

Abstract
Human cytomegalovirus (HCMV) infection is the most common congenital infection in developed countries and is responsible for a substantial fraction of sensorineural hearing impairment (SNHI) in children. The risk of hearing impairment is associated with viral load in urine and blood collected during the first postnatal month. However, although inner ear abnormalities are observed in some children with HCMV induced SNHI, the exact mechanism whereby congenital HCMV infection causes hearing impairment is unknown. Earlier studies using standard cytogenetic mapping techniques showed that infection of S-phase human fibroblast cells with HCMV resulted in two specific, site-directed, chromosome breaks at band positions 1q21 and 1q42 which include loci involved in dominantly and recessively inherited hearing impairment, respectively. These findings suggested that cells infected with HCMV might provide a reservoir for genetic damage and, in a clinical perspective, a scenario could be envisioned whereby hearing impairment could result from early DNA damage of dividing fetal cells rather than viral replication and cell lysis. In this work we demonstrate, using fine mapping techniques, that HCMV infection in S-phase fibroblast cells induces genetic damage at 1q23.3, within a maximal region of 37 kb, containing 5 low copy repeat (LCR) elements. The breakpoint is situated between two hearing impairment (HI) loci, DFNA49 and DFNA7, and in close proximity to the MPZ gene previously shown to be involved in autosomal dominant Charcot-Marie-Tooth syndrome (CMT1B) with auditory neuropathy.
Keywords: HCMV, cytomegalovirus, hearing impairment, DFNA7, DFNA49, MPZ, chromosome break, chromosome gap
INTRODUCTION
Human cytomegalovirus (HCMV), a member of the Herpesviridae family, is a common cause of virally induced congenital handicaps that include, among many other symptoms, deafness and blindness [1]. Based on a retrospective study, which included 130 children with hearing loss of >40 dB and PCR detection of HCMV DNA from corresponding neonatal blood spots, Barbi et al [2] suggested that more than 40% of deafness cases with an unknown cause are actually caused by congenital HCMV infection.
World wide the incidence of congenital HCMV infection varies between 0.2% and 2.5% in newborns [3]. Neonatal infection may be caused by re-activation in sero-positive mothers or by primo-infection of sero-negative mothers followed by transmission to the fetus. Ten percent of neonates infected suffer serious symptomatic infection with early development of sequelae. In addition, although most neonates infected trans-placentally are asymptomatic at birth, 10% of these children develop progressive, sensorineural, hearing impairment (HI) early in infancy (reviewed in [3]).
The exact mechanism whereby congenital HCMV infection causes HI is unclear. Cytomegalovirus and cytomegalovirus DNA have been detected in the perilymph of patients with congenital CMV infection [4-6]. Congenital CMV infection occurring in the first 7-8 weeks may cause malformations such as short cochlea, enlarged vestibular aqueduct and short and wide auditory canal (Mondini dysplasia). However, infection after the 12th week of pregnancy was postulated not to cause morphological changes in the temporal bone [7]. Fortunato et al [8] demonstrated that infection of S-phase human fibroblast cells with HCMV resulted in two specific, site-directed, chromosome breaks at positions 1q21 and 1q42. These chromosomal band locations include loci involved in dominantly and recessively inherited hearing impairment, respectively [8]. These findings suggested that cells infected with HCMV might provide a reservoir for genetic damage. Furthermore, in a clinical perspective, this opens up the possibility that disease symptoms may result from early DNA damage of dividing fetal cells rather than viral replication and cell lysis [8]. Chromosomal position 1q21-q23 contains non-syndromic hearing impairment (NSHI) loci DFNA7, DFNM1 and DFNA49 [9-11] whereas 1q42 is close to the Usher type IIa (USH2A) locus [12,13]. Thus, this spatial co-incidence of HCMV-mediated induction of site-specific chromosomal lesions with known hearing impairment loci may provide clues to understand the mechanisms involved in the development of HI in congenitally infected infants as well as providing leads to genes involved in inheritable hearing impairment.
The goals of this study were to 1) map the HCMV induced breakpoint at 1q21, 2) determine if the chromosome damage results in gaps or physical breakage and 3) investigate whether HCMV induced genetic damage potentially causes inactivation of genes at 1q21 that may be required for the development and maintenance of normal hearing.
To achieve these goals, fine mapping of the breakpoint was carried out by FISH analysis. This was followed by in silico investigation of the breakage region to see if HCMV potentially disrupts or affects specific genes, directly or indirectly, and whether any of these genes could be candidates for autosomal dominant, non-syndromic, sensorineural hearing impairment (ADNSSHI).
MATERIALS AND METHODS
Cells and virus
Primary human foreskin fibroblasts were obtained from the UCSD Medical Center and were propagated in MEM Earle's media in incubators maintained at 37°C and 5% CO2. Media was supplemented with 10% heat inactivated fetal bovine serum, L-glutamine (2 mM), penicillin (200 U/ml), streptomycin (200 μg/ml), amphotericin B (1.5 μg/ml) and gentamycin sulfate (50 μg/ml). The Towne (# VR 977) strain of HCMV was obtained from the ATCC. HCMV was propagated as described earlier [14]. Virus was used at a multiplicity of infection of five to ensure synchronous infection of all cells.
Cell cycle synchronization and infection conditions
Experiments were performed under G0 or S-phase infection conditions [15,16]. Cells were seeded into flasks and allowed to become confluent. After 2 to 3 days at confluence, cells were reseeded onto 10 cm dishes at 0.5 x 106 cells/dish. Approximately 2 h (G0) or 24 h (S-phase) after plating, media was removed and the infection inoculum (either virus or an equivalent amount of mock conditioned media, diluted in fresh culture media) was added to the cells. At 2 hours post infection (h pi), the inoculum was removed, cells were washed, and fresh culture media was added. At the indicated times pi cells were harvested for either interphase or metaphase chromosome analysis.
Pretreatment of cells in preparation for mitotic analysis
One hour prior to collection, 5 μg/ml ethidium bromide was added to the cells for 20 min to prevent over-condensation of chromosomes. Fresh media containing 0.1μg/ml demecolcine was added for an additional 20 minutes to block microtubule polymerization, and then cells were trypsinized and collected for mitotic analysis. Cells were swollen in a hypotonic buffer containing 75 mM KCl and 10 mM EDTA (to prevent nuclease activity) for 20 min at room temperature. They were then pelleted, fixed using a series of incubations in 3:1 MeOH: Acetic Acid, and metaphase spreads were prepared by standard cytogenetic methods.
Databases
BAC (Bacterial Artificial Chromosome) and PAC (P1-derived Artificial Chromosome) clones used for the chromosome breakpoint mapping were selected from the physical maps provided by the genome browsers of University of California, Santa Cruz (UCSC) (http://www.genome.ucsc.edu), the National Centre for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) and Ensembl (EMBL, WTSI) (http://www.ensembl.org/). Together with the NCBI BLAST service (http://www.ncbi.nlm.nih.gov/BLAST/) these databases were also employed to assess the extent of clone overlap as well as the gene content and marker localizations in the breakpoint region.
Fluorescent in situ hybridization (FISH) analysis
To map the HCMV induced breakpoint the following BAC/PAC clones, encompassing 25 Mb, were included in metaphase FISH analyses: ←cen 144.17 Mb - RP11-30I17, RP11-71L20, RP11-66D17, RP11-137M19, RP11-97G24, RP11-8D14, RP11-312J18, RP11-122G18, RP13-389K14, RP11-5K23, RP11-474I16, RP11-565P22, RP11-260G23, RP11-359K18, RP11-80D6, RP11-193J5, RP11-54B9, RP4-782G3, RP11-234M3, RP11-28D10, RP11-167M6, RP11-249C5, RP11- 171H24, RP11-184N12 and RP11-165A8 – 169.19 Mb tel→. Clones were purchased from Children's Hospital Oakland Research Institute (CHORI) (http://www.bacpac.chori.org/) and Research Genetics (http://www.resgen.com/index.php3). DNA was manually isolated (Nucleobond BAC 100 kit, Machery-Nagel GmbH & co. KG, Germany). Probes were labeled by nick translation with either Spectrum Orange dUTP or Spectrum Green dUTP (Vysis, Abbott Diagnostics, USA) and precipitated in the presence of Cot1 DNA (Vysis, Abbott Diagnostics, USA). In interphase FISH analysis nuclei were co-hybridized with RP11-5K23 (labeled with Spectrum Green) and RP11-122G18 (labeled with Spectrum Orange) or two adjacent Ewing sarcoma (EWS) probes R1 (Chr. 22q12), Cat. No. 30-190059 (Vysis, Abbott Diagnostics, USA). Hybridizations were performed by using 3μl probe and 18μl Hybrisol VI (Qbiogene, USA). The slides and probes were co-denatured in a HYBrite Denaturation/Hybridization System (Vysis, Abbott Diagnostics, USA) at 76°C for 5 minutes and hybridized overnight at 37°C in a humidified chamber. Slides were washed for 2 minutes (metaphase) or 8 minutes (interphase nuclei) at 76°C in 0.4 × SSC/0.3% NP-40 and 1 minute at room temperature in 2 × SSC/0.1% NP40. Slides were dried before counterstaining with DAPI (4', 6-diamidino-2-phenylindole) II (Vysis, Abbott Diagnostics, USA).
Digital-Imaging Microscopy
In each in situ hybridization (ISH) experiment >200 metaphases were analyzed. When analyzing bridging clones RP11-5K23, RP13-389K14 and RP11-122G18, 500 metaphases were scored. For interphase FISH, >300 nuclei were scored using a Nikon Eclipse E800 fluorescence microscope. In a small fraction (<10%) of interphase nuclei 3 or 4 pairs of fluorescent signals could be seen. These were disregarded in the analyses since they are unlikely to represent G0 cells but rather they represent cells that have escaped initial synchronization. Images were obtained using a CytoVision (Applied Imaging, San Jose, CA, USA) digital system equipped with a CCD camera (Cohu Inc., San Diego, USA).
RESULTS
HCMV induced breakpoint is defined by bridging clones mapping at 1q23.3
To map the HCMV induced chromosome breakpoint at 1q21 HCMV infected fibroblast cells were subjected to chromosome walks (25 Mb) by FISH analyses with BAC and PAC clones as listed in Materials and Methods. In accordance with the findings of Fortunato et al [8], inspection of metaphase chromosomes from HCMV infected cells demonstrated chromatid breaks at 1q21 and at 1q42. In mock-infected cells such breaks were absent.
Two clones, RP11-5K23 and RP13-389K14, were found to bridge the break point giving rise to split signals at 1q23.3. Hybridization with BAC clone RP11-5K23 (129.5 kb) gave rise to a strong signal at the distal side and a weak signal at the proximal side of the break point (Fig. 1a and b). Likewise, clone RP13-389K14 (8.8 kb), which overlaps with the proximal end of RP11-5K23 with 6.3 kb, mapped to both sides of the breakpoint giving a somewhat more intense FISH signal on the proximal side (Fig. 1c). Clone RP11-122G18, which overlaps with the proximal end of RP13-389K14 with 2 kb, gave rise to a discrete signal at the proximal side of the breakpoint (Fig. 1d). Thus, Fig. 1 demonstrates that the HCMV induced breakpoint corresponds to the distal end of clone RP13-389K14 and the proximal end of clone RP11-5K23, within a critical minimal interval that appears to be less than 6.3 kb, at 1q23.3. The relative location and clone order is shown in Fig. 2, lower panel.
Figure 1.
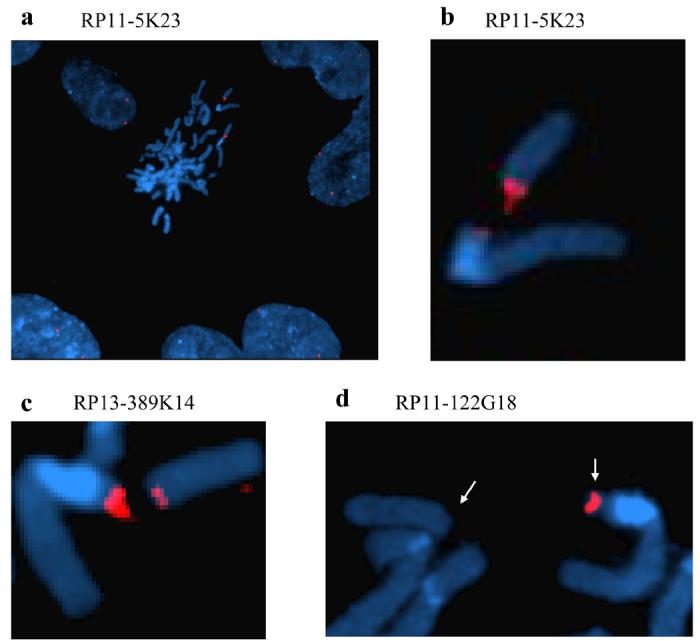
FISH analyses with BAC clones RP11- 5K23, RP11-122G18 and RP13-389K14 as probes on metaphases and nuclei from HCMV infected fibroblasts. a: A metaphase surrounded by interphase nuclei showing a chromosome 1q gap and a split fluorescent signal with probe RP11- 5K23. b: Close-up of a showing the close proximity of the two chromosome 1 segments and the RP11-5K23 FISH signal on both the distal and proximal side of the break. c: Like probe RP11-5K23, probe RP13-389K14 bridges the chromosome break giving rise to a split signal at 1q23.3, but produces a more intense signal at the proximal side of the breakpoint. d: FISH analysis with probe RP11-122G18 gives a signal at the proximal side of the break only. Note the displacement of the distal chromosome 1 segment (arrows). Blue color; interphase nuclei and metaphase chromosomes counterstained with DAPI II. Red color; BAC clones RP11- 5K23, RP11-122G18 and RP13-389K14 labeled with Spectrum Orange.
Figure 2.

An overview of the genetic structure of the HCMV induced breakpoint region is shown in the upper part of the figure with the location of the HI loci, critical markers and important candidate genes. A more detailed picture of the critical breakpoint region is shown in the lower part with positions of the BAC clones and genes mentioned in the text. The BAC clones were of the following sizes; RP11-312J18 (141 kb) RP11-122G18 (160 kb), RP13-389K14 (8.8 kb), RP11-5K23 (129.5 kb) and RP11-474I16 (106 kb). The stippled lines indicate the maximum breakpoint region of 37 kb. The minimal breakpoint region of 6.8 kb corresponds to the sequence overlap of clones RP13-389K14 and RP11-5K23.
The 1q23.3 breakpoint is located in a region with repetitive DNA elements
The breakpoint appears to be situated in a region with low gene and marker density (Fig. 2). The RP11-122G18 clone mapping to the proximal end of the breakpoint is 160 kb in length and contain two validated genes; the MPZ gene (myelin protein zero) involved in Charcot-Marie-Tooth neuropathy (CMT1B) and the SDHC gene encoding succinate dehydrogenase complex subunit C. The RP11-5K23 clone is approximately 129.5 kb in length. Four validated genes are contained within the distal end. These are FCGR2A encoding the Fc fragment of IgG low affinity receptor for CD32, HSPA6 encoding heat shock 70 kDa protein 6 (HSP70B), FCGR3A encoding the Fc fragment of IgG low affinity receptor for CD16 and FCGR3B encoding the low affinity immunoglobulin gamma Fc region.
In the proximal end of RP11-5K23, corresponding to the breakpoint, there is a region of complex repetitive primary DNA structure. Including one partially disrupted repeat segment situated in the 8.8 kb RP13-389K14 region, there are a total of five repetitive segments (>95% identity) of 7410 bp (+/− 30 bp) extending from RP13-389K14 into the proximal 37 kb of RP11-5K23 (Fig 2). Additional elements displaying a lesser degree of similarity are scattered throughout the proximal half of RP11-5K23. The low copy DNA repeat (LCR) core structure contains tandem repeat substructures; LTRs (MER11A, HERV9, LTR12C, HUERS-P3), type I transposons/LINEs (L1MA4A, L1M1) and RNA repeats. Interestingly, the RNA repeats are assembled in 5 clusters of altogether 23 putative tRNA genes encoding 9 tRNA-Gly-GGA/GGY, 4 tRNA-Glu-CAG, 5 tRNA-Asp-GAY and 5 tRNA-Leu-CTG.
The HCMV induced breakpoint maps between NSHI loci DFNA7 and DFNA49
As displayed in Fig. 2, DFNA7 encompasses 22cM (18.6Mb), between markers D1S104 and D1S466 (LOD score 7.65, θ=0) [9] an interval that also includes the DFNM1 locus (Lod score 4.32, θ=0) containing a dominant modifier that suppresses the DFNB26 phenotype [10]. The DFNA49 locus (Lod score 6.02 at θ=0) is situated 5.9 cM proximal to DFNA7, between markers D1S3786 and GDB:190880 embracing a region of 4 cM (0.9 Mb) [11]. The HCMV induced breakpoint is located 640 kb (0.64 Mb) distal to the DFNA49 locus and 2.22 Mb proximal to the DFNA7 locus.
HCMV induced chromosome 1 damage can occur early post G0 infection
Earlier results showed that DNA damage was observed in metaphase cells harvested 12 h after fibroblast cells were infected with HCMV in S-phase [8]. In order to assess whether the DNA damage could also be induced prior to cellular DNA synthesis, both G0 and S-phase infected cells were harvested early after infection (between 3 and 6 hpi) and interphase nuclei were monitored by dual color FISH analysis (Fig. 3) with BAC clones RP11-122G18/RP11-5K23 located on the proximal and distal side of the breakpoint, respectively (Fig. 2). Pair wise BLAST searches with clones RP11-122G18, RP11-5K23 and RP13-389K14 show that RP11-5K23 overlaps with RP13-389K14 and is physically separated from clone RP11-122G18 by only 2.5 kb (Fig. 2). Therefore, in interphase nuclei located in the G1 phase of the cell cycle (i.e.- prior to cellular DNA replication and having 2N DNA content), two pairs of juxtaposed signals were expected from RP11-122G18/RP11-5K23 dual-color fluorescence co-hybridization. In cells with no breaks present, partially overlapping green and red signals (with a small region of yellow overlap in between) should be observed. However, in cells that have acquired DNA damage, the red and green FISH signals should be separated, with no overlapping yellow region observable (see figure 3a for examples). In addition, since FACs analysis of prior [8] and recent experiments (data not shown) showed that between 30-50% of fibroblasts in an S phase infection were still within the G1 compartment at the time of infection (due to slow release from G0 arrest), we could also analyze cells with 2N DNA content at early times post S phase infection using this assay. To our surprise, we found that in a significant proportion of HCMV infected G0 and S-phase cells, hybridizing probes were split by distances corresponding to the width of one or more fluorescent signals. As seen in Figure 3b, DNA damage already occurred in G0/G1 cells and could be detected as early as 3 hpi in 14.2% of the nuclei and at 5 hpi in 15.2% of the nuclei, which is 7 fold above what is observed in mock-infected cells. Similar results were obtained for cells with 2N DNA content (i.e.- possessing 2 sets of signals in each nuclei for each probe) in the early times after S phase infection (Figure 3b). In a control experiment with adjacent Ewing sarcoma probes mapping to chromosome position 22q12, no significant difference was observed between HCMV infected cells and mock-infected cells (data not shown). These results confirm that the DNA damage is HCMV dependent and, furthermore, they demonstrate that HCMV induced genetic damage can occur very early post infection in cells with 2N DNA content and therefore prior to S-phase.
Figure 3.
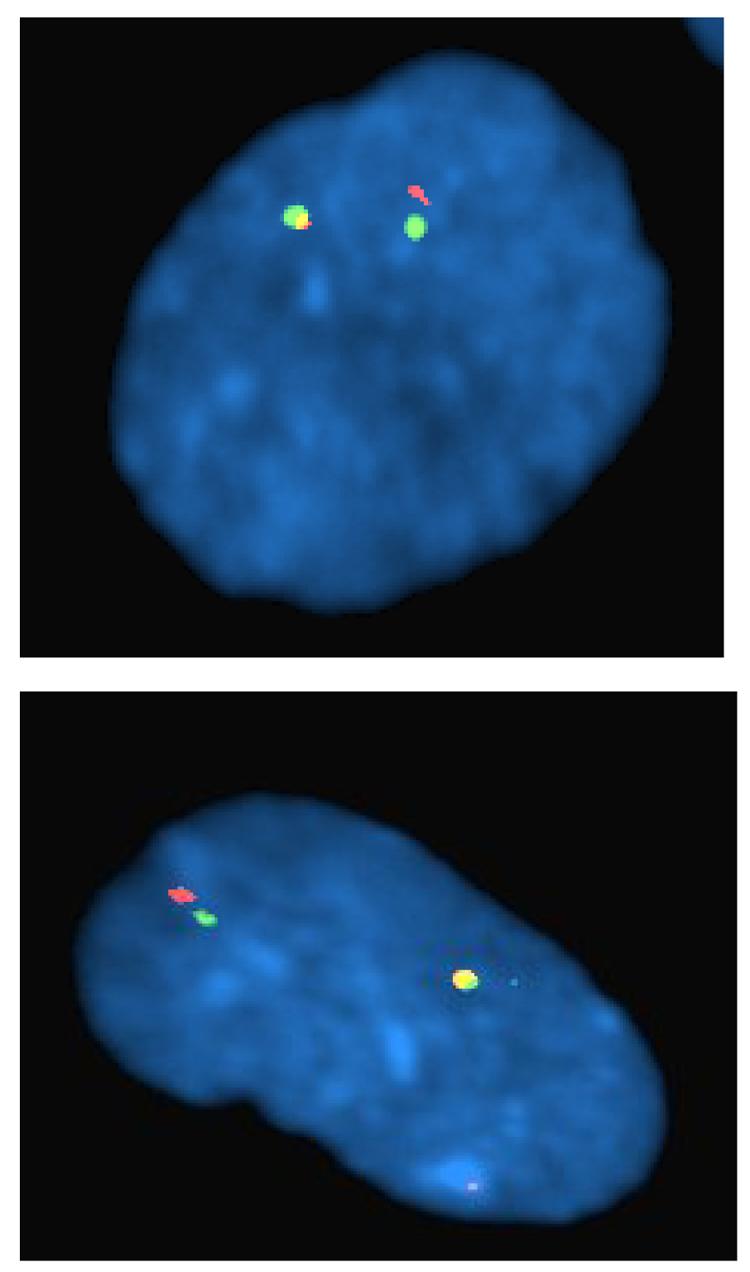

a: HCMV infected fibroblast cell at G0 showing displaced FISH signals upon hybridization with probes RP11-5K23 (green) and RP11-122G18 (red). b: FISH analyses with probes from BAC clones RP11- 5K23 and RP11-122G18 on nuclei from HCMV infected G0 and S-phase fibroblasts. Percentage of nuclei with split signals at 3 h pi, 5 h pi and 6 h pi in fibroblast cells infected in either G0 or S-phase with HCMV (filled bars) or mock supernatant (open bars).
HCMV induced 1q23.3 chromosome damage is sustained in S phase infected cells
The chromosomal lesions observed from metaphase karyograms [8] and FISH analyses appear as chromosomal discontinuity, seen as a gap, rather than displacement caused by physical breakage and separation of the distal 1q fragment. The infected cells were artificially arrested with the mitotic spindle inhibitor demecolcine prior to metaphase spread. To discriminate between gaps and breaks, FISH analysis has to be performed in the next interphase after chromosomes undergo separation as a consequence of cell division. Thus, to assess if HCMV mediated chromatid damage is sustained through G1 phase and also to assess whether physical chromosome breakage occurred, cells were infected with HCMV in S-phase and then allowed time for one round of cell division before they were harvested in interphase at 24 and 48 hrs pi. Two juxtaposed FISH signals were observed in the large majority of interphase cells (Fig. 4a). However, at 12, 24 and 48 h pi, 12.5 %, 9.2% and 16.5% of the FISH signals were separated by a distance corresponding to the width of one or more fluorescent signals, respectively (see Figures 4b and c for examples of 24 h pi hybridizations). The cell cycle profile of an S phase infection is complex, with 30-50% of cells in the G1 compartment at the time of infection. These cells express immediate early viral proteins quickly and do not proceed through the rest of the cell cycle, but arrest before dividing [17].
Figure 4.

FISH analyses of interphase nuclei at 24 h after HCMV infection with probes from clones RP11-5K23 (green) and RP11-122G18 (red). Three representative interphase nuclei are shown in which probe signals are juxtaposed (a), displaced with the width of at least one fluorescent signal (b), or showing a large displacement (c) (equivalent to the width of six fluorescent signals or more). Blue color; interphase nuclei counterstained with DAPI II.
As all of the cells showing displaced signals at 12 h pi had only 2 pairs of hybridization signals, we assume these cells sustained damage in this initial G1 phase, as 12 h is not long enough for these cells to have cycled through mitosis [17]. However, the nuclei showing displaced signals at 24 and 48 h pi, which also were predominantly showing 2 hybridization pairs, could very well be a combination of the latter cells damaged in the initial G1 as well as cells damaged in S and G2 that had subsequently cycled through mitosis. This is particularly important when we see an increase in displaced signals in the nuclei of 48 h pi cells. At this juncture, we cannot discriminate between these two populations. It is important however to realize that even if these do represent cells damaged quickly after their infection in the G1 compartment, the breaks are not resolved, even 48 h later, indicating an inherent inability to repair this damage. These results demonstrate that HCMV induced chromosome damage occurs early after infection and can potentially be sustained through metaphase and into the next interphase during cell cycling. The displacement observed may result from lack of heterochromatin condensation or from physical chromosome breakage. Indeed, in a small proportion of nuclei the extent of signal displacement was pronounced, equivalent to the width of two or more fluorescent signals, suggesting that physical chromosome breakage had occurred (Fig. 4c).
DISCUSSION
Although many viruses are known to induce generalized chromosome damage to their host cell's chromosomes, only three viruses induce locus-specific rather than random damage. These viruses are the oncogenic adenoviruses (Ad), herpes simplex virus (HSV) and human cytomegalovirus (HCMV) (reviewed in [18]).
Infection of S-phase fibroblast cells with HCMV causes locus-specific damage at 1q21 and 1q42, as seen in metaphase by G banding techniques. In contrast to Ad and HSV, induction of chromatid breakage only requires HCMV entry into the cell, not de novo viral protein synthesis [8]. While Ad and HSV have been thoroughly studied, little is known about HCMV induced damage with regard to the genetic and physical composition of the observed fragile sites, the mechanisms behind locus specificity, virus/host interactions and cellular downstream effects. Through fine-mapping techniques, we demonstrate in this work that one of the DNA lesions induced by HCMV is located at 1q23.3, in a region of five segments of repeated DNA (LCRs), encompassing ∼37 kb of DNA (Figs. 1 and 2). The lesion appears to be maintained through cell division as a fragile site, but in a small fraction of the nuclei we suspect covalent disruption seen as a large displacement of adjacent marker probes (Fig. 4c). Whether physical disruption is a consequence of mechanical strain during anaphase chromosome separation or caused by other mechanisms remains unknown. HCMV encodes its own endonuclease complex (terminase) required for the digestion of the a-sequence (pac-sites) during DNA packaging [19]. It is tempting to speculate if chromatid damage might be a result of HCMV encoded endonuclease activity. This would require DNA sequence motifs that are shared between the a-sequence and the breakpoint region. However, BLAST searches did not reveal such common DNA sequence motifs. Furthermore, HCMV has not been reported to insert its DNA into the host genome and, accordingly, metaphase FISH analysis using the HCMV genome as a probe revealed no integration of HCMV DNA (results not shown). Thus, breakage/reunion events based on recombination into the genome is not a plausible explanation for genetic damage.
Viruses have evolved elaborate interactions with the cellular repair, recombination and checkpoint machinery in order to create an environment advantageous for their own multiplication. In the host cell, cell cycle regulatory and DNA damage machinery scan for the introduction of DNA damage. Upon HCMV infection, components of this machinery are activated. However, due to improper localization of many of these components, the damage signals are not propagated [15,20]. Infection of primary fibroblasts with HCMV inhibits cell cycle progression and alters the expression of regulatory proteins such as Rb and p53 (reviewed in [21]). Interestingly in this respect, partial inhibition of DNA synthesis by chemical agents in cultured human cells induces the formation of common fragile sites seen as chromosome gaps and breaks [22]. However, even though HCMV induced alterations in replication timing could explain the emergence of random fragile sites it would not explain the site/locus specificity.
LCR segments are known to be involved in genome rearrangements such as non-allelic homologous recombination (NAHR), which mediate deletions, duplications, inversions and translocations. This property of LCR sequence is thought to be due, not to the DNA primary structure, but rather to disturbance of the complex genomic architecture that may create instability of the genome [23]. The LCR region subject to HCMV induced fragility is predicted to contain five clusters of 23 tRNA genes. Similarly, Adenovirus type 12 induces fragility at four specific loci of which three contain an array of highly transcribed small structural RNAs. Li et al [24] first demonstrated that fragility was dependent upon active RNA transcription levels in these regions.
The mechanisms by which chromosome damage/fragile sites may lead to phenotypic changes are diverse and include gene interruption, gene dosage and position effects. The HCMV induced breakpoint maps between two loci involved in autosomal dominant, non-syndromic, sensorineural hearing impairment. The breakpoint resides ∼640 kb (0.64 Mb) distal to the minimal critical region of the DFNA49 locus and ∼2.22 Mb proximal to the DFNA7 locus; thus, the HCMV induced genetic lesion does not directly disrupt genes harbored within DFNA49 and DFNA7. Affected individuals from the large DFNA7 family reported by Fagerheim et al. [9] have been investigated by DNA sequencing of the most obvious candidate genes in the 22cM interval (GJA5, GJA8, POU2F1 and including MPZ), however, no disease causing mutation has so far been identified (Fagerheim, unpublished results). Likewise, Moreno-Pelayo et al. [11] investigated candidate genes (KCNJ9, KCNJ10, ATP1A2, CASQ1) within DFNA49 without disclosing disease associated DNA sequence alterations. Long- range effects, caused by alterations in higher-order DNA structures have been shown to cause dysregulation of genes (reviewed in [25,26]. However, the question of fragile site mediated dysregulation of genes in DFNA7/DFNA49 can only be addressed by employing methods such as Northern or qPCR analysis of specific gene expression, in cell clones harboring permanent HCMV induced genetic damage. Experiments in the future will address these issues.
BAC clones, RP11-122G18, RP11-5K23 and RP13-389K14, defining the breakpoint, contain all together six validated genes of which the MPZ gene appears to be the most obvious candidate gene to be involved in hearing impairment. MPZ maps distal to DFNA49 and 130 kb proximal to the breakpoint (Fig. 2). It encodes myelin protein zero which is the major structural protein of peripheral myelin, accounting for more than 50% of the protein present in the sheath of peripheral nerves. Expression of the MPZ gene is restricted to Schwann cells and the protein is not found in the CNS [27]. Inherited mutations in MPZ are associated with Charcot-Marie-Tooth-syndrome type 1B, an autosomal dominant disorder characterized by demyelinating peripheral neuropathy with distal muscle weakness and atrophy, sensory loss, and slow nerve conduction velocity. Strikingly, some CMT1B families carrying MPZ mutations also feature auditory neuropathy (AN) characterized by abnormal auditory nerve function with hearing impairment in the presence of normal cochlear hair cell function [28-31]. Individuals with AN also suffer from impaired speech comprehension and sound localization similar to clinical features observed in HCMV congenitally infected infants. Moreover, in regard to MPZ function, neuroimaging of children with congenital HCMV infection reveals demyelination whereas grey matter abnormalities were not observed [29,32,33]. Hence, the potential relationship, between locus specific damage and hearing impairment deserves further characterization at the molecular level. Future experiments will test if gene dysregulation occurs as a consequence of altered chromatin architecture by measuring the expression levels of candidate genes, such as MPZ and those embedded in DFNA7/DFNA49. Finally, characterization of the HCMV induced breakpoint at 1q42 is underway (Fortunato et al. unpublished). Comparison of the genetic composition of the breakpoint at 1q23.3 with that of 1q42 may provide further clues to both mechanisms and phenotype.
Acknowledgements
MN, TF, VB and ØN were funded by the University Hospital of North-Norway. EAF was funded by NIH grants RO1-AI51463 and P20 RR015587.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ahlfors K, Ivarsson S-T, Harris S. Report on a long-term study of maternal and congenital cytomegalovirus infection in sweden. Review of prospective studies available in the literature. Scand J Infect Dis. 1999;31:443–457. doi: 10.1080/00365549950163969. [DOI] [PubMed] [Google Scholar]
- 2.Barbi M, Binda S, Caroppo S, Ambrosetti U, Corbetta C, Sergi P. A wider role for congenital cytomegalovirus infection in sensorineural hearing loss. Pediatr Infect Dis J. 2003;1:39–42. doi: 10.1097/00006454-200301000-00012. [DOI] [PubMed] [Google Scholar]
- 3.Lagasse N, Dhooge I, Govaert P. Congenital CMV-infection and hearing loss. Acta Otorhinolaryngol Belg. 2000;54:431–6. [PubMed] [Google Scholar]
- 4.Davis LE, James CG, Fiber F, McLaren LC. Cytomegalovirus isolation from a human inner ear. Ann Otol Rhinol Laryngol. 1979;88:424–426. doi: 10.1177/000348947908800322. [DOI] [PubMed] [Google Scholar]
- 5.Sugiura S, Yoshikawa T, Nishiyama Y, Morishita Y, Sato E, Hattori T, Nakashima T. Detection of human cytomegalovirus DNA in perilymph of patients with sensorineural hearing loss using real-time PCR. J Med Virol. 2003;69:72–5. doi: 10.1002/jmv.10263. [DOI] [PubMed] [Google Scholar]
- 6.Bauer PW, Parizi-Robinson M, Roland PS, Yegappan S. Cytomegalovirus in the perilymphatic fluid. The Laryngoscope. 2005;115:223–225. doi: 10.1097/01.mlg.0000154722.55044.fc. [DOI] [PubMed] [Google Scholar]
- 7.Bauman NM, Kirby-Keyser LJ, Dolan KD, Wexler D, Gantz BJ, McCabe BF, Bale JF., Jr Mondini dysplasia and congenital cytomegalovirus infection. J Pediatr. 1994;124:71–78. doi: 10.1016/s0022-3476(94)70256-x. [DOI] [PubMed] [Google Scholar]
- 8.Fortunato EA, Dell'Aquila ML, Spector DH. Specific chromosome 1 breaks induced by human cytomegalovirus. Proc. Natl. Acad Sci. 2000a;97:853–858. doi: 10.1073/pnas.97.2.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fagerheim T, Nilssen Ø, Raeymaekers P, Brox V, Moum T, Elverland HH, Teig E, Omland HH, Fostad GK, Tranebjærg L. Identification of a new locus for autosomal dominant non-syndromic hearing impairment (DFNA7) in a large Norwegian family. Hum Mol Genet. 1996;5:1187–1191. doi: 10.1093/hmg/5.8.1187. [DOI] [PubMed] [Google Scholar]
- 10.Riazuddin S, Castelein CM, Ahmed ZM, Lalwani AK, Mastroianni MA, Naz S, Smith TN, Liburd NA, Friedman TB, Griffith AJ, Riazuddin S, Wilcox ER. Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat Genet. 2000;26:431–434. doi: 10.1038/82558. [DOI] [PubMed] [Google Scholar]
- 11.Moreno-Pelayo MA, Modamio-Hoybjor S, Mencia A, del Castillo I, Chardenoux S, Fernandez-Burriel M, Lathrop M, Petit C, Moreno F. DFNA49, a novel locus for autosomal dominant non-syndromic hearing loss, maps proximal to DFNA7/DFNM1 region on chromosome 1q21-q23. J Med Genet. 2003;40:832–836. doi: 10.1136/jmg.40.11.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimberling WJ, Weston MD, Moller C, van Aarem A, Cremers CW, Sumegi J, Ing PS, Connolly C, Martini A, Milani M, Tamoyo ML, Bernal J, Greenberg J, Ayuso C. Gene mapping of Usher syndrome type IIa: localization of the gene to a 2.1-cM segment on chromosome 1q41. Am J Hum Genet. 1995;56:216–223. [PMC free article] [PubMed] [Google Scholar]
- 13.Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, Greenberg J, Ramesar R, Martini A, Moller C, Smith RJ, Sumegi J, Kimberling WJ. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66:1199–1210. doi: 10.1086/302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamashiro JC, Hock LJ, Spector DH. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169) J Virol. 1982;42:547–557. doi: 10.1128/jvi.42.2.547-557.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fortunato EA, Spector DH. p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J Virol. 1998;72:2033–2039. doi: 10.1128/jvi.72.3.2033-2039.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salvant BS, Fortunato EA, Spector DH. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol. 1998;72:3729–3741. doi: 10.1128/jvi.72.5.3729-3741.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fortunato EA, Sanchez V, Yen JY, Spector DH. Infection of cells with human cytomegalovirus during S phase results in a blockade to immediate-early gene expression that can be overcome by inhibition of the proteasome. J Virol. 2002;76:5369–5379. doi: 10.1128/JVI.76.11.5369-5379.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fortunato EA, Spector DH. Viral induction of site-specific chromosome damage. Rev. Med Virol. 2003;13:21–37. doi: 10.1002/rmv.368. [DOI] [PubMed] [Google Scholar]
- 19.Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res. 2002;30:1695–1703. doi: 10.1093/nar/30.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo MH, Rosenke K, Czornak K, Fortunato EA. Human cytomegalovirus disrupts both telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol. 2007;81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fortunato EA, McElroy AK, Sanchez I, Spector DH. Exploitation of cellular signalling and regulatory pathways by human cytomegalovirus. Trends Microbiol. 2000b;8:111–119. doi: 10.1016/s0966-842x(00)01699-1. [DOI] [PubMed] [Google Scholar]
- 22.Glover TW, Arlt MF, Casper AM, Durkin SG. Mechanisms of common fragile site instability. Human Molecular Genetics. 2005;14:R197–R205. doi: 10.1093/hmg/ddi265. [DOI] [PubMed] [Google Scholar]
- 23.Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet. 2004;13:R57–R64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- 24.Li YP, Tomanin R, Smiley JR, Bacchetti S. Generation of a new adenovirus type 12-inducible fragile site by insertion of an artificial U2 locus in the human genome. Mol Cell Biol. 1993;13:6064–6070. doi: 10.1128/mcb.13.10.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gericke GS. Chromosomal fragility, structural rearrangements and mobile element activity may reflect dynamic epigenetic mechanisms of importance in neurobehavioural genetics. Med Hypotheses. 2006;66:276–285. doi: 10.1016/j.mehy.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 27.Eylar EH, Uyemura K, Brostoff SW, Kitamura K, Ishaque A, Greenfield S. Proposed nomenclature for PNS myelin proteins. Neurochem Res. 1979;4:289–293. doi: 10.1007/BF00964152. [DOI] [PubMed] [Google Scholar]
- 28.Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditory neuropathy. Brain. 1996;119:741–753. doi: 10.1093/brain/119.3.741. [DOI] [PubMed] [Google Scholar]
- 29.Starr A, Michalewski HJ, Zeng FG, Fujikawa-Brooks S, Linthicum F, Kim CS, Winnier D, Keats B. Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145->Ser) Brain. 2003;126:1604–1619. doi: 10.1093/brain/awg156. [DOI] [PubMed] [Google Scholar]
- 30.Chapon F, Latour P, Diraison P, Schaeffer S, Vandenberghe A. Axonal phenotype of Charcot-Marie-Tooth disease associated with a mutation in the myelin protein zero gene. J Neurol Neurosurg Psychiatry. 1999;66:779–782. doi: 10.1136/jnnp.66.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Misu K, Yoshihara T, Shikama Y, Awaki E, Yamamoto M, Hattori N, Hirayama M, Takegami T, Nakashima K, Sobue G. An axonal form of Charcot-Marie-Tooth disease showing distinctive features in association with mutations in the peripheral myelin protein zero gene (Thr124Met or Asp75Val) J Neurol Neurosurg Psychiatry. 2000;69:806–811. doi: 10.1136/jnnp.69.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinlin MI, Nadal D, Eich GF, Martin E, Boltshauser EJ. Late intrauterine cytomegalovirus infection: clinical and neuroimaging findings. Ped Neurol. 1996;15:249–253. doi: 10.1016/s0887-8994(96)00170-1. [DOI] [PubMed] [Google Scholar]
- 33.Haginoya K, Ohura T, Kon K, Yag T, Sawashi Y, Ishii KK, Funato T, Higani S, Takahashi S, Iinuma K. Abnormal white matter lesions with sensorineural hearing loss caused by cytomegalovirus infection: retrospective diagnosis by PCR using Guthrie cards. Brain and Development. 2002;24:710–714. doi: 10.1016/s0387-7604(02)00088-8. [DOI] [PubMed] [Google Scholar]