

Abstract
Objective
To present the clinical, biochemical, and genetic features of a male pseudohermaphrodite due to 17beta-hydroxysteroid dehydrogenase 3 (17beta-HSD3) deficiency.
Design
Setting
University teaching hospital Gynecology practice
Patient(s)
A 15-year-old black American male pseudohermaphrodite with 17beta-HSD3 deficiency.
Intervention(s)
Laboratory evaluation, genetic mutation analysis, bilateral gonadectomy, hormone replacement.
Main Outcome Measure(s)
Endocrinologic evaluation and genetic analysis.
Result(s)
A diagnosis of 17beta-HSD3 deficiency made on the basis of hormone evaluation was confirmed through genetic mutation analysis of the HSD17B3 gene. Female phenotype was attained after gonadectomy, passive vaginal dilatation, and hormone therapy.
Conclusion(s)
17beta-HSD3 deficiency was diagnosed in this patient based on endocrinologic evaluation and confirmed with genetic mutation analysis. The patient was able to retain her female sexual identity after surgical and medical treatment.
Keywords: pseudohermaphrodite, 17beta hydroxysteroid dehydrogenase 3 deficiency, 17-ketosteroid reductase deficiency
Introduction
Male pseudohermaphroditism is defined by the presence of female or incompletely virilized external genitalia in a 46, XY individual. This condition is the result of a defect in testosterone biosynthesis or an impairment in the response of tissue to dihydrotestosterone and/or testosterone (1). Defective testosterone synthesis can be due to a mutation in any of the five enzymes involved in the conversion of cholesterol to testosterone (2). 17beta hydroxysteroid dehydrogenase 3 (17beta-HSD3) deficiency, also known as 17-ketosteroid reductase deficiency, is a rare autosomal recessive cause of male pseudohermaphroditism resulting from a defect in the final reversible step in testosterone synthesis in the testes, specifically, the conversion of androstenedione to testosterone (3,4). This enzymatic defect results in elevated levels of blood androstenedione (A) and either low or normal male levels of testosterone (T), usually reported as an increased plasma androstenedione-to-testosterone ratio (A/T) (1).
Originally described in 1971 by Saez and his colleagues, the characteristic phenotype at birth is an XY individual with female or ambiguous external genitalia (3,4). Testes may be undescended or located in the inguinal canal or labia majora. The majority of those affected have female external genitalia and consequently are raised as girls (1). Two interesting features of this disease are the presence of Wolffian duct derivatives (epididymus, vas deferens, and seminal vesicles) and the progressive virilization at the time of puberty, both of which depend on the action of testosterone (1,5,6).
The gene encoding 17beta-HSD3 contains 11 exons and has been cloned and mapped to chromosome 9q22. To date, a total of 20 mutations of the HSD17B3 gene have been identified, including sixteen missense mutations, three splice junction abnormalities, and one small deletion that results in a frame shift mutation. Genetically, patients can be homozygous or compound heterozygous (7–15).
We describe a case of male pseudohermaphroditism due to 17beta-HSD3 deficiency. Her clinical presentation and endocrinologic evaluation were classic for this disorder, and genetic mutation analysis identified a homozygous defect in the HSD17B3 gene.
Case report
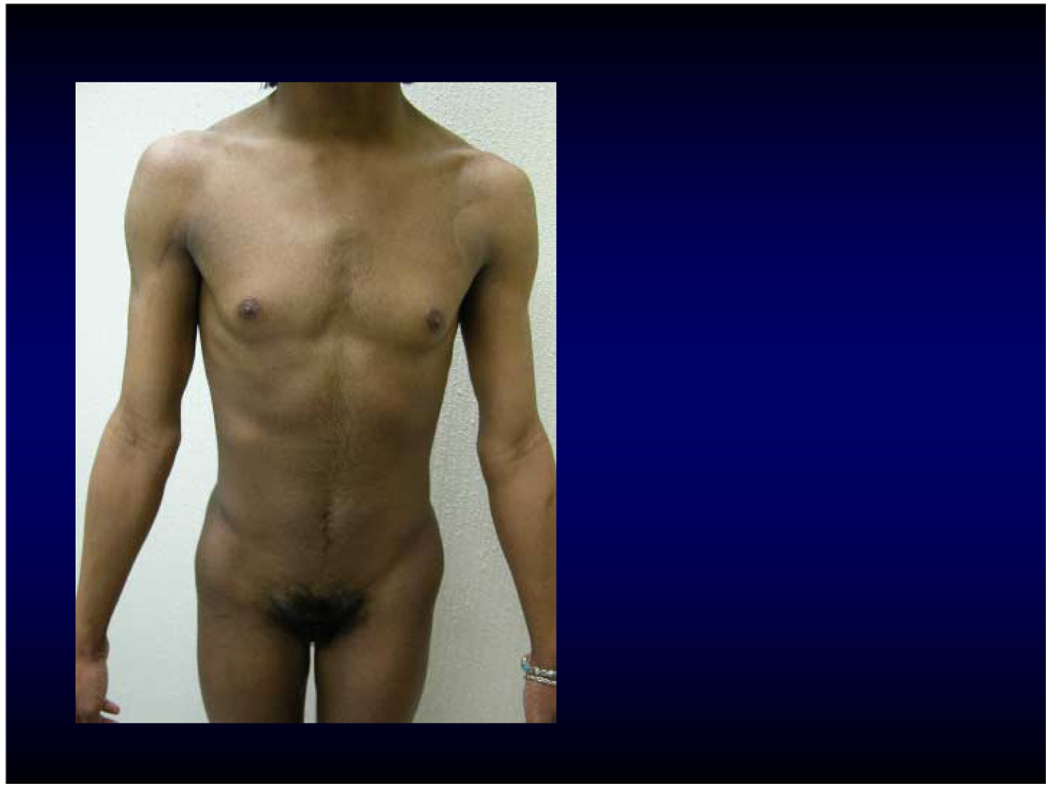
A 15 y.o. black phenotypic female patient presented to her gynecologist in New Orleans, Louisiana, with primary amenorrhea. The patient had no significant past medical or surgical history. Her external female genitalia had reportedly been considered normal at birth. She was a student in the tenth grade and lived with her mother. She was the second offspring of 18 year-old, healthy, non-consanguineous black American parents. The family history was negative in a 4 generation pedigree, including 3 normal siblings. On exam, patient was 171.5 cm tall and weighed 56.2 kg. Her breasts were Tanner stage 1. She was also noted to have hyperkeratotic skin with “orange peel” aspect, widow’s peak, and minor torus palatinus. She had increased hair growth on her abdomen and face as well as increased muscle mass and deepening of her voice (Figure 1). On pelvic exam, she had clitoromegaly measuring ~3 × 0.8 cm. Pubic hair was Tanner stage 4. Her vagina ended in a 1 cm pouch; uterus was absent. On pelvic exam she was found to have bilateral inguinal masses consistent with gonads.
Laboratory evaluation included a karyotype, which was found to be 46, XY. She was also noted to have an elevated androstenedione (A) of 2236 ng/dl (17–151 ng/dl) and decreased total testosterone (T) of 100 ng/dl (250–1100 ng/dl) compared to a normal pubertal male, resulting in an A/T ratio of 22.36. MRI of the abdomen and pelvis showed nonvisualization of the uterus or vagina with likely gonadal tissue in the inguinal canals bilaterally with an approximate 4.5 cm unilocular cystic structure contiguous to the gonadal tissue on the right. The patient was given a provisional diagnosis of male pseudohermaphroditism secondary to 17beta-hydroxysteroid dehydrogenase 3 deficiency based on her elevated A/T ratio. She was scheduled for surgery for bilateral gonadectomy in February of 2004.
Prior to her surgery, the patient was admitted to the General Clinical Research Center for further hormonal evaluation to confirm the enzymatic deficiency. Funding for this hospital admission and the blood testing was granted by the Tulane-Charity-LSU GCRC (grant #5M01 RR05096-10). Informed consent was obtained from the patient and her mother. Baseline testing magain revealed an elevated A of 973 ng/dl (35–100 ng/dl) and normal male T of 256.1 ng/dl (15–500 ng/dl) with an A/T ratio of 3.8. Results of additional laboratory evaluation are shown in Table 1.
Table 1.
Baseline laboratory testing
| Hormone | Value | Reference range |
|---|---|---|
| Androstenedione (A) (ng/dl) | 973 | 35–100 (male, 15–17 yo) |
| Testosterone, total (T) (ng/dl) | 256.1 | 15–500 (male, 13–15 yo) |
| A / T | 3.79 | |
| Androstenediol-delta5 a (ng/dl) | 76 | 70–190 (male) |
| Free testosterone b (pg/ml) | 29.4 | 35–155 (14–18 yo male) |
| 11-desoxycortisol (ng/dl) | 7 | <8 ng/dl (adult?) |
| DHEA (ng/dl) | 1640 | 240–520 (12–15 yo male) |
| Estradiol c (pg/ml) | 55 | <54 (males) |
| Estriol d (ng/ml) | <0.10 | <0.20 (adult male) |
| Aldosterone (ng/dl) | 10.8 | 1.0–18.0 (12–16 yo) |
| Estrone (pg/ml) | 144 | 17–44 (12–15 yo male) |
| Corticosterone a (ng/dl) | 248 | 135–1860 (1–16 yo) |
| 17-OH progesterone (ng/dl) | 239 | 0–240 (11–16 yo male) |
| Cortisol (mcg/dl) | 12.8 | 2.4–28.6 (14–15 yo male) |
| Progesterone (ng/ml) | 1.1 | 0.3–1.2 (male) |
| FSH (mIU/ml) | 10.1 | 1.4–18.1 (15 yo male) |
| LH (mIU/ml) | 14.7 | 0.5–5.3 (11–19 yo male) |
Assays performed at Laboratory Corporation of America (1447 York Court, Burlington, NC 27215-2230) except where indicated.
Esoterix Laborotory (4301 Lost Hills Road, Agour Hills, CA 91301).
Quest Diagnostics (4648 I-10 Service Road, Metarie, LA 70001).
Labcorp Birmingham (1801 First Avenue, Birmingham, AL 35233).
Quest Diagnostics/NI (33608 Ortega Hwy, San Juan Capistrano, CA 92675-2042).
A, androstenediol-delta5, 11-dexosoxycortisol, DHEA, estriol, aldosterone, estrone, corticosterone measured by radioimmunoassay (RIA).
T, estradiol, cortisol progesterone, FSH, and LH measured by immunochemiluminometric assay (ICMA).
Free testosterone measured by equilibrium dialysis.
17-OH progesterone measured by enzyme immunoassay (EIA).
Adrenal stimulation with cosyntropin (ACTH) showed no effect on cortisol, progesterone, 17-hydroxyprogesterone, and 11-desoxycortisone at 0, 20, 60, 90, and 120 minutes, though A rose one hour after stimulation from 973 to 1565 ng/dl. The patient also underwent hCG stimulation (1500 IU daily for 5 days) after which the patient showed an immediate rise in A to 2069 ng/dl by day 2 while T remained constant for 5 days, consistent with a defect in 17beta-HSD3.
In view of her female gender identity, it was decided to proceed with surgical removal of her gonads followed by manual vaginal dilation and estrogen replacement therapy. Intraoperatively, she was found to have bilateral inguinal gonads approximately 3 cm in diameter consistent with testes and a right-sided hydroceole. There was no evidence of an indirect inguinal hernia. Histologic examination showed bilateral testicles with Leydig cell hyperplasia and Sertoli cell hyperplasia. No evidence of malignancy was present. There were no complications. Diagnosis of 17beta-HSD3 deficiency was confirmed with genetic mutation analysis which showed the patient to be homozygous for the splice donor site mutation 325 + 4, A>T in intron 3 of the HSD17B3 gene.
Following surgery, patient was placed on conjugated estrogen 1.25 mg twice daily and instructed on passive dilatation to allow her to obtain a vagina of adequate capacity for sexual relations. At follow up 9 months later, she had no complaints except desire to have full breast development and was continued on her current dose of estrogen replacement. When seen again 18 months post-operatively, her clitoris had decreased in size, but she was still considering clitoroplasty in the future. Breasts at this time had developed to Tanner stage V. Vagina had developed with dilators to ~3.5 inches in length. Estrogen was decreased to 1.25 mg daily, and she was instructed to continue dilation.
Methods
Genomic DNA analysis
Genomic DNA was extracted from white blood cells using the Puregene DNA Isolation Blood Kit (Gentra Systems, Minneapolis, MN). The purified DNA was then used as a template in PCR using oligonucleotide pairs and PCR conditions as described by Andersson et al. (7). The nucleotide sequence of the 11 exons including splice junctions were determined by cycle sequencing using a thermostable DNA polymerase.
Discussion
The clinical presentation of 17beta-HSD3 deficiency typically consists of an XY individual with female phenotype at birth who presents at puberty with failure to menstruate and significant virilization. Differential diagnosis of this presentation includes a defect in the synthesis of testosterone, its conversion to dihydrotestosterone, or its receptor. However, endocrine evaluation showing an elevated androstenedione-to-testosterone ratio (A/T) narrows this differential to a defect in the enzyme 17beta-HSD3, resulting in an elevation of its substrate, A, and a deficiency of its product, T (16).
A molecular basis of this disorder has been shown to be the result of any of twenty mutations in the HSD17B3 gene, a gene that appears to be expressed predominately or exclusively in the testes (7,9). The majority of these mutations have been reconstituted by transient expression in cultured cells, and their functional consequences determined (7–15). All mutations so far identified appear to be inherited in an autosomal recessive pattern, requiring affected individuals to be homozygous or compound heterozygous for a mutation (17) (Table 2).
Table 2.
Reported mutations to date in patients with 17beta-HSD3 deficiency
| Mutation | Molecular lesion | Type |
|---|---|---|
| S65L | Exon 2 | Missense |
| 325+4, A>T | Intron 3 | Splice junction |
| 326−1;G>C | Intron 3 | Splice junction |
| R80Q / R80W a | Exon 3 | Missense |
| Q176P | Exon 8 | Missense |
| 655−1;G>A | Intron 8 | Splice junction |
| E215D | Exon 9 | Missense |
| F2081 | Exon 9 | Missense |
| A203V | Exon 9 | Missense |
| V205E | Exon 9 | Missense |
| Del 777-783 | Exon 10 | Deletion |
| S232L | Exon 10 | Missense |
| C268Y b | Exon 10 | Missense |
| P282L | Exon 11 | Missense |
| M235V | Exon 12 | Missense |
| G289S c | Exon 3 | SNP* |
| N130S c | Exon 5 | Missense |
| A188V d | Exon 8 | Missense |
| A56T c | Exon 2 | Missense |
| N74T d | Exon 3 | Missense |
Our patient illustrates the classical biochemical presentation of patients with this disorder. Although her plasma testosterone levels were within the normal range for a pubertal male, her plasma androstenedione concentration was over ten times greater than the normal upper limit for a pubertal male.
The homozygous mutation identified in our patient was 325 +4, A>T, a splice donor site mutation in intron 3 of the HSD17B3 gene. This mutation was reported on in detail in 1996 by Andersson et al. who found it present in homozygous form in three families and in heterozygous form in two others, making it one of the most prevalent mutations found in subjects with this disorder (7). This mutation was later shown to disrupt normal splicing by Boehmer et al. who sequenced cDNA prepared from RNA extracted from the testes of a homozygous patient and found that exon 3 was missing (and in a minority of cases, a transcript with deletion of exon 4 as well) (13).
This is the tenth reported case of 17beta-HSD3 deficiency in the United States and only the second reported case in a black American from the southern United States. The first case in a black American patient (17HSD3-Dallas) was found to be homozygous for the missense mutation on exon 10 (S232L) (9). Knowledge of the ethnic descent of a patient with 17beta-HSD3 deficiency has facilitated mutation analysis in other reported cases, as it can help predict the expected mutation for an individual. However, it appears that these two cases were the result of unrelated mutations.
In recent years, two interesting features of this disease have been the subject of investigation: first, the presence of Wolffian duct derivatives such as epididymus, vas deferens, and seminal vesicles whose development depends on the action of testosterone (1,5); second, typically at the time of puberty plasma testosterone can rise to levels that approximate the normal male range which results in progressive virilization. In some cases, this virilization leads to the spontaneous adoption of a male gender role (1,3–6). The presence of these features is thought to be the result of other isoenzymes of 17beta-HSD in extragonadal tissues that may not be affected by the genetic defect and are capable of converting A to T. The theory of extragonadal enzymatic activity is supported by recent work by Andersson et al. who found negligible amounts of testosterone (~5%) in spermatic venous blood of these patients (7).
17beta-HSD3 is the isoenzyme responsible for testosterone synthesis in the testes. However, 17beta-HSD1 is present in the ovary and placenta; 17beta-HSD2 and 4 are widely distributed in peripheral tissues (7,8). The specific isoenzyme responsible for extraglandular conversion of A to T is presently unknown. Why this process seems to be more efficient later in life than during embryogenesis is also unclear, but perhaps it is simply due to increased available substrate, A, produced in response to the pubertal surge of LH (7).
In summary, we have described a patient with classic clinical and biochemical features of 17beta-HSD3 deficiency in whom a mutation was identified in the HSD17B3 gene. Identification of affected individuals and molecular biologic studies may help elucidate the clinical conundrums of this disorder.
Acknowledgments
* Funding for this work was granted by the Tulane-Charity-LSU General Clinical Research Center (GCRC) of Charity Hospital in New Orleans, Louisiana, through grant 5M01 RR05096-10 and by the National Institute of Health through grant DK-52167.
Footnotes
Capsule: Diagnosis of 17beta-hydroxysteroid dehydrogenase 3 deficiency in a male pseudohermaphrodite was confirmed with genetic mutation analysis. Identification and treatment of this disorder allowed her to retain her female sexual identity.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Imperato-McGinley J. Male pseudohermaphroditism. In: Adashi EY, Rock JA, Rosenwaks Z, editors. Reproductive endocrinology, surgery, and technology. Philadelphia: Lippincott-Raven; 1996. pp. 936–955. [Google Scholar]
- 2.George FW, Wilson JD. Sex determination and differentiation. In: Knobil E, Neill JD, editors. The physiology of reproduction. New York: Raven Press; 1994. pp. 3–28. [Google Scholar]
- 3.Saez JM, de Peretti E, Morera AM, David M, Bertrand J. Familial male pseudohermaphroditism with gynecosmastia due to a testicular 17-ketosteroid reductase defect. J Clin Endocrinol Metab. 1971;32:604–610. doi: 10.1210/jcem-32-5-604. [DOI] [PubMed] [Google Scholar]
- 4.Saez JM, Morera AM, de Peretti E, Mertrand J. Further in vivo studies in male pseudohermaphroditism with gynecosmastia due to a testicular 17-ketosteroid reductase defect (compared to a case of testicular feminization) J Clin Endocrinol Metab. 1972;34:598–600. doi: 10.1210/jcem-34-3-598. [DOI] [PubMed] [Google Scholar]
- 5.Imperato-McGinley J, Peterson RE. Male pseudohermaphroditism: complexities of male sexual development. Am J Med. 1976;61:251–272. doi: 10.1016/0002-9343(76)90175-3. [DOI] [PubMed] [Google Scholar]
- 6.Virdis R, Saenger P. Adrenal diseases in childhood: pediatric adolescent endocrinology. Basel: Karger; 1984. 17beta-hydroxysteroid dehydrogenase deficiency; pp. 110–124. [Google Scholar]
- 7.Andersson S, Geissler WM, Wu L, Davis D, Grumbach M, New M. Molecular genetics and pathophysiology of 17beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1996;81:130–136. doi: 10.1210/jcem.81.1.8550739. [DOI] [PubMed] [Google Scholar]
- 8.Rosler A, Belanger A, Labrie F. Mechanisms of androgen production in male pseudohermaphroditism due to 17beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1992;75:773–778. doi: 10.1210/jcem.75.3.1325474. [DOI] [PubMed] [Google Scholar]
- 9.Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, et al. Male pseudohermaphroditism caused by mutations of testicular 17-betahydroxysteroid dehydrogenase 3. Nature Genetics. 1994;7:34–39. doi: 10.1038/ng0594-34. [DOI] [PubMed] [Google Scholar]
- 10.Eckstein B, Cohen S, Farkas S, Rosler A. The nature of the defect in familial male pseudohermaproditism in Arabs of Gaza. J Clin Endocrinol Metab. 1989;64:447–485. doi: 10.1210/jcem-68-2-477. [DOI] [PubMed] [Google Scholar]
- 11.Lindqvist A, Huges I, Andersson S. Substitution Mutation C268Y Causes 17beta-Hydroxysteroid Dehydrogenase 3 Deficiency. J Clin Endocrinol Metab. 2001;86:921–926. doi: 10.1210/jcem.86.2.7172. [DOI] [PubMed] [Google Scholar]
- 12.Bilbao J, Loridan L, Audi L, Gonzalo E, Castano L. A novel missense (R80W) mutation in 17-beta-hydroxysteroid dehydrogenase type 3 gene associated with male pseudohermaphroditism. Eur J Endorinol. 1998;139:330–333. doi: 10.1530/eje.0.1390330. [DOI] [PubMed] [Google Scholar]
- 13.Boehmer A, Brinkmann A, Sandkuijl L, Halley D, Niermeijer M, Andersson S, et al. 17Beta-hydroxysteroid dehydrogenase-3 deficiency: diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J Clin Endocrinol Metab. 1999;84:4713–4721. doi: 10.1210/jcem.84.12.6174. [DOI] [PubMed] [Google Scholar]
- 14.Moghrabi N, Hughes I, Dunaif A, Andersson S. Deleterious missense mutations and silent polymorphism in the human 17beta-hydroxysteroid dehydrogenase 3 gene. J Clin Endocrinol Metab. 1998;83:2855–2860. doi: 10.1210/jcem.83.8.5052. [DOI] [PubMed] [Google Scholar]
- 15.Andersson S, Moghrabi N. Physiology and molecular genetics of 17 beta-hydroxysteroid dehydrogenases. Steroids. 1997;62:143–147. doi: 10.1016/s0039-128x(96)00173-0. [DOI] [PubMed] [Google Scholar]
- 16.Can S, Zhu Y, Cai L, Ling Q, Katz M, Akgun S, et al. The identification of 5alpha-reductase-2 and 17beta-hydroxysteroid dehydrogenase-2 gene defects in male pseudohermaphrodites from a Turkish kindred. J Clin Endrocrinol Metab. 1998;83:560–576. doi: 10.1210/jcem.83.2.4535. [DOI] [PubMed] [Google Scholar]
- 17.Mendonca B, Inacio M, Arnhold I, Costa E, Bloise W, Martin R, et al. Male pseudohermaphroditism due to 17 beta-hydroxysteroid dehydrogenase 3 deficiency. Diagnosis, psychological evaluation, and management. Medicine. 2000;79:299–309. doi: 10.1097/00005792-200009000-00003. [DOI] [PubMed] [Google Scholar]