Å2, much larger than those of the receptor-bound structures of B/HK HA (Protein Data Bank [PDB] accession no. 2RFT and 2RFU, with average B factors of 47.3 and 52.5 Å2, respectively), probably indicative of a certain degree of crystal disordering.
is the mean square displacement of the atomic vibration) is 93.5 Å2, much larger than those of the receptor-bound structures of B/HK HA (Protein Data Bank [PDB] accession no. 2RFT and 2RFU, with average B factors of 47.3 and 52.5 Å2, respectively), probably indicative of a certain degree of crystal disordering.
TABLE 1.
Statistics of data collection of unliganded influenza B virus HAa
| Parameterb | Result for data set:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Heavy atom (concn [mM]) | Native | K2PtCl4 (0.5) | KAuBr4 (2) | KAu(CN)2 (20) | K2PtCl4 (0.5) | K2PtCl4 (1.5) | KAuBr4 (2) | KAuBr4 (4) |
| Unit cell (Å) of space group P321 | a, b = 98.38; c = 135.98 | a, b = 98.06; c = 135.85 | a, b = 98.31; c = 135.86 | a, b = 98.19; c = 135.68 | a, b = 98.20; c = 135.65 | a, b = 98.23; c = 135.76 | a, b = 98.44; c = 135.44 | a, b = 98.26; c = 135.13 |
| No. of unique reflections | 17,606 | 14,882 | 14,590 | 14,716 | 13,362 | 13,497 | 12,988 | 12,612 |
| Resolution (Å) | 23.8-2.6 | 20.2-3.0 | 20.3-3.0 | 21.6-3.0 | 29.0-3.0 | 29.1-3.0 | 31.3-3.0 | 29.0-3.0 |
| Completeness (%)c | 85.0 (50.2) | 92.5 (88.6) | 93.7 (89.6) | 88.8 (83.2) | 82.1 (38.5) | 83.1 (40.3) | 78.3 (42.0) | 76.9 (35.0) |
| I/σIc | 16.6 (1.9) | 10.6 (1.7) | 9.4 (1.7) | 9.3 (1.7) | 11.3 (1.9) | 12.7 (2.2) | 10.3 (1.4) | 11.8 (1.8) |
| Rmerge (%)c | 6.7 (32.6) | 6.5 (40.0) | 7.9 (48.6) | 8.1 (45.7) | 7.0 (39.0) | 6.9 (35.4) | 7.0 (42.9) | 5.9 (43.7) |
| Riso (%) (10-3 Å) | 13.3 | 11.5 | 10.6 | 12.6 | 13.5 | 16.6 | 17.5 | |
| Rcullis acentric (centric, anomalous) | 0.86 (0.84, 0.91) | 0.73 (0.69, 0.86) | 0.87 (0.77, 0.94) | 0.92 (0.94, 0.99) | 0.87 (0.88, 0.98) | 0.77 (0.79, 0.95) | 0.75 (0.76, 0.93) | |
| Phasing power acentric (centric) | 0.90 (0.64) | 1.49 (1.25) | 0.98 (0.92) | 0.54 (0.43) | 0.75 (0.56) | 1.15 (0.95) | 1.24 (1.03) | |
The figure of merit for all data sets is 0.66.
Rmerge, ∑|I − <I>|/∑<I>; Riso, ∑|FPH − FP|/∑FP, where FP and FPH are native and derivative structure amplitudes, respectively; Rcullis, ∑|||FPH| ± ∑|FP‖ − |FH||/∑‖FPH| ± |FP||.
Numbers in parentheses are for the highest-resolution shell.
TABLE 2.
Statistics of structural refinement of unliganded influenza B virus HA
| Parameter | Value |
|---|---|
| Resolution (Å) | 23.8-2.8 |
| Rcryst (%) | 28.0 |
| Rfree (%) | 30.9 |
| Average B factor (Å2) | 93.5 |
| RMSD bond length (Å) | 0.01 |
| RMSD angle (°) | 1.4 |
| Ramachandran value (core/allowed/generously allowed/disallowed) (%) | 80.7/16.6/2.3/0.5 |
For structural analysis, the accessible surface area (ASA) was calculated using AREAIMOL in CCP4 (18). Structural alignment was performed using LSQKAB in CCP4 (18). For the ionizable residues, we used a distance of 4.0 Å as a cutoff in identifying the interresidue interactions. All of the interactions described for B/HK HA are supported by electron density maps used for structural refinement. In cases where two residues were close enough in the structure to interact with each other but the electron density of the side chains was not very strong, we generally gave them low confidence and did not include them in our discussion.
Computational normal mode analysis.
Computational normal mode analysis was performed on the monomeric form of H5 HA (PDB accession no. 1JSM). An improved method of elastic normal mode analysis was used for computing the Cα-based motional pattern (50). The reason for computing the modes based on the monomeric subunit instead of the trimeric HA is that the intrinsic deformational pattern of the individual subunit is more likely utilized in evolution to achieve different structures within the HA family (46, 54).
Protein structure accession number.
The structure factors and coordinates have been deposited in the PDB under accession number 3BT6.
RESULTS AND DISCUSSION
Structure of the unliganded influenza virus B/HK HA.
The asymmetric unit contains one monomeric subunit of the unliganded influenza B/HK HA, encompassing residues 1 through 342 in HA1 [HA1(1-342)] and HA2(1-169) (Fig. 1a), which gives rise to the biological molecule of a trimeric HA through symmetry operations (Fig. 1b). Despite many insertions and deletions in the HA1 chain, B/HK HA shares an overall similar fold with influenza A virus HAs (28, 33, 78, 87, 88, 100). It has an elongated fusion domain (F) composed of the central coiled-coil structure from HA2, the extended regions from HA1(1-42) and HA1(288-342), a globular membrane-distal domain containing the receptor-binding subdomain (R), HA1(116-274), and a vestigial esterase subdomain (E′), HA1(43-115) and HA1(275-287) (Fig. 1a). Each subunit of B/HK HA has seven disulfide bridges (Fig. 1a), and five of them have counterparts in influenza A virus H3 HA: Cys41-Cys1372, Cys601-Cys721, Cys941-Cys1431, Cys2921-Cys3181, and Cys1442-Cys1482 (45). However, the disulfide bridge Cys521-Cys2771 (H3 numbering) conserved in influenza A virus HAs that connects the ascending N-terminal and descending C-terminal segments of HA1 is absent in influenza B/HK HA. Instead, it has two unique disulfide bridges: Cys541-Cys571 and Cys1781-Cys2721.
FIG. 1.

Structure of B/HK HA. (a) Structure of a B/HK HA subunit. The elongated fusion domain (F) is composed of HA2 (green) and the N-terminal (pink) and C-terminal (blue) segments of HA1. The membrane-distal domain contains a receptor-binding subdomain (R, yellow) and a vestigial esterase subdomain (E′, cyan). The two major helices on HA2, helix A and helix B, are also labeled. Disulfide bonds on each subunit are shown as space-filling models in purple, and 11 sugar residues are in ball-and-stick format. The RBS is highlighted in red. (b) Structure of a B/HK HA trimer, colored as described above. (c) RMSDs between the unliganded and avian receptor-bound structures (solid black line) and between the avian and human receptor-bound structures (dashed red line), shown for Cα atoms only. Some of the regions with large RMSDs (spikes) are implicated as antigenic sites.
On each HA1/HA2 subunit, there are a total of 10 potential glycosylation sites, 7 on HA1 (HA125, 59, 123, 145, 163, 301, and 330) and 3 on HA2 (HA2145, 171, and 184). Oligosaccharides at seven sites (HA125, 123, 145, 163, 301, and 330 and HA2145) were built in the model (Fig. 1a). Interestingly, the glycosylation at HA1145 first appeared in the influenza B/Great Lakes/54 (B/GL/54) strain and has been maintained ever since. It is possible that the presence of such a glycan is advantageous for influenza B virus to evade human immunity, given its location on a prominent neutralizing epitope loop, the 150 loop. Moreover, the gain or loss of the glycosylation at HA1194 (95) is frequently found to be responsible for antigenic variation in monoclonal antibody (mAb) escape mutants (5, 6), in egg-adapted variants (27, 68, 69, 73, 74, 79, 80), or in field isolates (38, 58, 59). Thus, influenza A and B viruses appear to share a common mechanism of utilizing the addition or removal of glycosylation for modulating antigenicity (14, 21, 82, 84, 85).
The overall structure of unliganded B/HK HA is very similar to the receptor-bound structures reported earlier (95). However, several regions [HA1(43-62), HA1(73-79), HA1(139-152), HA1(226-241), HA1(283-289), and HA2(15-21)] display large deviations between the unliganded and avian receptor-bound structures and between the avian and human receptor-bound structures when the full-length proteins are aligned (Fig. 1c). Interestingly, regions HA1(73-79), HA1(139-152), and HA1(226-241) are part of the antigenic site of influenza B virus HAs surrounding the receptor-binding site (RBS) (see Fig. 3a). It is possible that structural plasticity of these regions is an important factor for efficient binding of antibodies and receptors (24, 25, 47, 91).
FIG. 3.

Antigenic structure of influenza B virus HAs. (a) Antigenic structure of B/HK HA. Mutations in four overlapping regions, the 120 loop (cyan), the 150 loop (green), the 160 loop (blue), and the 190 helix (red), have been found to cause antigenicity variation. Together they form a single large antigenic site with overlapping epitopes. The RBS is labeled. (b) Sequence alignment of influenza B virus HAs in the region of residues 161 to 166, highlighting the insertion or deletion in this region. A dot indicates a deletion in the sequence. By using the sequence of B/HK HA as a reference, the extra residues between HA1162 and HA1163 present in other strains are treated as insertions and numbered accordingly. For example, B/VI has a 3-residue insertion numbered Asn162′, Asp162′′, and Asn162′′′. Thus, all of the sequences mentioned in the text are based on B/HK HA numbering. B/MD/59, B/Maryland/59; B/SI/64, B/Singapore/64; B/SI/79, B/Singapore/222/79; B/OR/80, B/Oregon/5/80; B/ID/86, B/Idaho/1/86; B/GA/86, B/Georgia/1/86.
Structural comparison of HA2/HEF2.
The structures of influenza A and B virus HAs and influenza C virus HEF are readily superimposed on the HA2/HEF2 chain (Fig. 2). While the root mean square deviation (RMSD) between different subtypes of influenza A virus HAs is typically less than 1.0 Å (78), the RMSD between influenza B/HK and influenza A virus HA2 subunits is averaged at 1.72 Å for helices A and B only and at 3.59 Å for the entire HA2 chain (residues 1 to 160) (Table 3; Fig. 2a). The differences between influenza B/HK HA2 and influenza C virus HEF2 are considerably larger, 5.30 Å for helices A and B only and 6.89 Å for the entire chain (residues 1 to 160). These structural differences are in agreement with the suggested evolutionary history that influenza A and B virus HAs diverged from influenza C virus HEF about 8,000 years ago and from each other about 4,000 years ago and that influenza A virus HA started to diverge into different subtypes about 2,000 years ago (92).
FIG. 2.

Comparison of different HA structures. (a) Comparison of HA2 subunit of B/HK (green) with H3 (blue) and H5 (white) superimposed on helices A and B. (b) Comparison of HA2 subunit of B/HK and H3 HAs in the region of the fusion peptide and its neighboring structures on helices A and B. The three subunits of B/HK HA are shown in red, green, and blue. The corresponding three subunits of H3 HA are shown in the respective lighter colors. The threefold axis of the trimer is roughly perpendicular to the paper. It is clear that the fusion peptide of B/HK HA is shifted away from helix B of the same subunit and toward the trimer interface with the neighboring subunit. (c) Detailed interactions in the fusion peptide region of B/HK HA. Interactions within a 4.0-Å distance are shown as dashed lines in cyan. HA1 is in green and HA2 in white. (d) The R subdomains of influenza A and B virus HAs are related by large rotation and in some cases small translation. The R subdomains of B/HK HA and H5 HA (red for HA1 and black for HA2) differ by ∼60° rotation. By following a single normal mode calculated on an H5 HA subunit (third normal mode), the R subdomain of H5 HA reaches the position of that of B/HK HA (the 190 helix and the β-sheets can serve as references for comparison).
TABLE 3.
Structural comparison of influenza A and B virus HAs and influenza C virus HEF
| HA or HEF (PDB accession no.) | RMSDa (Å)
|
Buried ASAb (Å2) | |
|---|---|---|---|
| B/HK (helices A and B) | B/HK (HA2 chain) | ||
| H1 (1RUZ) | 1.65 | 3.68 | 2128 |
| H3 (1HGE) | 1.68 | 3.73 | 2758 |
| H5 (1JSM) | 1.61 | 3.31 | 2450 |
| H9 (1JSD) | 1.95 | 3.64 | 2739 |
| HEF (1FLC) | 5.30 | 6.89 | 2759 |
The RMSD between influenza B/HK and influenza A virus HA2 subunits averages 1.72 Å for helices A and B only and 3.59 Å for the entire HA2 chain (1-160).
Total buried ASA at three R-R subdomain interfaces. The buried ASA for B/HK HA is 4,090 Å2.
Fusion peptide.
The N-terminal fusion peptide of HA2 has the most conserved sequence across the influenza family in the entire HA1/HA2 subunit. All influenza A virus HAs with known structures have similar conformations for the fusion peptide, while B/HK HA displays one of the largest deviations in this region (Fig. 2a). Different from those of influenza A virus HAs, the fusion peptide in B/HK HA exists as a short α-helix and is shifted away from helix B of the same subunit and toward the trimer interface (Fig. 2b). The region HA2(1-8) has an average shift of about 7 Å, with the largest displacement at HA26 (∼12 Å Cα-Cα), and is also located at a lower position (toward the viral membrane). It has been suggested previously that in influenza A virus HAs, two highly conserved aspartic acids, Asp1092 and Asp1122, together with the less conserved His171, form a surface cavity in which the newly generated N terminus of HA2 is buried upon cleavage between HA1 and HA2 (16). Within this cavity, Asp1092 and Asp1122 are completely buried and make a total of five hydrogen bonds with the main-chain amide groups of HA2(1-6) (97). In contrast, B/HK HA has Asn1092 and Asp1122 instead, which are more exposed, with a combined ASA of 94 Å2 (H3 HA has an ASA of 10 Å2 for Asp1092 and Asp1122 combined). The fusion peptide of B/HK HA is also more exposed, with an ASA of 172 Å2 for HA2(1-6), in comparison to the ASA of 102 Å2 for H3 HA in the same region. The lower position and outward shift of the fusion peptide of B/HK HA seem to have caused the loss of interactions with Asn1092. Instead, the N termini of HA2 and Asp1122 are now located close to one another (with a distance of 3.4 Å between the carboxylate group of Asp1122 and the main-chain amide of Gly12), possibly via electrostatic interactions (Fig. 2c). Additionally, the side chain of Asp1122 forms a hydrogen bond with the main-chain amide of Gly42 (Fig. 2c). The fact that helix B of B/HK HA is deformed locally between HA2108 and HA2114, apparently in order to stay in contact with the outward-shifted N terminus, suggests the importance of the interactions between them. As a result of the large displacement, the fusion peptide of B/HK HA interacts extensively with the neighboring subunit (with 55 interactions of less than 4.0-Å distance with its neighboring subunit, in sharp contrast to 31 interactions in H3 HA).
Interhelix loop and membrane-distal domain.
In comparison with those of influenza A virus HAs, the interhelix loop of B/HK HA2 appears to adopt a hybrid conformation: its N terminus is in close interaction with helix B, similarly to those of H5 and H9 HAs, whereas its C terminus is a sharp turn, more closely resembling those of H3 and H7 HAs (Fig. 2a; B/HK, H3, and H5 are shown). Furthermore, the membrane-distal domain of B/HK HA differs from those of H1, H5, and H9 HAs by a rotation of about 60° without translation (Fig. 2d) and from those of H3 and H7 HAs by about 35° in rotation and 5 Å in translation. Interestingly, the current position of the membrane-distal domain of B/HK HA can be achieved by rotating the equivalent domain of H5 HA following a single normal mode (the third mode), hinged at the thin connecting neck between the membrane-distal domain and the F domain (Fig. 2d). Importantly, this hinge region coincides with the site where functional receptor-binding and esterase proteins were hypothesized to have inserted into an ancestral membrane fusion protein to give rise to the modern version of HA (75). Thus, the seemingly large structural difference between the membrane-distal domains of influenza A and B HAs is within the intrinsic mechanical elasticity (as manifested by low-frequency normal modes) of the individual subunit of HA. Evolution of the HA family might have chosen to alter the key structural features along the directions of those soft modes (for a review, see reference 54). Similar en bloc deformation has also been observed in the functional cycle of the molecular chaperonin GroEL (55).
Antigenic structure of influenza B virus HAs.
To date, about two dozen amino acid substitutions of influenza B virus HAs have been confirmed to cause antigenicity changes (Fig. 3) in site-specific mutants, mAb-selected escape variants, or field isolates (5, 6, 10, 35, 44, 45, 51, 76, 77, 94, 96). These mutations can roughly be divided into four groups and their respective surrounding regions on the structure of unliganded B/HK HA: the 120 loop, HA1(116-137); the 150 loop, HA1(141-150); the 160 loop, HA1(162-167); and the 190 helix, HA1(194-202).
The 120 loop and its surrounding regions.
The 120 loop (Fig. 3a) appears to be one of the most frequently mutated regions in field isolates (94). Indeed, virus variants of a B/VI-like strain with specific substitutions at HA1 residues 129 and 137, produced by using reverse genetics, were found to cause altered antigenicity (53). Similarly, HA1 residues 75, 77, 116, 118, and 122 were found to present an epitope that is unique to B/VI-like strains (64). Collectively, these residues constitute part of the antigenic site that partially overlaps with site E in H3 HA (98) and sites Sa and Cb in H1 HA (14, 30).
The 150 loop.
The 150 loop (Fig. 3a) is an unusually long protruding loop, the tip of which (Thr1471) is pointed away from the main body of the structure by ∼9 Å. This site is one of the most obvious antibody-binding sites on B/HK HA. On multiple occasions, laboratory-selected escape variants have been shown to harbor single substitutions in this region: Gly1411→Val (96), Gly1411→Arg, Thr1471→Ile, Ser1481→Gly, Arg1491→Gly (62), and Asn1501→Ser (5, 6). In strains isolated from fields, a single substitution of Arg1491→Lys (60) has been shown to cause the strains not to react with Arg strain-specific sera, and the Lys strains rapidly became predominant in subsequent epidemic seasons (1). Moreover, some other variants of influenza B virus, such as those grown in embryonated eggs, were found to display altered antigenic properties due to a single amino acid substitution at Gly1411 (52, 53, 68, 69). In recent isolates, the 150 loop region appears to be the neutralizing epitope specific for B/YM-like strains (62). Comparing with influenza A virus HAs, the 150 loop partially overlaps with site A in H3 HA (99) and site Ca2 in H1 HA (14, 30).
The 160 loop.
The 160 loop (Fig. 3a) is the only region in influenza B virus HAs where insertion and deletion have repeatedly been detected in field isolates from different epidemic seasons (Fig. 3b) (56, 65). The protruding nature of the 160 loop may make it easy to accommodate even multiple-residue insertions or deletions. Similarly, in mAb escape mutants, single deletions at HA1162‴ or HA1167 (see the legend for Fig. 3b for an explanation of the numbering) (60) and a single insertion of Asn following HA1163 (96) were also observed. Moreover, single amino acid mutations were detected in a number of mAb escape variants: Lys1641→Ile or Asn (5, 6), Asp1621"→Tyr, and Asn1621‴→Ser or Thr (61). Given the fact that B/HK/73 HA has the shortest 160 loop among all known influenza B virus HAs, further extension of the 160 loop in other influenza B virus HAs will make it a particularly strong antigenic site (Fig. 3b). In fact, field isolates with substitutions in this region, for instance, Asp1641→Glu or Asn1651→Lys, were found to have much weaker reactivity with human antibodies, which may impose a future epidemic threat (63). Thus, alterations of the pattern of insertion and deletion and amino acid substitutions in this region appear to be effective ways for influenza B virus to vary (56), in particular to survive a long period of time without antigenic shifts, as observed with influenza A virus (65). The 160 loop partially overlaps with site B in H3 HA (99) and site Sa in H1 HA (14, 30).
The 190 helix and its surrounding regions.
All of the residues at the external face of the 190 helix have important antigenic roles (Fig. 3a). Single mutations in this region were repeatedly found in mAb escape variants: Glu1951→Lys; Gln1971→Lys; Val1991→Ala, Leu, or Glu (5, 6); Lys2001→Asn, Arg, or Thr (5, 6, 61, 96); and Ser2051→Leu or Pro (5, 6). In addition, a mutagenesis study of B/HK HA found that Thr1961→Pro and Gln1971→Ile completely abolished the binding of an mAb raised against influenza B/HK/73 virus (72).
The hot spot on the 190 helix is HA1(194-196), a potential glycosylation site. A single mutation of Ala1961→Thr, which potentially creates a new glycosylation site at HA1(194-196), rendered the virus epidemic (59). On the other hand, the loss of the glycosylation site at this region in mAb escape mutants (Asn1941→Asp or Lys) (5, 6), in egg-adapted variants (27, 68, 69, 73, 74, 79, 80), or in field isolates (38, 58) was found to cause antigenic variation. Furthermore, a single difference at residue HA1194, Asn versus Ser, caused distinct differences in antigenic reactivity between two cocirculating B/YM-like strains (57). Thus, as observed for influenza A virus HAs (14, 21, 82, 84, 85), influenza B virus HAs also utilize the addition or removal of glycosylation as a mechanism for antigenicity alteration. Moreover, a substitution in egg-adapted influenza B virus, Asp1941→Tyr, was found to substantially alter antigenic properties, stressing the importance of the 190 helix in antigenicity independent of the glycans that are attached to it (53). The 190 helix partially overlaps with site B in H3 HA (99) and site Sb in H1 HA (14, 30).
Furthermore, the residue HA1238 appears to be part of the antigenic site of influenza B virus HA, which is partially overlapping with site D in H3 HA (99) and site Ca1 in H1 HA (14, 30). Substitutions at HA1 238, Pro2381→Ser (61), Gln, or Thr (5, 6), were observed in mAb escape variants of undiverged influenza B virus (5, 6) as well as in B/VI-like strains (61). Nevertheless, similar substitutions have not yet been observed for B/YM-like strains (62). Moreover, given the critical role of HA1238 in receptor binding (95), it is not clear from this single structure how amino acid substitutions at HA1238 are accommodated without compromising the binding of receptors.
Ionizable residues in HA/HEF.
Extensive structural studies of H3 HA (12, 16, 100) revealed that, upon exposure to low pH, extensive structural rearrangement takes place along the HA molecule. The overall stability of the molecule appears to play a central role in this process, as mutants of H3 HA with elevated fusion pH all harbored destabilizing mutations at chain or subunit interfaces (22). Consistent with this finding, a close correlation was noted between the fusion pH and the stability of the HA ectodomain, maintained by electrostatic interactions between HA1 and HA2 (36, 37, 43). The removal of a highly conserved tetrad salt bridge network (Glu891, Arg1091, Arg2691, and Glu672) at the HA1/HA2 interface destabilized H3 HA and resulted in membrane fusion at a higher pH (71). Our structural analysis of B/HK HA extended this finding by noting that there are ionizable residues at all regions where structural rearrangement is expected upon exposure to low pH, particularly at HA1/HA2 and the subunit interfaces (Fig. 4a, regions A to D). Although individual residues are generally not conserved among different types and subtypes, similar observations have been made with all types of influenza virus HA/HEF. These ionizable residues are likely the structural basis for the pH dependence and sensitivity to ionic strength of influenza virus HA/HEF (71). We next focus on these ionizable residues on B/HK HA.
FIG. 4.

Ionizable residues in neutral-pH structure of B/HK HA. (a) Structure of a subunit of B/HK HA, with regions A to D highlighted. (b to e) Ionizable residues at regions A, B, C, and D, respectively. All ionizable residues are shown explicitly in ball-and-stick format. Basic residues (Lys/Arg/His) are shown by black bonds and acidic residues (Glu/Asp) are shown by white bonds. Residues from a neighboring subunit are labeled with a “(2)” after the residue, for instance, Lys612(2). For clarity, various parts of the structure were omitted in the figures.
Region A.
Region A encompasses the intersubunit R-R subdomain interface between the subdomains at the membrane-distal end of the trimeric HA molecule (Fig. 4a). In this region of B/HK HA, we found two layers of buried residues, Lys2091 and His2201, one from each subunit, to interact at the threefold axis with pairwise distances of 2.9 Å and 3.7 Å, respectively. We did not find any electron density for counterions near these residues. The distance between Lys2091 and His2201 in the same subunit is 3.4 Å. Underneath His2201 is a cluster of five ionizable residues: Arg1051, His991, Asp1001, and Glu2331, with Lys2541 on a neighboring subunit (Fig. 4b). This is the only region that is limited to influenza B virus HAs.
Region B.
Region B covers the interface of the interhelix loop of HA2 with the R subdomain of HA1 and helix B of HA2 of the same subunit (Fig. 4a). Four clusters of ionizable residues are found in B/HK HA: Arg652 and Asp712; Asp812, Asp852, Lys3131, and Lys832 on a neighboring subunit; Glu762 with His742 on a neighboring subunit; and Asp862 with Lys612 on a neighboring subunit. Moreover, Arg882 is buried without pairing with any other ions (Fig. 4c). Of particular significance, a previous mutagenesis study has demonstrated that a cluster similarly located in H3 HA (Glu891, Arg1091, Arg2691, and Glu672) contributes substantially to the overall stability of the neutral-pH protein (71).
Region C.
Region C covers the fusion peptide and its surrounding regions (Fig. 4a). In influenza A virus HAs, His171, Asp1092, and Asp1122 have been implicated in low-pH-induced membrane fusion (16, 33, 40, 85, 98). In B/HK HA, in the absence of residues His171 and Asp1092, two histidine residues, His222 and His1142, are buried at a location very close to His171 in H3 HA without pairing with any other ions (Fig. 4d). Moreover, the extreme N terminus of HA2 and the residue Asp1122 are found to be part of an ionizable residue cluster that also includes Glu1112 and Arg3351 (Fig. 2C). In addition, there is another ionizable cluster composed of Glu1192 with Arg1202 on a neighboring subunit (Fig. 4d).
Region D.
Region D encompasses the N- and C-terminal segments of both HA1 and HA2 (Fig. 4a). In B/HK HA, we found two ionizable residue clusters: (i) Arg21 and Glu1392 and (ii) Asp1322 and Lys1232 (Fig. 4e). In the latter cluster, Asp1322 and Lys1232 interact at a pairwise distance of 3.2 Å, while the three Lys1232 residues, one from each subunit, coordinate a sulfate ion at the threefold axis. The presence of the sulfate ion in this structure could be the result of crystallization, as similar clusters found in the structures of influenza A virus HAs do not contain a counterion, even though some of them are at much higher crystallographic resolutions (for instance, H5 and H9 HAs are at resolutions of 1.9 and 1.8 Å, respectively [33]). In those cases, three pairs of HA2123 and nearby acidic residues interact near the threefold axis. Thus, the interactions between the three pairs of basic residues at HA2123 and nearby acidic residues might be a general phenomenon for this region. In addition, His1422 is buried in the molecule without pairing with any other ion.
Concluding remarks.
Here we present the crystal structure of unliganded influenza B virus HA (B/HK HA), which has been determined to a 2.8-Å resolution. The structure provides a framework for our detailed understanding of the antigenic variation of influenza B virus HAs. Structurally, there exist four major epitopes and their surrounding regions on influenza B virus HAs: the 120 loop, the 150 loop, the 160 loop, and the 190 helix. Consistent with previous operational and topological mapping using mAbs (5, 6), these four major epitopes are located close in space to form a large, continuous antigenic site. Similarly to that of influenza A virus HAs, the antigenic site of influenza B HAs is composed of various surface loops on the membrane-distal domains that are located in the vicinity of the RBS. Therefore, neutralizing antibodies are likely to function by interfering with the binding of receptors, as has been observed for influenza A virus HAs (4, 7, 8, 26, 31, 32, 42).
One important step of influenza infection is the conformational change of HA induced by the endosomal low pH that mediates the fusion of the influenza viral envelope membrane with the endosomal membrane, causing the release of influenza genetic materials into cytosols of infected host cells. Previous studies suggested that the overall stability of HA is central to this process, as mutants of H3 HA with elevated fusion pH all harbored destabilizing mutations at chain or subunit interfaces (22). Moreover, it was found that electrostatic interactions between HA1 and HA2 affect the stability of HA (36, 37, 43, 71). Our new structure of B/HK HA has allowed a systematic examination of possible interactions that play a common role in membrane fusion of influenza A and B virus HAs and influenza C virus HEF. Along this line, we have located ionizable residues at all regions where structural rearrangement is expected upon exposure to low pH, particularly at chain and subunit interfaces (Fig. 4a). Although individual residues are not generally conserved among different types and subtypes, this phenomenon is repeatedly observed with all influenza HA/HEF. These ionizable residues are likely the structural basis for the pH dependence and sensitivity to ionic strength of these proteins.
It is interesting to note that, among the ionizable residues found in B/HK HA, some are fully buried in the molecule without pairing with any other ions, such as Lys2091, His2201 (region A), Arg882 (region B), His222 and His1142 (region C), and His1422 (region D). Previous studies have demonstrated that unpaired ionizable residues tend to stay neutral when buried, because buried charges without pairing can be destabilizing (23, 29, 89). Therefore, it is possible that the fully buried unpaired ionizable residues are uncharged and make hydrogen bonds with neighboring atoms in the neutral-pH structure of HA. On the other hand, clusters containing both acidic and basic residues, although individually charged, are likely well balanced with no net charges at a neutral pH.
For the purpose of discussion, if we assume that Lys2091, His2201, Arg882, His222, His1142, and His1422 are not protonated (thus uncharged) in neutral-pH structure of HA, the drop in pH could protonate them to become positively charged. The positive charges on these buried residues could destabilize the overall HA structure, just as what was expected for His171 in H3 HA (16). Similarly, if we assume that those clusters containing both acidic and basic residues are well balanced with no net charges at a neutral pH, the low pH may protonate the acidic residues to be neutral and the basic residues to become positively charged. If the cluster has more than one basic residue, the positive charges they carry may repel each other to push apart chains and subunits (16, 85). This may be an explanation for the observation that the ionizable residue clusters in region A are found only in influenza B HAs and not in any other types of HA/HEF proteins. The total surface area buried at three R-R subdomain interfaces is averaged at ∼2520 Å2 for influenza A virus HAs and is 2759 Å2 for influenza C virus HEF. In contrast, B/HK HA buries an ASA of 4090 Å2 (Table 3). Thus, the considerably more extensive interactions at the R-R subdomain interfaces of B/HK HA may be harder to separate in the low-pH-induced conformational change, requiring a specific set of destabilizing forces in place.
However, it is important to emphasize that the degrees to which the ionizable residues are actually protonated at neutral pH and fusion pH are currently unknown. Further theoretical and experimental investigations are needed to gain a deeper understanding of this critical step in influenza virus infection.
Acknowledgments
We are indebted to D. J. Stevens at the MRC National Institute for Medical Research (Mill Hill, London, United Kingdom) for the protein samples. We thank D. C. Wiley, J. J. Skehel, and S. C. Harrison for guidance and stimulating discussions. Staff members at APS beamlines BM14C and BM8 are greatly acknowledged for their wonderful technical assistance. We are also grateful to the anonymous reviewers for their expert comments that helped to improve the manuscript.
J.M. is thankful for support from the National Institutes of Health (GM067801) and the Robert Welch Foundation (Q-1512).
Footnotes
Published ahead of print on 9 January 2008.
Dedicated to the memory of Don C. Wiley for his enlightenment and guidance.
REFERENCES
- 1.Abed, Y., M. B. Coulthart, Y. Li, and G. Boivin. 2003. Evolution of surface and nonstructural-1 genes of influenza B viruses isolated in the province of Quebec, Canada, during the 1998-2001 period. Virus Genes 27125-135. [DOI] [PubMed] [Google Scholar]
- 2.Air, G. M., and R. W. Compans. 1983. Influenza B and influenza C viruses, p. 21-69. In P. Palese and D. W. Kingsbury (ed.), Genetics of influenza viruses. Springer-Verlag, Vienna, Austria.
- 3.Air, G. M., A. J. Gibbs, W. G. Laver, and R. G. Webster. 1990. Evolutionary changes in influenza B are not primarily governed by antibody selection. Proc. Natl. Acad. Sci. USA 873884-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbey-Martin, C., B. Gigant, T. Bizebard, L. J. Calder, S. A. Wharton, J. J. Skehel, and M. Knossow. 2002. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology 29470-74. [DOI] [PubMed] [Google Scholar]
- 5.Berton, M. T., C. W. Naeve, and R. G. Webster. 1984. Antigenic structure of the influenza B virus hemagglutinin: nucleotide sequence analysis of antigenic variants selected with monoclonal antibodies. J. Virol. 52919-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berton, M. T., and R. G. Webster. 1985. The antigenic structure of the influenza B virus hemagglutinin: operational and topological mapping with monoclonal antibodies. Virology 143583-594. [DOI] [PubMed] [Google Scholar]
- 7.Bizebard, T., C. Barbey-Martin, D. Fleury, B. Gigant, B. Barrere, J. J. Skehel, and M. Knossow. 2001. Structural studies on viral escape from antibody neutralization. Curr. Top. Microbiol. Immunol. 26055-64. [DOI] [PubMed] [Google Scholar]
- 8.Bizebard, T., B. Gigant, P. Rigolet, B. Rasmussen, O. Diat, P. Bosecke, S. A. Wharton, J. J. Skehel, and M. Knossow. 1995. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 37692-94. [DOI] [PubMed] [Google Scholar]
- 9.Blasco, R., and B. Moss. 1995. Selection of recombinant vaccinia viruses on the basis of plaque formation. Gene 158157-162. [DOI] [PubMed] [Google Scholar]
- 10.Bootman, J. S., and J. S. Robertson. 1988. Sequence analysis of the hemagglutinin of B/Ann Arbor/1/86, an epidemiologically significant variant of influenza B virus. Virology 166271-274. [DOI] [PubMed] [Google Scholar]
- 11.Brünger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54905-921. [DOI] [PubMed] [Google Scholar]
- 12.Bullough, P. A., F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 37137-43. [DOI] [PubMed] [Google Scholar]
- 13.Carrat, F., and A. J. Valleron. 1995. Influenza mortality among the elderly in France, 1980-90: how many deaths may have been avoided through vaccination? J. Epidemiol. Community Health 49419-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caton, A. J., G. G. Brownlee, J. W. Yewdell, and W. Gerhard. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31417-427. [DOI] [PubMed] [Google Scholar]
- 15.Chakraverty, P. 1971. Antigenic relationship between influenza B viruses. Bull. W. H. O. 45755-766. [PMC free article] [PubMed] [Google Scholar]
- 16.Chen, J., K. H. Lee, D. A. Steinhauer, D. J. Stevens, J. J. Skehel, and D. C. Wiley. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95409-417. [DOI] [PubMed] [Google Scholar]
- 17.Chi, X. S., T. V. Bolar, P. Zhao, R. Rappaport, and S. M. Cheng. 2003. Cocirculation and evolution of two lineages of influenza B viruses in Europe and Israel in the 2001-2002 season. J. Clin. Microbiol. 415770-5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50760-763. [DOI] [PubMed] [Google Scholar]
- 19.Cox, N. J., and C. A. Bender. 1995. The molecular epidemiology of influenza viruses. Semin. Virol. 6359-370. [Google Scholar]
- 20.Cox, N. J., and K. Subbarao. 2000. Global epidemiology of influenza: past and present. Annu. Rev. Med. 51407-421. [DOI] [PubMed] [Google Scholar]
- 21.Daniels, R. S., A. R. Douglas, J. J. Skehel, and D. C. Wiley. 1983. Analyses of the antigenicity of influenza haemagglutinin at the pH optimum for virus-mediated membrane fusion. J. Gen. Virol. 641657-1662. [DOI] [PubMed] [Google Scholar]
- 22.Daniels, R. S., J. C. Downie, A. J. Hay, M. Knossow, J. J. Skehel, M. L. Wang, and D. C. Wiley. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40431-439. [DOI] [PubMed] [Google Scholar]
- 23.Dao-pin, S., D. E. Anderson, W. A. Baase, F. W. Dahlquist, and B. W. Matthews. 1991. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry 3011521-11529. [DOI] [PubMed] [Google Scholar]
- 24.Davies, D. R., and G. H. Cohen. 1996. Interactions of protein antigens with antibodies. Proc. Natl. Acad. Sci. USA 937-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyson, H. J., and P. E. Wright. 1995. Antigenic peptides. FASEB J. 937-42. [DOI] [PubMed] [Google Scholar]
- 26.Fleury, D., B. Barrere, T. Bizebard, R. S. Daniels, J. J. Skehel, and M. Knossow. 1999. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat. Struct. Biol. 6530-534. [DOI] [PubMed] [Google Scholar]
- 27.Gambaryan, A. S., J. S. Robertson, and M. N. Matrosovich. 1999. Effects of egg-adaptation on the receptor-binding properties of human influenza A and B viruses. Virology 258232-239. [DOI] [PubMed] [Google Scholar]
- 28.Gamblin, S. J., L. F. Haire, R. J. Russell, D. J. Stevens, B. Xiao, Y. Ha, N. Vasisht, D. A. Steinhauer, R. S. Daniels, A. Elliot, D. C. Wiley, and J. J. Skehel. 2004. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 3031838-1842. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Moreno, B., J. J. Dwyer, A. G. Gittis, E. E. Lattman, D. S. Spencer, and W. E. Stites. 1997. Experimental measurement of the effective dielectric in the hydrophobic core of a protein. Biophys. Chem. 64211-224. [DOI] [PubMed] [Google Scholar]
- 30.Gerhard, W., J. Yewdell, M. E. Frankel, and R. Webster. 1981. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 290713-717. [DOI] [PubMed] [Google Scholar]
- 31.Gigant, B., C. Barbey-Martin, T. Bizebard, D. Fleury, R. Daniels, J. J. Skehel, and M. Knossow. 2000. A neutralizing antibody Fab-influenza haemagglutinin complex with an unprecedented 2:1 stoichiometry: characterization and crystallization. Acta Crystallogr. D 561067-1069. [DOI] [PubMed] [Google Scholar]
- 32.Gigant, B., D. Fleury, T. Bizebard, J. J. Skehel, and M. Knossow. 1995. Crystallization and preliminary X-ray diffraction studies of complexes between an influenza hemagglutinin and Fab fragments of two different monoclonal antibodies. Proteins 23115-117. [DOI] [PubMed] [Google Scholar]
- 33.Ha, Y., D. J. Stevens, J. J. Skehel, and D. C. Wiley. 2002. H5 avian and H9 swine influenza virus haemagglutinin structures: possible origin of influenza subtypes. EMBO J. 21865-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hay, A. J., V. Gregory, A. R. Douglas, and Y. P. Lin. 2001. The evolution of human influenza viruses. Philos. Trans. R. Soc. Lond. B 3561861-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hovanec, D. L., and G. M. Air. 1984. Antigenic structure of the hemagglutinin of influenza virus B/Hong Kong/8/73 as determined from gene sequence analysis of variants selected with monoclonal antibodies. Virology 139384-392. [DOI] [PubMed] [Google Scholar]
- 36.Huang, Q., R. Opitz, E. W. Knapp, and A. Herrmann. 2002. Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys. J. 821050-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang, Q., R. P. Sivaramakrishna, K. Ludwig, T. Korte, C. Bottcher, and A. Herrmann. 2003. Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: stability and energetics of the hemagglutinin. Biochim. Biophys. Acta 16143-13. [DOI] [PubMed] [Google Scholar]
- 38.Ikonen, N., R. Pyhala, T. Axelin, M. Kleemola, and H. Korpela. 2005. Reappearance of influenza B/Victoria/2/87-lineage viruses: epidemic activity, genetic diversity and vaccination efficacy in the Finnish Defence Forces. Epidemiol. Infect. 133263-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47110-119. [DOI] [PubMed] [Google Scholar]
- 40.Kampmann, T., D. S. Mueller, A. E. Mark, P. R. Young, and B. Kobe. 2006. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 141481-1487. [DOI] [PubMed] [Google Scholar]
- 41.Kanegae, Y., S. Sugita, A. Endo, M. Ishida, S. Senya, K. Osako, K. Nerome, and A. Oya. 1990. Evolutionary pattern of the hemagglutinin gene of influenza B viruses isolated in Japan: cocirculating lineages in the same epidemic season. J. Virol. 642860-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knossow, M., M. Gaudier, A. Douglas, B. Barrere, T. Bizebard, C. Barbey, B. Gigant, and J. J. Skehel. 2002. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology 302294-298. [DOI] [PubMed] [Google Scholar]
- 43.Korte, T., K. Ludwig, Q. Huang, P. S. Rachakonda, and A. Herrmann. 2007. Conformational change of influenza virus hemagglutinin is sensitive to ionic concentration. Eur. Biophys. J. 36327-335. [DOI] [PubMed] [Google Scholar]
- 44.Krystal, M., R. M. Elliott, E. W. Benz, Jr., J. F. Young, and P. Palese. 1982. Evolution of influenza A and B viruses: conservation of structural features in the hemagglutinin genes. Proc. Natl. Acad. Sci. USA 794800-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krystal, M., J. F. Young, P. Palese, I. A. Wilson, J. J. Skehel, and D. C. Wiley. 1983. Sequential mutations in hemagglutinins of influenza B virus isolates: definition of antigenic domains. Proc. Natl. Acad. Sci. USA 804527-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leo-Macias, A., P. Lopez-Romero, D. Lupyan, D. Zerbino, and A. R. Ortiz. 2005. An analysis of core deformations in protein superfamilies. Biophys. J. 881291-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li, Y., H. Li, S. J. Smith-Gill, and R. A. Mariuzza. 2000. Three-dimensional structures of the free and antigen-bound Fab from monoclonal antilysozyme antibody HyHEL-63. Biochemistry 396296-6309. [DOI] [PubMed] [Google Scholar]
- 48.Lin, Y. P., V. Gregory, M. Bennett, and A. Hay. 2004. Recent changes among human influenza viruses. Virus Res. 10347-52. [DOI] [PubMed] [Google Scholar]
- 49.Lindstrom, S. E., Y. Hiromoto, H. Nishimura, T. Saito, R. Nerome, and K. Nerome. 1999. Comparative analysis of evolutionary mechanisms of the hemagglutinin and three internal protein genes of influenza B virus: multiple cocirculating lineages and frequent reassortment of the NP, M, and NS genes. J. Virol. 734413-4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu, M., B. Poon, and J. Ma. 2006. A new method for coarse-grained elastic normal-mode analysis. J. Chem. Theory Comput. 2464-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lubeck, M. D., J. L. Schulman, and P. Palese. 1980. Antigenic variants of influenza viruses: marked differences in the frequencies of variants selected with different monoclonal antibodies. Virology 102458-462. [DOI] [PubMed] [Google Scholar]
- 52.Lugovtsev, V. Y., G. M. Vodeiko, and R. A. Levandowski. 2005. Mutational pattern of influenza B viruses adapted to high growth replication in embryonated eggs. Virus Res. 109149-157. [DOI] [PubMed] [Google Scholar]
- 53.Lugovtsev, V. Y., G. M. Vodeiko, C. M. Strupczewski, Z. Ye, and R. A. Levandowski. 2007. Generation of the influenza B viruses with improved growth phenotype by substitution of specific amino acids of hemagglutinin. Virology 365315-323. [DOI] [PubMed] [Google Scholar]
- 54.Ma, J. 2005. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure 13373-380. [DOI] [PubMed] [Google Scholar]
- 55.Ma, J., P. B. Sigler, Z. Xu, and M. Karplus. 2000. A dynamic model for the allosteric mechanism of GroEL. J. Mol. Biol. 302303-313. [DOI] [PubMed] [Google Scholar]
- 56.McCullers, J. A., G. C. Wang, S. He, and R. G. Webster. 1999. Reassortment and insertion-deletion are strategies for the evolution of influenza B viruses in nature. J. Virol. 737343-7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muyanga, J., Y. Matsuzaki, K. Sugawara, K. Kimura, K. Mizuta, I. Ndumba, Y. Muraki, E. Tsuchiya, S. Hongo, F. C. Kasolo, Y. Numazaki, and K. Nakamura. 2001. Antigenic and genetic analyses of influenza B viruses isolated in Lusaka, Zambia in 1999. Arch. Virol. 1461667-1679. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa, N., R. Kubota, A. Maeda, T. Nakagawa, and Y. Okuno. 2000. Heterogeneity of influenza B virus strains in one epidemic season differentiated by monoclonal antibodies and nucleotide sequences. J. Clin. Microbiol. 383467-3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakagawa, N., R. Kubota, A. Maeda, and Y. Okuno. 2004. Influenza B virus Victoria group with a new glycosylation site was epidemic in Japan in the 2002-2003 season. J. Clin. Microbiol. 423295-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakagawa, N., R. Kubota, S. Morikawa, T. Nakagawa, K. Baba, and Y. Okuno. 2001. Characterization of new epidemic strains of influenza B virus by using neutralizing monoclonal antibodies. J. Med. Virol. 65745-750. [DOI] [PubMed] [Google Scholar]
- 61.Nakagawa, N., R. Kubota, T. Nakagawa, and Y. Okuno. 2001. Antigenic variants with amino acid deletions clarify a neutralizing epitope specific for influenza B virus Victoria group strains. J. Gen. Virol. 822169-2172. [DOI] [PubMed] [Google Scholar]
- 62.Nakagawa, N., R. Kubota, T. Nakagawa, and Y. Okuno. 2003. Neutralizing epitopes specific for influenza B virus Yamagata group strains are in the ‘loop’. J. Gen. Virol. 84769-773. [DOI] [PubMed] [Google Scholar]
- 63.Nakagawa, N., R. Kubota, and Y. Okuno. 2005. Variation of the conserved neutralizing epitope in influenza B virus Victoria group isolates in Japan. J. Clin. Microbiol. 434212-4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakagawa, N., J. Suzuoki, R. Kubota, S. Kobatake, and Y. Okuno. 2006. Discovery of the neutralizing epitope common to influenza B virus Victoria group isolates in Japan. J. Clin. Microbiol. 441564-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nerome, R., Y. Hiromoto, S. Sugita, N. Tanabe, M. Ishida, M. Matsumoto, S. E. Lindstrom, T. Takahashi, and K. Nerome. 1998. Evolutionary characteristics of influenza B virus since its first isolation in 1940: dynamic circulation of deletion and insertion mechanism. Arch. Virol. 1431569-1583. [DOI] [PubMed] [Google Scholar]
- 66.Osterhaus, A. D., G. F. Rimmelzwaan, B. E. Martina, T. M. Bestebroer, and R. A. Fouchier. 2000. Influenza B virus in seals. Science 2881051-1053. [DOI] [PubMed] [Google Scholar]
- 67.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode, p. 307-326. In C. W. Carter, Jr., and R. M. Sweet (ed.), Methods in enzymology, vol. 276. Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- 68.Oxford, J. S., R. Newman, T. Corcoran, J. Bootman, D. Major, P. Yates, J. Robertson, and G. C. Schild. 1991. Direct isolation in eggs of influenza A (H1N1) and B viruses with haemagglutinins of different antigenic and amino acid composition. J. Gen. Virol. 72185-189. [DOI] [PubMed] [Google Scholar]
- 69.Oxford, J. S., G. C. Schild, T. Corcoran, R. Newman, D. Major, J. Robertson, J. Bootman, P. Higgins, W. al-Nakib, and D. A. Tyrrell. 1990. A host-cell-selected variant of influenza B virus with a single nucleotide substitution in HA affecting a potential glycosylation site was attenuated in virulence for volunteers. Arch. Virol. 11037-46. [DOI] [PubMed] [Google Scholar]
- 70.Puzelli, S., F. Frezza, C. Fabiani, F. Ansaldi, L. Campitelli, Y. P. Lin, V. Gregory, M. Bennett, P. D'Agaro, C. Campello, P. Crovari, A. Hay, and I. Donatelli. 2004. Changes in the hemagglutinins and neuraminidases of human influenza B viruses isolated in Italy during the 2001-02, 2002-03, and 2003-04 seasons. J. Med. Virol. 74629-640. [DOI] [PubMed] [Google Scholar]
- 71.Rachakonda, P. S., M. Veit, T. Korte, K. Ludwig, C. Bottcher, Q. Huang, M. F. Schmidt, and A. Herrmann. 2007. The relevance of salt bridges for the stability of the influenza virus hemagglutinin. FASEB J. 21995-1002. [DOI] [PubMed] [Google Scholar]
- 72.Rivera, K., H. Thomas, H. Zhang, P. Bossart-Whitaker, X. Wei, and G. M. Air. 1995. Probing the structure of influenza B hemagglutinin using site-directed mutagenesis. Virology 206787-795. [DOI] [PubMed] [Google Scholar]
- 73.Robertson, J. S., J. S. Bootman, C. Nicolson, D. Major, E. W. Robertson, and J. M. Wood. 1990. The hemagglutinin of influenza B virus present in clinical material is a single species identical to that of mammalian cell-grown virus. Virology 17935-40. [DOI] [PubMed] [Google Scholar]
- 74.Robertson, J. S., C. W. Naeve, R. G. Webster, J. S. Bootman, R. Newman, and G. C. Schild. 1985. Alterations in the hemagglutinin associated with adaptation of influenza B virus to growth in eggs. Virology 143166-174. [DOI] [PubMed] [Google Scholar]
- 75.Rosenthal, P. B., X. Zhang, F. Formanowski, W. Fitz, C. H. Wong, H. Meier-Ewert, J. J. Skehel, and D. C. Wiley. 1998. Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 39692-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rota, P. A., M. L. Hemphill, T. Whistler, H. L. Regnery, and A. P. Kendal. 1992. Antigenic and genetic characterization of the haemagglutinins of recent cocirculating strains of influenza B virus. J. Gen. Virol. 732737-2742. [DOI] [PubMed] [Google Scholar]
- 77.Rota, P. A., T. R. Wallis, M. W. Harmon, J. S. Rota, A. P. Kendal, and K. Nerome. 1990. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 17559-68. [DOI] [PubMed] [Google Scholar]
- 78.Russell, R. J., S. J. Gamblin, L. F. Haire, D. J. Stevens, B. Xiao, Y. Ha, and J. J. Skehel. 2004. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 325287-296. [DOI] [PubMed] [Google Scholar]
- 79.Saito, T., Y. Nakaya, T. Suzuki, R. Ito, H. Saito, S. Takao, K. Sahara, T. Odagiri, T. Murata, T. Usui, Y. Suzuki, and M. Tashiro. 2004. Antigenic alteration of influenza B virus associated with loss of a glycosylation site due to host-cell adaptation. J. Med. Virol. 74336-343. [DOI] [PubMed] [Google Scholar]
- 80.Schild, G. C., J. S. Oxford, J. C. de Jong, and R. G. Webster. 1983. Evidence for host-cell selection of influenza virus antigenic variants. Nature 303706-709. [DOI] [PubMed] [Google Scholar]
- 81.Schild, G. C., M. S. Pereira, P. Chakraverty, M. T. Coleman, W. R. Dowdle, and W. K. Chang. 1973. Antigenic variants of influenza B virus. Br. Med. J. 4127-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schulze, I. T. 1997. Effects of glycosylation on the properties and functions of influenza virus hemagglutinin. J. Infect. Dis. 176(Suppl. 1)S24-S28. [DOI] [PubMed] [Google Scholar]
- 83.Shaw, M. W., X. Xu, Y. Li, S. Normand, R. T. Ueki, G. Y. Kunimoto, H. Hall, A. Klimov, N. J. Cox, and K. Subbarao. 2002. Reappearance and global spread of variants of influenza B/Victoria/2/87 lineage viruses in the 2000-2001 and 2001-2002 seasons. Virology 3031-8. [DOI] [PubMed] [Google Scholar]
- 84.Skehel, J. J., D. J. Stevens, R. S. Daniels, A. R. Douglas, M. Knossow, I. A. Wilson, and D. C. Wiley. 1984. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc. Natl. Acad. Sci. USA 811779-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69531-569. [DOI] [PubMed] [Google Scholar]
- 86.Steinhauer, D. A., S. A. Wharton, J. J. Skehel, and D. C. Wiley. 1995. Studies of the membrane fusion activities of fusion peptide mutants of influenza virus hemagglutinin. J. Virol. 696643-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stevens, J., O. Blixt, T. M. Tumpey, J. K. Taubenberger, J. C. Paulson, and I. A. Wilson. 2006. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312404-410. [DOI] [PubMed] [Google Scholar]
- 88.Stevens, J., A. L. Corper, C. F. Basler, J. K. Taubenberger, P. Palese, and I. A. Wilson. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 3031866-1870. [DOI] [PubMed] [Google Scholar]
- 89.Stites, W. E., A. G. Gittis, E. E. Lattman, and D. Shortle. 1991. In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. J. Mol. Biol. 2217-14. [DOI] [PubMed] [Google Scholar]
- 90.Sullivan, K. M., A. S. Monto, and I. M. Longini, Jr. 1993. Estimates of the US health impact of influenza. Am. J. Public Health 831712-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sundberg, E. J., and R. A. Mariuzza. 2002. Molecular recognition in antibody-antigen complexes. Adv. Protein Chem. 61119-160. [DOI] [PubMed] [Google Scholar]
- 92.Suzuki, Y., and M. Nei. 2002. Origin and evolution of influenza virus hemagglutinin genes. Mol. Biol. Evol. 19501-509. [DOI] [PubMed] [Google Scholar]
- 93.Terwilliger, T. 2004. SOLVE and RESOLVE: automated structure solution, density modification and model building. J. Synchrotron Radiat. 1149-52. [DOI] [PubMed] [Google Scholar]
- 94.Verhoeyen, M., L. Van Rompuy, W. M. Jou, D. Huylebroeck, and W. Fiers. 1983. Complete nucleotide sequence of the influenza B/Singapore/222/79 virus hemagglutinin gene and comparison with the B/Lee/40 hemagglutinin. Nucleic Acids Res. 114703-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang, Q., X. Tian, X. Chen, and J. Ma. 2007. Structural basis for receptor specificity of influenza B virus hemagglutinin. Proc. Natl. Acad. Sci. USA 10416874-16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Webster, R. G., and M. T. Berton. 1981. Analysis of antigenic drift in the haemagglutinin molecule of influenza B virus with monoclonal antibodies. J. Gen. Virol. 54243-251. [DOI] [PubMed] [Google Scholar]
- 97.Weis, W. I., S. C. Cusack, J. H. Brown, R. S. Daniels, J. J. Skehel, and D. C. Wiley. 1990. The structure of a membrane fusion mutant of the influenza virus haemagglutinin. EMBO J. 917-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wiley, D. C., and J. J. Skehel. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56365-394. [DOI] [PubMed] [Google Scholar]
- 99.Wiley, D. C., I. A. Wilson, and J. J. Skehel. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289373-378. [DOI] [PubMed] [Google Scholar]
- 100.Wilson, I. A., J. J. Skehel, and D. C. Wiley. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289366-373. [DOI] [PubMed] [Google Scholar]
- 101.Yamashita, M., M. Krystal, W. M. Fitch, and P. Palese. 1988. Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology 163112-122. [DOI] [PubMed] [Google Scholar]
- 102.Zhang, X., P. B. Rosenthal, F. Formanowski, W. Fitz, C. H. Wong, H. Meier-Ewert, J. J. Skehel, and D. C. Wiley. 1999. X-ray crystallographic determination of the structure of the influenza C virus haemagglutinin-esterase-fusion glycoprotein. Acta Crystallogr. D 55945-961. [DOI] [PMC free article] [PubMed] [Google Scholar]