

Abstract
We study gene family coevolution on a tree of life based on a large-scale ancestral gene content reconstruction, which includes gene duplication and deletion events. The insights obtained from this study are threefold: (1) Global properties, such as the distribution of coevolution partners and the formation of disconnected clusters of coevolving families, can be an inevitable consequence of evolution along a tree. (2) Concerted family expansion (gene duplication) and contraction (gene deletion) reflect functional constraints and therefore lead to better function prediction. (3) “Long-range” coevolutionary relationships, caused mostly by large family expansions or contractions, reveal high-level evolutionary organization of cellular processes in prokaryotes.
Coevolution can be seen as the interdependency between evolutionary histories. In the context of genome evolution, when we consider gene families as evolutionary units, we expect those families that take part in the same complex, pathway, or process to show a nonzero correlation of their histories. This means that inferring coevolutionary relationships should help not only to predict new direct interactions but also to elucidate the concerted evolution of gene content as reflected by “long-range” interdependencies between families occurring in different but related pathways or processes.
To infer a map of coevolutionary relationships, we compare the reconstructed evolutionary histories of gene families along a tree of life. Similar approaches have been applied in automated function prediction (Vert 2002; Barker and Pagel 2005; Gabaldon and Huynen 2005; Barker et al. 2007), showing that the results obtained from correlating the process of losses and gains (de novo creation of a gene or horizontal gene transfer) along a tree of life improve those of standard phylogenetic profiles, based on correlating presence–absence patterns across species.
Our work is based on a full-parsimony reconstruction, which in addition to losses and gains of gene families also includes deletions and duplications of their genes. We thereby obtain a more complete description of a gene family’s evolutionary history. Here we show that the inclusion of gene duplication and deletion events improves the prediction of functional relationships. This means that patterns of gene family expansion and contraction reflect functional dependencies, just like gains and losses have been shown to do.
Previous studies of large-scale evolution of gene families have shown that only small fragments of cellular pathways are coinherited in evolution (Glazko and Mushegian 2004). Here we analyze coevolution between pathways and the global organization that emerges from those interactions.
Results
Making a coevolution map
We measure coevolution based on two alternative codings, a quantitative measure, in which the actual number of duplications and deletions are taken into account, and a binary measure, in which only the sign of the change is used. In both cases the scores are calculated for all pairs of gene families. Given the 4595 COGs with at least one representative in our 163 prokaryotic species, we calculate 1.05 × 107 ([45952 × 4595]/2) scores.
For the quantitative coevolutionary score, we calculate the correlation of changes along the tree (see Methods). As Figure 1A shows, the distribution of simple Pearson correlations looks unexpectedly bimodal, with a large peak at zero and another smaller peak close to 0.1. We correct this by calculating partial correlations relative to the vector of total changes in genome size per branch (see Methods). As can be seen in Figure 1A, this corrects the shape of the distribution and centers it around zero. This shows that the bimodality seen with simple Pearson correlations is caused by the force of genome expansion and contraction, i.e., large genome expansions or contractions produce concerted events that affect the observed correlations. Notice that partial correlation makes a difference, especially for low correlation values. This is also the case for the function prediction benchmark, which will be discussed later in this article.
Figure 1.

Distribution of correlations and global network properties. (A) Distributions of correlation values. We see that Pearson correlations are bimodally distributed, with a peak at zero and otherwise a bell-shaped distribution centered at the right side of zero. The partial correlation corrects this and centers the distribution around zero, showing that spurious correlations occur as a result of genome expansions and contractions. The inset shows the distribution in log scale. (B) Clustering behavior for partial correlations (pcor) and sign score (sign)-based networks, where clusters are simply connected components of size ≥2. (C) Degree distributions for different thresholds. For the thresholds shown here, the best fit is given by a power law with exponential cutoff (see Supplementary material S6).
For the binary coevolutionary score, we take into account only the sign of the change in family size. The score equals the number of branches where both family sizes increase (+, +) or decrease (−, −), minus those in which the change is not concerted: (+, −), (−, +), (+, 0), (0, +), (−, 0), (0, −). This means that the score is limited by the number of events in the history. This score is the same as used previously for gain–loss reconstructions (Gabaldon and Huynen 2005; Barker et al. 2007) but applied on data including duplications and deletions (see Supplementary material S3–S4 for an extensive discussion about the score and other alternatives).
Coevolution map topology is an emergent property of evolution
We studied the topological properties of the coevolution map and tried to understand the observed features from an evolutionary point of view. Networks of coevolving families are built by considering all pairs of families with a minimum coevolutionary score. Figure 1B shows that the coevolution network is a disconnected graph formed by separate clusters. As this cutoff is decreased, new clusters start to appear, reaching a maximum between partial correlation 0.6 and 0.725 (sign scores 0 and 1). After this the cluster starts to percolate into a connected component. As for the degree distribution, we find that it is best described by a power law with exponential cutoff (see Supplementary material S6).
We study the extent to which these global network properties can be explained as side effects of the mutational dynamics of genomes. To this end, we make use of null models based on a simulation of genome evolution (see Methods). The simulation is based on the fact that, given the reconstructed history, we can replay evolution stochastically under different constraints. We use two different models: In the first, more stringent model, we keep the same number of duplications and deletions (including losses) per branch and reproduce gains exactly as they appear on the reconstruction. That is to say, we only simulate the occurrence of each duplication and deletion on a given branch by assuming that the chance an event happens on a family is simply proportional to its size. Although the resulting coevolution map conveys almost no functional information (at its best, only four links coincide with data from KEGG), the degree of distribution reflects closely that of the real coevolution map (Fig. 2). On the other hand, the clustering behavior is at most about two-thirds of the number of clusters seen in the data. This means that functional constraints increase the clustering of coevolutionary interactions.
Figure 2.

Global properties of the coevolution map. (A,B) Similar degree distributions as seen in the data can be obtained for simulations of genome evolution, even for the most relaxed conditions, when only the number of events and the tree structure is maintained (low resemblance). In the high resemblance case, the simulation shows similar clustering behavior at a lower level (C), while the low resemblance simulation has almost no connections for high correlations and therefore no clustering. The compared networks were selected for having similar numbers of edges.
A second, less stringent null model, only maintains the total number of events inferred by the reconstruction. In this model, all families are present at the root of the tree of life, so no gain occurs during the simulation. The number of events is equally divided among all branches where duplications or deletions occur with equal chance. Hence, resemblance with the real reconstructed process is only on the fact that the same tree is used. Surprisingly, even for these extremely unrealistic assumptions, we observe that the degree of distribution of the coevolution map with a similar number of edges (at a lower threshold) still resembles the class of distribution found in the data (both for simulation and reconstruction of simulation). Notice that the same process on a star tree produces a truly random graph with a Poisson degree distribution, since the probability of finding a high correlation between two vectors that share no common history decreases exponentially. On the other extreme, without speciation (one descendant per ancestor) the degree of distribution becomes degenerate as a result of fast family extinctions. This shows that global connectivity properties can be explained by the dynamics of the duplication–deletion process on a tree. The specific connections do, however, bear functional significance.
Gene family expansions and contractions reflect functional dependencies
In this section we wish to obtain a measure of “correctness” for the predicted coevolutionary relationships, which allow us to compare the results from the gain–loss and full reconstructions as well as our two different scoring methods. We do this by measuring the overlap between coevolution and known functional interactions as contained in the KEGG database (see Methods). If the inferred coevolutionary link overlaps with one inferred from KEGG, the prediction is labeled as a “true positive” (TP). Otherwise, it represents an interpathway interaction between COGs, i.e., a “false positive” (FP) according to the KEGG definition of pathways.
We applied both our quantitative and sign-based scores on the gain–loss reconstruction data and full reconstruction data. Figure 3, A and B, shows that the coevolution map built from the full reconstruction predicts known interactions better than the gain–loss reconstruction. This holds for both the quantitative and nonquantitative scores used in this paper. The prediction performance for the full reconstruction improves mainly because of two reasons: (1) Concerted gene duplication and deletion allow better discrimination of TPs from FPs, which means that family expansions and contractions do reflect functional constraints. (2) Presence–absence reconstructions or phylogenetic profiles fail when gene families have few losses or gains, since spurious scores are produced when only few events are shared. In contrast, completion of the evolutionary history by inclusion of duplication and deletion events helps to distinguish these cases from long periods of simultaneous family size conservation that do reflect positive coevolution.
Figure 3.

Function prediction with full and gain–loss reconstructions. (A) Accuracy vs. coverage plot shows the number of TP that can be obtained in relationship to the accuracy of the predictions, TP/(TP + FP). (B) ROC (receiver operating characteristics) curve shows the trade off between the false-negative and false-positive rates. The Y-axis can be interpreted as sensitivity, or the ratio of TP to all links that should have been predicted. The X-axis is 1 − specificity, where specificity measures the ratio of true negatives (TN) with respect to all links that are not present in KEGG. Both plots show that the full reconstruction method with partial correlations is a better predictor. (C) Relationship between scores as a smoothed 2D histogram of COG pairs. We see that the scores differ the most when one of the COGs in the predicted link contains a large expansion or contraction.
The sign score does better than the quantitative one, in particular because the former reaches a high percentage of true positives while the partial correlation remains bounded at ∼80%. Figure 3C shows a comparison of both scoring methods. We see that there are many pairs of families for which there is a low correspondence between the quantitative and the nonquantitative scores. We have found that these discrepancies are caused mainly by the occurrence of large duplication or deletion events: A high sign score can result in a low partial correlation when a large family expansion or contraction in one family does not occur in the other. Conversely, when the large expansion or contraction occurs in both families at the same branch, a high partial correlation could be paired with a low sign score, since the size of the event is invisible for the latter. This analysis allows us to filter our different classes of coevolutionary relationships. In the rest of this paper, we will focus on those cases in which the quantitative data produces a new coevolutionary prediction, that is to say, when fast family expansions or contractions co-occur.
Major cases of coevolution
We have manually surveyed our coevolution map in the regions where high partial correlations (≥0.7) are paired with low sign scores. We find not only that these discrepancies are caused by large family expansions and contractions but also that “false positives” have in many cases a biological interpretation in terms of “long-range” interactions.
Most of these cases are seen in the cluster of ATP-binding-cassette (ABC) transporters, one of the largest and most ancient families of genes (Locher et al. 2002; Locher 2004; Dawson and Locher 2006). It is in fact the largest group of paralogs in bacteria and Archaea (Tatusov et al. 1997). The map of ABC transporters reflects coevolution according to the type of transported substrate (Fig. 4A). More specifically, at partial correlation ≥0.7 we observe six groups containing interpathway links corresponding to different substrates. Moreover, some of these links reflect coevolution between transporters and families involved in metabolism of the transported substrates. For example, COG0687, a periplasmic polyamine (spermidine/putrescine) transport component coevolves with COG0665, a deaminating oxidoreductase family. In Escherichia coli, one of the members of the oxidoreductase family, PuuB, is involved in the putrescine utilization pathway (Kurihara et al. 2005), while PotF, a member of the periplasmic component family, binds to putrescine to allow its import to the cell (Pistocchi et al. 1993). See Supplemental Figure S7 for a detailed representation of the evolution of ABC transporter families.
Figure 4.

Coevolution of ABC transporter (A) and information processing (B) families. Gray lines correspond to the KEGG-based COG network constructed from genes, i.e., a link is colored gray when both COGs have a gene in the same pathway. In panel A the large cluster corresponds to the general KEGG group of ABC transporters, while the ribosome is shown in panel B. Links colored in orange are TP, i.e., links that are both in the KEGG-based COG network and in the coevolution map. Green links are false positives, links that are only in our coevolution map.
TonB-dependent outer membrane receptors, involved in transport of cobalamin iron siderophore complexes and colicins, have recently been found to scavenge sucrose in Xanthomonas campestris. This is proposed to play an important role in the adaptation of phytopathogenic bacteria to host plants as well as in the uptake of plant-derived polysaccharides in aquatic bacteria (Blanvillain et al. 2007). In accordance with this view, we observe coevolution of two TonB family receptor families, COG4206 and COG4771, with a number of other families involved in metabolism of different types of saccharides. Moreover, these metabolic families coevolve as well with endopolygalacturonase (COG5434), a wall-degrading enzyme produced by plant pathogens (Collmer and Keen 1986).
In all these cases, coevolution is strongly determined by a few concerted massive duplications or deletions rather than by many small events. The same explanation holds for true positives predicted by partial correlation but not predicted by the sign score.
Another interesting case of coevolution is found between the different “systems” involved in different stages of information processing, from DNA replication and RNA polymerization to protein synthesis. Figure 4B shows three independently coevolving clusters of ribosomal proteins corresponding each to archaeal–eukaryotic (AE), bacterial–eukaryotic (BE), and universal (U) families. A group of proteins, outside the protein-synthesis pathway, coevolves with AE-specific ribosomal proteins. In these groups, we find subunits of AE DNA-directed RNA polymerase as well as subunits of the AE-specific DNA replication machinery. On the other hand, the U and BE protein synthesis machinery is linked to a number of transcription initiation factors. Other families, like prefoldins and RNA binding proteins, which are involved at different stages of post-translational and post-transcriptional control, are found in the part of the coevolution map that contains COGs not present in KEGG pathways (not shown in Fig. 4).
The evolution of information-processing systems is marked by strong conservation, which means that the number of events from which we can infer coevolution is minimal. However, given that our reconstruction allows for the complete description of the evolutionary history, the simultaneous conservation of family size is in this case the information used to infer coevolution.
Structure of cellular organization as revealed by coevolution
Most true positives are links between COGs that fall in the same functional category. At ∼50% accuracy ((TP/TP+FP)), 80% of the TP are within category links (>90% for accuracies ≥70%). This can be observed in the over-representation of self-loops in Figure 5, which provides a more global view of the coevolutionary interactions, depicting relationships between COG functional categories rather than between COGs (see Methods). On the other hand, false positives (at least 75% intercategory links) reveal a distributed structure corresponding to the organization of cell processes as revealed by the intercategory links in Figure 5. This shows that false positives are not just missed true positives or prediction mistakes but that they have a different structure corresponding to a higher-level organization.
Figure 5.

Network of coevolutionary relationships between functional categories. Links are P = 0.05 significant coevolutionary relationships between COG functional categories. Significance is calculated in relationship to an ensemble of 2500 randomized coevolutionary maps (see Methods). The colored lines correspond to links found only with partial correlation and not with sign score. The rest of the links are found both by sign and partial correlation scores. Dotted circles enclose information processing and carbohydrate metabolism modules. Most of the underlying coevolutionary relationships relate to the cases shown in Fig. 4 e.g., antimicrobial peptides (defense mechanisms) and uncharacterized signal transduction families, TonB outer membrane receptors (ion transport), and metabolism of sugars, etc.
Figure 5 also shows that those false positives resulting from the match of a few large expansions or contractions, as detected by the partial correlation score, help to create a more complete image of long-range interactions between cellular pathways, revealing an evolutionary module around carbohydrate metabolism and pinning signal transduction families as central role players in evolution.
Discussion
We have seen that considering the full description of a gene family’s history in terms of gain, deletion, and duplication events increases the extent to which coevolution reflects functional interactions in prokaryotes. This holds for both our quantitative and nonquantitative scoring schemes. By comparing the predictions made by these two methods, we have shown that large expansions and contractions reflect interpathway coevolution. Our results also show that the sign score, and in particular our extended version that includes duplication and deletion events, produces the best function prediction results.
We should keep in mind that only a small fraction of the families show high scores: less than half of all the COGs show a correlation ≥0.6. About 50% of these high coevolutionary score values are predicted as well by the STRING (Von Mering et al. 2003) gene neighborhood score, which suggests that about half of the detected coevolutionary relationships coincide with a physical linkage in the chromosome.
Global topology properties of the coevolution map arise neutrally (without intervention of selection) as a result of the dynamics of family expansion and contraction on a tree. Our simulated genome evolution results in distributions of coevolution partners, which resembles the data closely; however, the coevolution interactions rendered by the simulation have no functional value. This is in line with the idea that such global properties are more universal than functional content (Wolf et al. 2002) and that they may come about as a result of the evolutionary dynamics (Koonin et al. 2002), as seen in the cases of family size distribution (Karev et al. 2002) and transcription and coexpression network structure (Van Noort et al. 2004; Cordero and Hogeweg 2006).
On the interaction scale, the study of coevolution via expansions and contractions along a tree of life allows us to look at functional interactions. In this article we have paid special attention to well-conserved groups of families that evolve under large expansions and contractions. One such group is transcription regulation. In recent work, we have pointed out that large expansions of contractions of regulatory families occur at the onset of major prokaryotic lineages (Cordero and Hogeweg 2007). It is, however, unknown what the functional role of those dramatic changes is on regulome size. Our coevolution map reveals that expansions of LysR and TetR families of regulators (the largest and second-largest families of transcription factors in prokaryotes) correlate with expansions of efflux systems such as the threonine/LysE-like transporters, drug/metabolite transporter (DMT) (Jack et al. 2001), and resistance-nodulation-division (RND) (Tseng et al. 1999) multidrug efflux families. This suggests that large duplications of regulators could be related to the sensing and subsequent export of metabolites and toxins.
More generally speaking, the coevolution map and the analysis of functional categories show that FP are not only just missed interactions or prediction mistakes but interpathway links that help us reveal the global structure of long-range interactions between cellular processes.
As the number of well-sequenced eukaryotic species increases, the question of to what extent gene duplication reflects functional interaction in eukaryotes as well could be addressed in more detail.
Methods
Maximum parsimony reconstruction
The reconstruction of ancestral gene content is performed per family with maximum parsimony, i.e., finding the evolutionary scenario that renders the least number of (weighted) events. This is done by minimizing the cost function S = δ + λd + γg, where δ is the number of deletions, d the number of duplications, λ the duplication cost, g the number of gains, and γ the gain cost (see Mirkin et al. 2003 for related methods). This is implemented as an extension of the PAUP generalized parsimony algorithm (Swofford 1998; Mirkin et al. 2003). The algorithm proceeds in two steps: (1) Given a family species distribution, it calculates all possible paths that connect the family sizes on the leaves (species) of the tree down to the root (last common ancestor), passing through all intermediate ancestor sizes. (2) Starting from the root, the path that minimizes the cost function is selected. Our reports refer to λ = 2 and γ = 3 (see Supplementary material S1 for more information on the algorithm, and Supplementary material S3 for an extensive discussion about the use of other gain costs).
As a definition of gene family, we use the COG database (Tatusov et al. 2000) as defined in STRING, v6.3 (Von Mering et al. 2003), on 163 prokaryotic species, which leaves 4595 nonempty COGs to work with. As a tree of life we use a recently published tree (Ciccarelli et al. 2006), rooted between archaea–eukarya and eubacteria and pruned down to our 163 species. An outlier leaf, which contains “1” when the family is present in eukaryotes and a “0” otherwise, was added to distinguish kingdom specificity.
Calculating scores
For a given COG, its reconstructed history is represented as a vector where each entry contains the signed difference in COG size between the descendant and ancestor of a branch. Pearson correlations between reconstructed history vectors of COGs X and Y are defined as ρXY = (1/N)(∑(X − X)(Y − Y)/σXσY), where X and Y are the corresponding average values, σX and σY are the standard deviations, and N is the number of branches in the tree. Partial correlations are calculated with respect to the vector of total changes in genome size, G. Each element, gi, of this vector is calculated per branch, i, as the sum of all changes in COG size, Δ, i.e., gi = ∑jΔ(i,j), where j is an index that runs over all COGs. The partial correlation is calculated then as ΨXY/G = (ρXY − 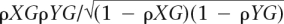 ).
).
To calculate the sign score, we consider only branches in which Xi or Yi≠0. The score equals the number of cases in which sign (Xi) = sign (Yi) minus the rest.
Benchmarking
To benchmark our results, we constructed a network of COG interactions based on the KEGG v41.1 database (Kanehisa and Goto 2000). To do this, we use KO (Kanehisa et al. 2004) entries in KEGG. The KEGG-based COG network is constructed by establishing a link between two COGs when their corresponding KOs participate in the same pathway. Alternatively, one can build the COG network directly from the co-occurrence of COG members in KEGG pathways, which results in larger coverage of COGs by the addition of less “significant” links (1933 COGs and 78,874 links). To estimate false positives, we refer only to the subset of COGs and COG interactions that are contained in the network; i.e., links between COGs outside the KEGG-based COG network are not counted as false positives. The benchmarking results reported in Figure 3 refer to the KO-based network, while, for its larger coverage, the gene-based network is used to color links in Figure 4. See Supplemental Fig. S6 for the benchmarking results on the gene-based network, and Supplementary material S7 for more details on the methods.
Genome evolution simulation
We simulate genome evolution by reproducing the reconstructed number of duplication and deletion events stochastically. We do this in the following way: On each branch of the tree we reproduce N = δ + d events, where δ is the number of deletions on the branch and d the number of duplications on the branch. Assume the ancestor on a branch has G genes. For each event until reaching N, we chose a random COG, of size x, with probability equal to x/G, and update the COG size as x = x + 1 with probability d/N or as x = x − 1 otherwise. This is done starting from the first ancestor until reaching the leaves.
For our null models, we have implemented this in two ways: A stringent model where gains and COG deletions are not simulated but added exactly as they are found in the data. The number of per-branch duplications, d, and deletions, δ, as well as the COG sizes in the last common ancestor are taken directly from the reconstruction. Another less stringent alternative is implemented by performing the same number of events per branch with equal chance of a duplication or a deletion and all families being present in the last common ancestor, so no gain occurs. Using the simulation result or its reconstruction yields the same result in our analysis, which shows that the global topological properties of the coevolution map are not an artifact of parsimony.
Functional category network
Given two functional categories, the statistical significance of intercategory coevolution is calculated by comparison with 2500 randomized coevolution maps, keeping the degree per COG. The results shown in Figure 5 refer to a P-value of 0.05, i.e., 125 out of the 2500 cases show equal or higher numbers of links between the two categories.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6815508
References
- Barker D., Pagel M., Pagel M. Predicting functional gene links from phylogenetic–statistical analyses of whole genomes. PLoS Comput. Biol. 2005;1:e3. doi: 10.1371/journal.pcbi.0010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D., Meade A., Pagel M., Meade A., Pagel M., Pagel M. Constrained models of evolution lead to improved prediction of functional linkage from correlated gain and loss of genes. Bioinformatics. 2007;23:14–20. doi: 10.1093/bioinformatics/btl558. [DOI] [PubMed] [Google Scholar]
- Blanvillain S., Meyer D., Boulanger A., Lautier M., Guynet C., Denance N., Vasse J., Lauber E., Arlat M., Meyer D., Boulanger A., Lautier M., Guynet C., Denance N., Vasse J., Lauber E., Arlat M., Boulanger A., Lautier M., Guynet C., Denance N., Vasse J., Lauber E., Arlat M., Lautier M., Guynet C., Denance N., Vasse J., Lauber E., Arlat M., Guynet C., Denance N., Vasse J., Lauber E., Arlat M., Denance N., Vasse J., Lauber E., Arlat M., Vasse J., Lauber E., Arlat M., Lauber E., Arlat M., Arlat M. Plant carbohydrate scavenging through TonB-dependent receptors: A feature shared by phytopathogenic and aquatic bacteria. PLoS ONE. 2007;2:e224. doi: 10.1371/journal.pone.0000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli F.D., Doerks T., Von Mering C., Creevey C.J., Snel B., Bork P., Doerks T., Von Mering C., Creevey C.J., Snel B., Bork P., Von Mering C., Creevey C.J., Snel B., Bork P., Creevey C.J., Snel B., Bork P., Snel B., Bork P., Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Collmer A., Keen N.T., Keen N.T. The role of pectic enzymes in plant pathogenesis. Annu. Rev. Phytopathol. 1986;24:383–409. [Google Scholar]
- Cordero O.X., Hogeweg P., Hogeweg P. Feed-forward loop circuits as a side effect of genome evolution. Mol. Biol. Evol. 2006;23:1931–1936. doi: 10.1093/molbev/msl060. [DOI] [PubMed] [Google Scholar]
- Cordero O.X., Hogeweg P., Hogeweg P. Large changes in regulome size herald the main prokaryotic lineages. Trends Genet. 2007;10:10. doi: 10.1016/j.tig.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Dawson R.J., Locher K.P., Locher K.P. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- Gabaldon T., Huynen M.A., Huynen M.A. Lineage-specific gene loss following mitochondrial endosymbiosis and its potential for function prediction in eukaryotes. Bioinformatics. 2005;21:ii144–ii150. doi: 10.1093/bioinformatics/bti1124. [DOI] [PubMed] [Google Scholar]
- Glazko G.V., Mushegian A.R., Mushegian A.R. Detection of evolutionarily stable fragments of cellular pathways by hierarchical clustering of phyletic patterns. Genome Biol. 2004;5:R32. doi: 10.1186/gb-2004-5-5-r32. http://genomebiology.com/2004/5/5/R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack D.L., Yang N.M., Saier M.H., Yang N.M., Saier M.H., Saier M.H. The drug/metabolite transporter superfamily. Eur. J. Biochem. 2001;268:3620–3639. doi: 10.1046/j.1432-1327.2001.02265.x. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M., Goto S., Kawashima S., Okuno Y., Hattori M., Kawashima S., Okuno Y., Hattori M., Okuno Y., Hattori M., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karev G.P., Wolf Y.I., Rzhetsky A.Y., Berezovskaya F.S., Koonin E.V., Wolf Y.I., Rzhetsky A.Y., Berezovskaya F.S., Koonin E.V., Rzhetsky A.Y., Berezovskaya F.S., Koonin E.V., Berezovskaya F.S., Koonin E.V., Koonin E.V. Birth and death of protein domains: A simple model of evolution explains power law behavior. BMC Evol. Biol. 2002;2:18. doi: 10.1186/1471-2148-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E.V., Wolf Y.I., Karev G.P., Wolf Y.I., Karev G.P., Karev G.P. The structure of the protein universe and genome evolution. Nature. 2002;420:218–223. doi: 10.1038/nature01256. [DOI] [PubMed] [Google Scholar]
- Kurihara S., Oda S., Kato K., Kim H.G., Koyanagi T., Kumagai H., Suzuki H., Oda S., Kato K., Kim H.G., Koyanagi T., Kumagai H., Suzuki H., Kato K., Kim H.G., Koyanagi T., Kumagai H., Suzuki H., Kim H.G., Koyanagi T., Kumagai H., Suzuki H., Koyanagi T., Kumagai H., Suzuki H., Kumagai H., Suzuki H., Suzuki H. A novel putrescine utilization pathway involves gamma-glutamylated intermediates of Escherichia coli K-12. J. Biol. Chem. 2005;280:4602–4608. doi: 10.1074/jbc.M411114200. [DOI] [PubMed] [Google Scholar]
- Locher K.P. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 2004;14:426–431. doi: 10.1016/j.sbi.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Locher K.P., Lee A.T., Rees D.C., Lee A.T., Rees D.C., Rees D.C. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- Mirkin B.G., Fenner T.I., Galperin M.Y., Koonin E.V., Fenner T.I., Galperin M.Y., Koonin E.V., Galperin M.Y., Koonin E.V., Koonin E.V. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol. Biol. 2003;3:2. doi: 10.1186/1471-2148-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistocchi R., Kashiwagi K., Miyamoto S., Nukui E., Sadakata Y., Kobayashi H., Igarashi K., Kashiwagi K., Miyamoto S., Nukui E., Sadakata Y., Kobayashi H., Igarashi K., Miyamoto S., Nukui E., Sadakata Y., Kobayashi H., Igarashi K., Nukui E., Sadakata Y., Kobayashi H., Igarashi K., Sadakata Y., Kobayashi H., Igarashi K., Kobayashi H., Igarashi K., Igarashi K. Characteristics of the operon for a putrescine transport system that maps at 19 minutes on the Escherichia coli chromosome. J. Biol. Chem. 1993;268:146–152. [PubMed] [Google Scholar]
- Swofford D. Phylogenetic analysis using parsimony (PAUP) Sinauer; Sunderland, MA: 1998. version 4.0b10. [Google Scholar]
- Tatusov R.L., Koonin E.V., Lipman D.J., Koonin E.V., Lipman D.J., Lipman D.J. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V., Galperin M.Y., Natale D.A., Koonin E.V., Natale D.A., Koonin E.V., Koonin E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng T.T., Gratwick K.S., Kollman J., Park D., Nies D.H., Goffeau A., Saier M.H., Gratwick K.S., Kollman J., Park D., Nies D.H., Goffeau A., Saier M.H., Kollman J., Park D., Nies D.H., Goffeau A., Saier M.H., Park D., Nies D.H., Goffeau A., Saier M.H., Nies D.H., Goffeau A., Saier M.H., Goffeau A., Saier M.H., Saier M.H. The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- Van Noort V., Snel B., Huynen M.A., Snel B., Huynen M.A., Huynen M.A. The yeast coexpression network has a small-world, scale-free architecture and can be explained by a simple model. EMBO Rep. 2004;5:280–284. doi: 10.1038/sj.embor.7400090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vert J.P. A tree kernel to analyse phylogenetic profiles. Bioinformatics. 2002;18:S276–S284. doi: 10.1093/bioinformatics/18.suppl_1.s276. [DOI] [PubMed] [Google Scholar]
- Von Mering C., Huynen M., Jaeggi D., Schmidt S., Bork P., Snel B., Huynen M., Jaeggi D., Schmidt S., Bork P., Snel B., Jaeggi D., Schmidt S., Bork P., Snel B., Schmidt S., Bork P., Snel B., Bork P., Snel B., Snel B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y.I., Karev G., Koonin E.V., Karev G., Koonin E.V., Koonin E.V. Scale-free networks in biology: New insights into the fundamentals of evolution? BioEssays. 2002;24:105–109. doi: 10.1002/bies.10059. [DOI] [PubMed] [Google Scholar]