

Abstract
Recent studies demonstrated that a synthetic fusion peptide of HIV-1 self-associates in phospholipid membranes and inhibits HIV-1 envelope glycoprotein-mediated cell fusion, presumably by interacting with the N-terminal domain of gp41 and forming inactive heteroaggregates [Kliger, Y., Aharoni, A., Rapaport, D., Jones, P., Blumenthal, R. & Shai, Y. (1997) J. Biol. Chem. 272, 13496–13505]. Here, we show that a synthetic all d-amino acid peptide corresponding to the N-terminal sequence of HIV-1 gp41 (D-WT) of HIV-1 associates with its enantiomeric wild-type fusion (WT) peptide in the membrane and inhibits cell fusion mediated by the HIV-1 envelope glycoprotein. D-WT does not inhibit cell fusion mediated by the HIV-2 envelope glycoprotein. WT and D-WT are equally potent in inducing membrane fusion. D-WT peptide but not WT peptide is resistant to proteolytic digestion. Structural analysis showed that the CD spectra of D-WT in trifluoroethanol/water is a mirror image of that of WT, and attenuated total reflectance–fourier transform infrared spectroscopy revealed similar structures and orientation for the two enantiomers in the membrane. The results reveal that the chirality of the synthetic peptide corresponding to the HIV-1 gp41 N-terminal sequence does not play a role in liposome fusion and that the peptides’ chirality is not necessarily required for peptide–peptide interaction within the membrane environment. Furthermore, studies along these lines may provide criteria to design protease-resistant therapeutic agents against HIV and other viruses.
Membrane fusion is an essential step in fundamental biological processes, such as exocytosis, fertilization, protein trafficking, membrane recycling, and enveloped virus infection. In the case of enveloped viruses, the fusion of viral and target cell membranes is mediated by envelope glycoproteins that undergo conformational changes during the fusion process (1). The envelope glycoprotein gp160 of the HIV-1, consisting of two noncovalently associated subunits, gp120 and gp41 (2), mediates the membrane fusion activity of the virus. The outer surface subunit, gp120, contains sites necessary for viral binding to target cells containing the CD4 receptor (3) whereas the transmembrane gp41 is responsible for the fusion process between viral and cell membranes (4). The binding of gp120 to the CD4 receptor induces a conformational change in the glycoprotein that results in exposure of a previously hidden hydrophobic N-terminal domain of gp41 and its penetration into the target cell membrane. This hydrophobic domain, named “fusion peptide,” shares high homology with equivalent domains of other enveloped viruses (5). Evidence for the role of the fusion peptide domain in mediating membrane fusion comes from mutagenesis studies in intact envelope proteins of several viruses (6–8) and also from studies on synthetic fusion peptides. These included fusion peptides derived from influenza virus (9–12), Simian immunodeficiency virus (13), Sendai virus (14), and HIV (15–17). A fundamental event in the fusion process is the interaction of the fusion peptide with the target membrane. It has been shown that the ability of a particular peptide to induce membrane fusion is highly dependent on its sequence, and most mutations, even of only one amino acid, severely reduced or abolished its fusogenic activity (8, 18). This is different from other families of membrane-binding peptides such as antibacterial peptides, pore-forming cytolytic peptides, and surfactants, which also exert their activities by direct interaction with lipid constituents of the cell membrane, whose sequences can be altered significantly without affecting their biological function. For example, many studies confirmed that an amphipathic α-helical structure is a sufficient criteria for a polypeptide to lyse cells and that amino acid substitutions that preserve the amphipathic structure or even slightly disrupt it do not significantly alter polypeptide activity (19–22). All d-amino acid amphipathic peptides had similar activities compared with their corresponding l-amino acid enantiomers, confirming that lysis of cell membranes does not require receptors or a stereospecific recognition between the peptide and the phospholipid head group (23–25).
The recent release of crystallographically determined structures of fragments of the fusion protein of moloney murine leukemia virus (26) and gp41 from HIV-1 (27, 28) is helpful for formulating hypotheses concerning mechanisms of membrane fusion. Comparison with the crystal structure of the influenza HA2 subunits in a low pH-induced conformation (29) reveals common structural motifs, which provide growing support for the “spring-loaded” type of mechanistic models (30–32). In this scenario, activation of the fusion protein results in release of the fusion peptide and extension of the central coiled-coil structure. The new positioning of the fusion peptides at the tip of the stalk provides for easy contact with the target cell membrane. A small group of proximal fusion proteins that simultaneously are inserted into both the viral and target membranes would constitute a potential fusion site. A concerted collapse of this protein complex, actuated by the bending-in-half of the stalks at a central hinge region, presumably would position the C-terminal transmembrane anchors and N-terminal fusion peptides on top of each other in the center, would bring the two membranes into contact, and thus would allow for formation of the fusion pore (33). According to these proposed models, protein–lipid interactions underlie biological membrane fusion. However, it is still unknown whether a specific molecular recognition between proteins and lipids is required or whether nonspecific forces at protein–membrane interfaces are responsible. Chirality characterizes specific molecular recognition, and the natural lipids are chiral compounds found only in the L-form.
To examine whether the chirality of the fusion peptide has a role in membrane fusion, we synthesized WT, the fusion peptide of HIV-1, and WT-D, its d-amino acid enantiomer, and structurally and functionally characterized them. CD and attenuated total reflectance–fourier transform infrared (ATR-FTIR) spectroscopy were used to determine the secondary structure and organization of the peptides in trifluoroethanol/water and in l-α-phosphatidyl-dl-glycerol, β-oleyl-γ-palmitoyl (POPG) vesicles. The peptides also were labeled with fluorescent probes to facilitate determination of their mode of interaction with membranes. We found that WT-D is indistinguishable from WT in all of the assays done but is not susceptible to enzymatic degradation. The most intriguing finding was that the two enantiomers, WT and WT-D, could coassemble in the membrane and that WT-D inhibits cell–cell fusion mediated by the HIV-1, but not the HIV-2, envelope glycoprotein. The results are discussed in light of proposed models for the mechanism of membrane fusion and the mechanism of the inhibitory effect.
MATERIALS AND METHODS
Materials.
Butoxycarbonyl-amino acid phenylacetamidomethyl-resin was purchased from Applied Biosystems, and butoxycarbonyl-amino acids were obtained from Peninsula Laboratories. POPG, N-[7-nitrobenz-2-oxa-1,3-diazole-4-yl] (NBD)-fluoride, and the reagents for peptide synthesis were obtained from Sigma. N-[lissamine-rhodamine B-sulfonyl]-dioleoylphosphatidylethanolamine (Rho-PE), NBD-PE, and 5-(and 6)-carboxytetra-methyl-Rho were purchased from Molecular Probes. All other reagents were of analytical grade. Buffers were prepared by using double glass-distilled water. PBS was composed of 8 g/l NaCl, 0.2 g/l KCl, 0.2 g/l KH2PO4, and 1.09 g/l Na2HPO4 (pH 7.4).
Peptide Synthesis, Fluorescent Labeling, and Purification of Peptides.
The peptides were synthesized by a solid phase method on phenylacetamidomethyl-amino acid resin (0.15 milliequivalent) as described (17, 34). Labeling of the N terminus of the peptides was achieved as described (35, 36) The synthetic peptides were purified (>95% homogenicity) by reverse-phase HPLC on a C4 reverse phase Bio-Rad analytical column (250 × 4 mm, 300-Å pore size, 5-μm particle size) by using a linear gradient of 25–80% acetonitrile in 0.05% trifluoroacetic acid for 40 min. The peptides were subjected to amino acid analysis and mass spectroscopy to confirm their composition.
Preparation of Lipid Vesicles.
Small unilamellar vesicles (SUV) were prepared by sonication of POPG. Large unilamellar vesicles (LUV) also were prepared from POPG and, when necessary, with 0.6% (molar ratio) of Rho-PE and NBD-PE as described (17).
Peptide-Induced Lipid Mixing.
Lipid mixing of POPG-LUV was measured by using a fluorescence probe dilution assay based on resonance energy transfer measurements (37), as described in details elsewhere (17).
Fluorescence Video Imaging Microscopy.
Fusion of HIV envelope glycoprotein-expressing cells with susceptible target cells was scored by fluorescence microscopy of cells labeled with membrane and aqueous dyes as described (17, 33, 38, 39). We used the TF228.1.16 cell line (40), which constitutively expresses HIV-1 envelope protein (LAI strain). Alternatively, HIV envelope proteins were expressed transiently on the surface of HeLa cells by using the recombinant vaccinia constructs vSC50, expressing the full-length envelope gene from HIV-2SBL/ISY, or vSC60, expressing the full-length envelope gene from HIV-1BH10 envelope. The infections were done after the protocol described in Jones et al. (39). The envelope-expressing cells were labeled with DiI, a lipophilic dye. SupT1 cells, labeled with the aqueous dye calcein, were used as targets. Different concentrations of WT or WT-D peptides were applied to the envelope-expressing cells before addition of SupT1 cells. After 2-hr coculture at 37°C/5% CO2, video fluorescence images were taken. The proportion of effector cells, in contact with SupT1 cells, which had fused to SupT1 cells as indicated by being labeled with both dyes, was counted.
NBD Fluorescence Measurements.
The fluorescence of an NBD-labeled peptide increases on transfer from a solution to the hydrophobic environment. NBD-labeled peptide (0.1 μM) was added to 2 ml PBS, alone or containing SUV (400 μM), and the fluorescence was recorded with excitation set at 467 nm (10 mm slit) and emission at 530 nm (8 mm slit). The emission spectra were recorded after the establishment of a constant fluorescence intensity. Under these conditions, most of the peptide was bound to the vesicles (17).
Accessibility of the Peptides to Proteolytic Cleavage in the Membrane Bound State.
An NBD-labeled peptide (0.1 μM) was added to 2 ml of PBS solution containing SUV (400 μM). The proteolytic enzyme Proteinase-K (50 μg/ml) then was added, and the fluorescence intensity was monitored at 530 nm (8 mm slit) with the excitation set at 467 nm (10 mm slit). In a control experiment, a labeled peptide was added first to the solution containing the enzyme, followed by the addition of SUV.
CD Spectroscopy.
CD spectra were obtained by using a Jasco J-500A spectropolarimeter (Jasco, Easton, MD). The spectra were scanned with a quartz optical cell with a pathlength of 2 mm at room temperature at wavelengths of 250 to 195 nm. Fractional helicities (41) were calculated as follows:
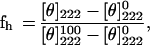 |
where [θ]222 is the experimentally observed mean residue ellipticity at 222 nm, and values for θ2220 and θ222000, corresponding to 0 and 100% helix content at 222 nm, were estimated at −2,000 and −32,000 deg⋅cm2/dmol, respectively.
ATR-FTIR Spectroscopy.
Spectra were obtained with a Perkin–Elmer 1600 FTIR spectrometer coupled with an ATR device. For each spectrum, 512 scans were collected with a resolution of 4 cm−1. The preparation of the samples was as follows. Lipid/peptide mixtures were prepared by dissolving them together in a 1:2 MeOH/CH2CL2 mixture. A mixture of phospholipids alone or with a peptide was deposited on a germanium prism (52 × 20 × 2 mm) and dried under a stream of nitrogen. The aperture angle of 45 yielded 25 internal reflections. Polarized spectra were recorded, and the respective pure phospholipid in each polarization was subtracted to yield the difference spectra to determine the amide I absorption peaks of the peptides. For each spectrum, the background was that of a clean germanium prism under the same experimental conditions. The spectra were analyzed as reported (42). Curve fitting of the amide I band area (1,600–1,700 cm−1), assuming Voight line shapes for the IR peaks, was performed to determine the relative contents of the different secondary structure elements.
ATR-FTIR data analysis.
The ATR electric fields of incident light were calculated as follows (43, 44, 42):
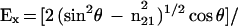 |
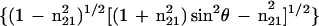 |
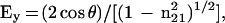 |
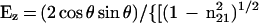 |
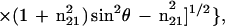 |
where θ is the angle of incidence between the light beam and the prism normal (45°) and n21 is the reflective index of the germanium (taken to be 4.03) divided by the reflective index of the membrane (taken to be 1.5). Under these conditions, Ex, Ey, and Ez are 1.40, 1.52, and 1.64, respectively. The electric field components together with the dichroic ratio [defined as the ratio between absorption of parallel (A∥) and perpendicular (A⊥) polarized light, RATR = A∥/A⊥] are used to calculate the orientation order parameter, f, by the following formula:
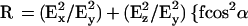 |
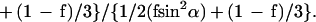 |
Lipid order parameters were obtained from the lipid symmetric (2,852 cm−1) and asymmetric (2,924 cm−1) stretching mode by using α = 90° (44).
Determining the Oligomeric States of the Peptides by SDS/PAGE.
The experiments were done as described (17, 45). The peptides were dissolved in a buffer composed of 0.065M Tris⋅HCl (pH 6.8), 2% SDS, and 10% glycerol by sonication. Fixing, staining, and destaining times were shorted to 1 min, 1 hr, and overnight, respectively, to decrease diffusion effects.
Resonance Energy Transfer Measurements.
Resonance energy transfer experiments were performed by using NBD-labeled peptides serving as energy donors and Rho-labeled peptides serving as energy acceptors as described in details elsewhere (46, 17).
RESULTS
A peptide representing the N-terminal 33-aa segment of gp41 of HIV-1 (LAV1a), WT, and its all [smcap]d-amino acid enantiomer, WT-D, were synthesized and were labeled fluorescently at their N-terminal amino acid with either NBD (an environmentally sensitive probe and an energy donor) or Rho (an energy acceptor). The sequence of the WT peptide is:
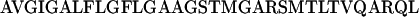 |
Liposome Fusion Induced by WT and WT-D.
In separate experiments, increasing amounts of each peptide were added to a fixed amount of vesicles, and the maximal values of the fluorescence intensity, reached after the addition of the peptides, were plotted as a function of the peptide/lipid molar ratio (Fig. 1). The data reveal that WT and WT-D have similar potency and kinetics in inducing lipid mixing.
Figure 1.

Dose dependence of lipid mixing of POPG LUV induced by WT and WT-D peptides. Peptide aliquots were added to mixtures of LUV (22 μM phospholipid concentration) containing 0.6% (molar ratio) each of NBD-PE and Rho-PE and unlabeled LUV (88 μM phospholipid concentration) in PBS. The increase of the fluorescence intensity of NBD-PE was measured at 10 min after the addition of the peptide, and the percentage from maximum was plotted versus the peptide/lipid molar ratio. The fluorescence intensity on the addition of Triton X-100 (Triton Biosciences) (0.25% vol/vol) was referred to as 100%. Squares, WT; triangles, WT-D. (Inset) Time profile of the lipid mixing at peptide/lipid ratio of 0.075.
Secondary Structure of the Peptides in trifluoroethanol/Water.
The α-helical content of WT in 40% trifluoroethanol/water was found to be 48% (Fig. 2A). WT-D showed spectra characteristic of all d-amino acids containing α-helix peptides with the mirror ellipticity at 222 corresponding to 47% α-helical content (Fig. 2A) similarly to other enantiomeric peptides (23, 24, 47).
Figure 2.

(A) CD spectra of WT (solid line) and WT-D (dashed line) peptides in 40% trifluoroethanol. (B) ATR-FTIR spectra of the peptides in POPG multibilayers. The samples were prepared as described in Material and Methods; peptide/lipid molar ratio was 1:80. The spectra were analyzed by using the curve fitting of the amide I band area assuming Voight line shapes for the IR peaks. Solid line, WT; dashed line, WT-D.
Secondary Structure of the Peptides in Lipid Multibilayers Determined by ATR-FTIR Spectroscopy.
The amide I region between 1,648 and 1,660 cm−1 is characteristic of α-helical structure (48). A spectrum of the amide I region for WT and WT-D bound to dry POPG multibilayers is shown in Fig. 2B. The contributions of the various secondary structure elements to the amide I peak were obtained by curve fitting by using peakfit (Jandel Scientific, San Rafael, CA) and the values from ref. 48. Two main peaks, located at 1,660 and 1,627 cm−1, represent α-helix and β-sheet, respectively. The α-helical and the β-sheet contents of WT and WT-D peptides were calculated at two lipid/peptide ratios. At 50:1 lipid/peptide ratio, WT and WT-D have 18 and 19% α-helix and 58 and 56% β-sheet, respectively, whereas at 80:1 lipid/peptide ratio, WT and WT-D have 25 and 26% α-helix and 46 and 50% β-sheet, respectively. There is an increase in α-helix/β-sheet ratio with an increase in the lipid/peptide ratio [which causes an increase in the fraction of membrane inserted peptide (17)], as has been shown also with short versions of HIV-1 fusion peptide (49).
Incorporation of the Peptides into Bilayers.
The fluorescence emission intensity of the NBD-labeled peptides was monitored in PBS alone or in the presence of POPG-SUV. In the buffer, the peptides (0.1 μM) exhibited emission spectra similar to the NBD moiety dissolved in water, with low intensity and maximum emission wavelength at 549 ± 1 nm (35, 50). However, in the presence of SUV (400 μM), the fluorescence emission intensity increased ≈10-fold concomitant with a blue shift of the emission maxima to 520 ± 1 nm (data not shown), indicating that the NBD probe of WT and WT-D is located equally within the hydrophobic environment of the membrane (35, 50, 51).
WT but not WT-D Is Accessible to Proteolytic Cleavage in the Membrane-Bound State.
The addition of Proteinase-K to a mixture containing NBD-WT and vesicles caused a decrease in the NBD fluorescence, demonstrating release of the probe from the hydrophobic environment of the vesicles (Fig. 3A). The level of the final fluorescence intensity was equal to that obtained when the enzyme first was added to NBD-WT, causing its cleavage, and then vesicles were added (Fig. 3B), indicating that the WT peptide is accessible to complete proteolytic cleavage in its membrane-bound state. As expected, NBD-WT-D was not accessible to enzymatic cleavage (Fig. 3C).
Figure 3.

Proteolytic digestion of membrane-bound NBD-labeled peptides. The fluorescence emission spectra of the NBD-labeled peptide was monitored at 530 nm with the excitation set at 467 nm: (1) addition of 0.1 μM NBD-labeled peptides; (2) addition of 400 μM SUV; and (3) addition of Proteinase-K (50 μg/ml). In A and B, WT was used, and in C, WT-D was used.
Orientation of the Lipid Multibilayers and the Effect of the Peptides on Lipid Acyl Chains Order.
The symmetric (νsym ≈ 2,850 cm−1) and antisymmetric (νantisym ≈ 2,920 cm−1) vibrations of lipid methylene C–H bond are perpendicular to the molecular axis of a fully extended hydrocarbon chain. Thus, measurements of the infrared absorbance dichroism show the order and the orientation of the lipid sample relative to the prism surface. The R values for the lipid and lipid/peptide samples were calculated from the stronger νantisym(CH2) peaks. The results are summarized in Table 1. The data show that the lipid samples were well ordered and that the peptides induce similar small changes in the ordering of the acyl chains (Table 1; graph not shown), as shown with other fusion peptides (49). This result, together with the finding that the N-termini of the peptides are located at similar positions [NBD fluorescence studies (50−51)], suggest similar orientation in the membrane for WT and WT-D.
Table 1.
The ATR dichroic analysis of lipid multibilayers
| Sample | lipid/peptide | R of νantisym (≈2853 cm−1) | f of lipid |
|---|---|---|---|
| Pure POPG | — | 1.28 | 0.52 |
| POPG + WT | 50 | 1.32 | 0.49 |
| POPG + WT | 80 | 1.29 | 0.52 |
| POPG + WT-D | 50 | 1.30 | 0.50 |
| POPG + WT-D | 80 | 1.28 | 0.52 |
Oligomerization of the Peptides in SDS/PAGE.
The aggregation state of several membrane proteins was determined in SDS, a membrane mimetic environment (52, 53). We have reported that the dominant form of WT fusion peptide in SDS/PAGE is a monomer and a higher order oligomer, presumably a tetramer (17). Similar results were obtained with WT-D in the present study (data not shown).
Self-Association and Coassembly of WT and WT-D in Their Membrane-Bound State: Resonance Energy Transfer Measurements.
WT peptide has been shown to self-associate in phosphatidylcholine/phosphatidylserine vesicles (17). In the present study, we have used POPG vesicles. When Rho-WT (final concentration of 0.025 to 0.5 μM) was added to a mixture of NBD-WT (0.05 μM) and POPG-SUV (400 μM), dose-dependent quenching of the donor’s emission, consistent with energy transfer, was observed. Similar experiments were done with NBD-WT/Rho-WT-D and NBD-WT-D/Rho-WT-D pairs. The energy transfer was calculated and was plotted versus the acceptor/lipid molar ratio (Fig. 4). The acceptor peptide was added only after the donor peptide already was bound to the membrane, thus decreasing peptides’ association in solution. The lipid/peptide molar ratio in these experiments was kept high to create low surface density of donors and acceptors to reduce energy transfer between unassociated peptide monomers. To confirm that the observed energy transfer was caused by peptide association, the transfer efficiencies observed in the experiments were compared with the energy transfer expected for randomly distributed membrane-bound donors and acceptors (Fig. 4, dashed line). The random distribution was calculated as described (54), assuming that 51 Å is the R0 value for the NBD/Rho donor/acceptor pair (46). The levels of energy transfer between all pairs of peptides are significantly higher then those expected for randomly distributed donors and acceptors.
Figure 4.

Fluorescence energy transfer dependence on NBD-labeled peptide acceptor concentration. The spectra were obtained for the donor peptide alone or in the presence of various amounts of an acceptor peptide. Each spectrum was recorded in the presence of 400 μM POPG-SUV in PBS. The excitation wavelength was set at 467 nm; emission was scanned from 500 to 600 nm. Transfer efficiencies between donor- and acceptor-WT (squares), donor- and acceptor-WT-D (triangles), and donor-WT and acceptor-WT-D (circles) are plotted versus the bound-acceptor/lipid molar ratio. A theoretical plot showing energy transfer efficiency as a function of the surface density of the acceptors, assuming random distribution of donors and acceptors and assuming R0 = 51Å, is given for comparison (dashed line).
Inhibition of Cell–Cell Fusion Induced by WT-D.
Fig. 5 shows fusion of HIV-1 envelope glycoprotein-expressing cells with a CD4+ cell line as a function of WT-D concentration. The pattern of inhibition is very similar to that shown for the WT peptide (17). Fitting the data to Equation 1 (see Discussion) yields an inhibition constant of 0.4 μM. As controls for specificity of the inhibition effect, we monitored inhibition by D-WT of HIV-2 envelope glycoprotein-mediated fusion (inset of Fig. 5). To directly compare the effects of the peptide on HIV-1 versus HIV-2 envelope glycoprotein-induced fusion in the same system, we expressed the envelope glycoproteins in HeLa cells by using vaccinia recombinants. The inset in Fig. 5 shows no inhibition of HIV-2 envelope glycoprotein-mediated cell fusion at concentrations of up to 3 μM peptide whereas HIV-1 envelope glycoprotein-mediated fusion was reduced significantly at this concentration. In fact, HIV-2 envelope glycoprotein-mediated cell fusion was enhanced at the relatively low concentration of peptide.
Figure 5.

Percentage fusion of TF228 cells to SupT1 cells versus peptide concentration for WT-D. The two cell types were incubated together for 2 hr before video fluorescence microscopy images were taken. Only TF228 cells in contact with SupT1 cells were counted. Empty circles, curve–fit to the lipid mixing data according to Equation 1, with ɛ = 0.81 μM and KI = 0.4 μM. (Inset) Histograms of percentage fusion of HeLa cells expressing HIV-2 gp41 (filled columns) and HIV-1 gp41 (empty columns) versus WT-D concentration.
DISCUSSION
Our findings that D-WT coassembles with the WT fusion peptide in the membrane is intriguing and seems to be unexpected. This result indicates that peptides’ chirality is not necessarily required for peptide–peptide interaction within the membrane environment. Peptide chirality was found to be crucial for peptide–protein interaction in solution in the case of the ribonuclease S-peptide/S-protein complex (55) and HIV-1 protease/substrate complex (56) but not for the binding of amphipathic peptides to calmodulin (47). Furthermore, peptide chirality was not required for antimicrobial peptides binding to bacteria (23, 24).
A second interesting observation found in this study was that the stereospecificity of the fusion peptide is not important for peptide–lipid interaction during the fusion process. To clarify the nature of the molecular interactions during the fusion process, we compared the properties of the wild type HIV-1 fusion peptides and its enantiomer WT-D by using model phospholipid systems. The natural phospholipids are all L-forms and therefore provide the biological membrane interface with stereospecificity. It has been shown that cell surfaces and phospholipid monolayers can recognize each other stereospecifically (57, 58). If there is a specific molecular recognition between peptides and lipids during the fusion process, two peptide enantiomers should show a difference in their ability to induce fusion of vesicles consisting of one enantiomer of POPG. We found that WT and WT-D have equal potency in inducing liposome fusion (Fig. 1). To understand the molecular basis for this activity, the peptides were labeled with fluorescent probes, and their structure and organization in membranes were investigated. As expected, the CD spectra of WT-D is the mirror image of that of WT (Fig. 2A). ATR-FTIR spectroscopy further demonstrates that the two enantiomers have similar secondary structures in lipid multibilayers (Fig. 2B). Both peptides also cause similar changes in the lipid order (Table 1). NBD blue shifts reveal that the N-termini of the two peptides equally penetrate into the hydrophobic core of the membrane. However, as expected, only the WT peptide is susceptible to enzymatic digestion in its membrane-bound state (Fig. 3). SDS/PAGE and resonance energy transfer revealed that both peptides self-associate to the same extent, presumably as trimers or tetrameres, as has been shown for WT peptide (17).
The most intriguing observation in this study is the inhibition of HIV-1 envelope glycoprotein-mediated cell fusion by WT-D peptide (Fig. 5). We have shown that WT fusion peptide is a potent inhibitor of cell–cell fusion (17). Our data suggest that the inhibitory effect of WT-D is derived from its ability to associate with the analogous domain in the intact fusion protein and to interfere with its action, similar to the mode of action of WT (17). This interpretation is based on the finding that WT and WT-D can coassemble in their membrane-bound state (Fig. 4). Furthermore, we labeled HIV-1 envelope glycoprotein-expressing cells with a Rho-conjugated fusion peptide and with an fluorescein isothiocyanate-conjugated anti-gp41 antibody (unpublished results). We then examined the same cells in a given field for fluorescein and Rho fluorescence by video microscopy. The micrographs indicated that the Rho fluorescence was brightest on cells with high levels of gp41 expression, as indicated by fluorescein isothiocyanate fluorescence, and that there was negligible Rho fluorescence on cells that did not express gp41. These observations indicate that the fusion peptide binds to the envelope glycoprotein and not to some putative receptor on the cell membrane. The inability of D-WT to completely inhibit cell–cell fusion may be the result of the balance between enhancing membrane fusion by promoting lipid curvature (because D-WT can induce liposome fusion by itself; see Fig. 1) and inhibiting membrane fusion by the binding of the D-WT to the fusion peptide domain of the envelope protein. The notion that the peptide enhances membrane fusion by inducing lipid curvature is supported by our data with HIV-2 envelope glycoprotein (inset of Fig. 5).
We have developed a model for inhibition of fusion pore formation by peptide inhibitors resulting in the following equation (33):
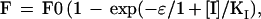 |
1 |
where F and F0 are fusion yields in the presence and absence of inhibitor, [I] is the concentration of inhibitor, and KI is the concentration of I where half of the fusion sites are inhibited and reflects the efficiency of conversion from the fusion site to the fusion pore. Peptides derived from the leucine zipper and membrane-proximal C-terminal domains of gp41 did not affect membrane curvature, and the inhibition of cell fusion was complete (33). However, to account for membrane curvature-inducing properties of the peptides derived from the N-terminal sequence of gp41, we hypothesize that the efficiency, ɛ, is enhanced linearly as a function of peptide concentration, according to
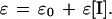 |
2 |
Incorporating this modification into Equation 1 results in the fit to the data shown in Fig. 5.
Future design of such anti-HIV agents involves peptides that are inhibitory by virtue of their molecular recognition of cognate sequences in gp41 but that are not membrane active. Our detailed knowledge concerning the properties of lipids involved in fusion and the properties of proteins that mediate the fusion as well as the interactions between the two will aid in the design of inhibitory peptides. The potent inhibitory effect of WT-D together with the fact that it is protected totally from enzymatic digestion suggests that it and its derivatives may form a basis for the development of new protease-resistant anti-AIDS drugs.
Acknowledgments
We are grateful to Dr. Zdenka Jonak for the TF228.1.16 line and Drs. Patricia Earl and Bernard Moss for the recombinant vaccinia viruses. This work was supported by the Henri and Francoise Glasberg Foundation. P.J. and R.B. are supported by the Intramural AIDS Targeted Antiviral Program.
ABBREVIATIONS
- ATR-FTIR
attenuated total reflectance–fourier transform infrared
- Rho
rhodamine
- PE
dioleoylphosphatidylethanolamine
- POPG
l-α-phosphatidyl-dl-glycerol, β-oleyl-γ-palmitoyl
- NBD
N-[l7-nitrobenz-2-oxa-1,3-diazole-4-yl]
- SUV
small unilamellar vesicles
- LUV
large unilamellar vesicles
References
- 1.White J M. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 2.Veronese F D, DeVico A L, Copeland T D, Oroszlan S, Gallo R C, Sarngadharan M G. Science. 1985;229:1402–1405. doi: 10.1126/science.2994223. [DOI] [PubMed] [Google Scholar]
- 3.Lasky L A, Nakamura G, Smith D H, Fennie C, Shimasaki C, Patzer E, Berman P, Gregory T, Capon D J. Cell. 1987;50:975–985. doi: 10.1016/0092-8674(87)90524-1. [DOI] [PubMed] [Google Scholar]
- 4.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh W C, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 5.Gallaher W R. Cell. 1987;50:327–328. doi: 10.1016/0092-8674(87)90485-5. [DOI] [PubMed] [Google Scholar]
- 6.Bosch M L, Earl P L, Fargnoli K, Picciafuoco S, Giombini F, Wong-Staal F, Franchini G. Science. 1989;244:694–697. doi: 10.1126/science.2541505. [DOI] [PubMed] [Google Scholar]
- 7.Freed E O, Myers D J, Risser R. Proc Natl Acad Sci USA. 1990;87:4650–4654. doi: 10.1073/pnas.87.12.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delahunty M D, Rhee I, Freed E O, Bonifacino J S. Virology. 1996;218:94–102. doi: 10.1006/viro.1996.0169. [DOI] [PubMed] [Google Scholar]
- 9.Burger K N, Wharton S A, Demel R A, Verkleij A J. Biochemistry. 1991;30:11173–11180. doi: 10.1021/bi00110a022. [DOI] [PubMed] [Google Scholar]
- 10.Clague M J, Knutson J R, Blumenthal R, Herrmann A. Biochemistry. 1991;30:5491–5497. doi: 10.1021/bi00236a023. [DOI] [PubMed] [Google Scholar]
- 11.Lear J D, DeGrado W F. J Biol Chem. 1987;262:6500–6505. [PubMed] [Google Scholar]
- 12.Epand R M, Epand R F. Biochem Biophys Res Commun. 1994;202:1420–1425. doi: 10.1006/bbrc.1994.2089. [DOI] [PubMed] [Google Scholar]
- 13.Martin I, Defrise Q F, Mandieau V, Nielsen N M, Saermark T, Burny A, Brasseur R, Ruysschaert J M, Vandenbranden M. Biochem Biophys Res Commun. 1991;175:872–879. doi: 10.1016/0006-291x(91)91646-t. [DOI] [PubMed] [Google Scholar]
- 14.Rapaport D, Shai Y. J Biol Chem. 1994;269:15124–15131. [PubMed] [Google Scholar]
- 15.Rafalski M, Lear J D, DeGrado W F. Biochemistry. 1990;29:7917–7922. doi: 10.1021/bi00486a020. [DOI] [PubMed] [Google Scholar]
- 16.Slepushkin V A, Melikyan G B, Sidorova M S, Chumakov V M, Andreev S M, Manulyan R A, Karamov E V. Biochem Biophys Res Commun. 1990;172:952–957. doi: 10.1016/0006-291x(90)90768-i. [DOI] [PubMed] [Google Scholar]
- 17.Kliger Y, Aharoni A, Rapaport D, Jones P, Blumenthal R, Shai Y. J Biol Chem. 1997;272:13496–13505. doi: 10.1074/jbc.272.21.13496. [DOI] [PubMed] [Google Scholar]
- 18.Durell S R, Martin I, Ruysschaert J M, Shai Y, Blumenthal R. Mol Membr Biol. 1997;14:97–112. doi: 10.3109/09687689709048170. [DOI] [PubMed] [Google Scholar]
- 19.White S H, Wimley W C. Curr Opin Struct Biol. 1994;4:79–86. doi: 10.1016/0959-440x(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 20.Shai Y. Trends Biochem Sci. 1995;20:460–464. doi: 10.1016/s0968-0004(00)89101-x. [DOI] [PubMed] [Google Scholar]
- 21.Epand R M, Shai Y, Segrest J P, Anantharamaiah G M. Biopolymers. 1995;37:319–338. doi: 10.1002/bip.360370504. [DOI] [PubMed] [Google Scholar]
- 22.Marsh, D. (1996) Biochem. J. 345–361. [DOI] [PMC free article] [PubMed]
- 23.Wade D, Boman A, Wahlin B, Drain C M, Andreu D, Boman H G, Merrifield R B. Proc Natl Acad Sci USA. 1990;87:4761–4765. doi: 10.1073/pnas.87.12.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bessalle R, Kapitkovsky A, Gorea A, Shalit I, Fridkin M. FEBS Lett. 1990;274:151–155. doi: 10.1016/0014-5793(90)81351-n. [DOI] [PubMed] [Google Scholar]
- 25.Merrifield E L, Mitchell S A, Ubach J, Boman H G, Andreu D, Merrifield R B. Int J Pept Protein Res. 1995;46:214–220. doi: 10.1111/j.1399-3011.1995.tb00592.x. [DOI] [PubMed] [Google Scholar]
- 26.Fass D, Harrison S C, Kim P S. Nat Struct Biol. 1996;3:465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- 27.Chan D C, Fass D, Berger J M, Kim P S. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 28.Weissenhorn W, Dessen A, Harrison S C, Skehel J J, Wiley D C. Nature (London) 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 29.Bullough P A, Hughson F M, Treharne A C, Ruigrok R W, Skehel J J, Wiley D C. J Mol Biol. 1994;236:1262–1265. doi: 10.1016/0022-2836(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 30.Carr C M, Kim P S. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 31.Hughson F M. Curr Opin Struct Biol. 1995;5:507–513. doi: 10.1016/0959-440x(95)80036-0. [DOI] [PubMed] [Google Scholar]
- 32.Gaudin Y, Ruigrok R W, Brunner J. J Gen Virol. 1995;76:1541–1556. doi: 10.1099/0022-1317-76-7-1541. [DOI] [PubMed] [Google Scholar]
- 33.Muñoz-Barroso I, Durrel S, Sakaguchi K, Appella E, Blumenthal R. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Merrifield R B, Vizioli L D, Boman H G. Biochemistry. 1982;21:5020–5031. doi: 10.1021/bi00263a028. [DOI] [PubMed] [Google Scholar]
- 35.Rapaport D, Shai Y. J Biol Chem. 1991;266:23769–23775. [PubMed] [Google Scholar]
- 36.Rapaport D, Shai Y. J Biol Chem. 1992;267:6502–6509. [PubMed] [Google Scholar]
- 37.Struck D K, Hoekstra D, Pagano R E. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 38.Puri A, Morris S J, Jones P, Ryan M, Blumenthal R. Virology. 1996;219:262–267. doi: 10.1006/viro.1996.0244. [DOI] [PubMed] [Google Scholar]
- 39.Jones P, Korte T, Blumenthal R. J Biol Chem. 1998;273:404–409. doi: 10.1074/jbc.273.1.404. [DOI] [PubMed] [Google Scholar]
- 40.Fu Y K, Hart T K, Jonak Z L, Bugelski P J. J Virol. 1993;67:3818–3825. doi: 10.1128/jvi.67.7.3818-3825.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu C S, Ikeda K, Yang J T. Biochemistry. 1981;20:566–570. doi: 10.1021/bi00506a019. [DOI] [PubMed] [Google Scholar]
- 42.Gazit E, Miller I R, Biggin P C, Sansom M S P, Shai Y. J Mol Biol. 1996;258:860–870. doi: 10.1006/jmbi.1996.0293. [DOI] [PubMed] [Google Scholar]
- 43.Harrick N J. Internal Reflection Spectroscopy. New York: Inter-science; 1967. [Google Scholar]
- 44.Ishiguro R, Kimura N, Takahashi S. Biochemistry. 1993;32:9792–9797. doi: 10.1021/bi00088a034. [DOI] [PubMed] [Google Scholar]
- 45.Schägger H, Jagow G V. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 46.Gazit E, Shai Y. Biochemistry. 1993;32:12363–12371. doi: 10.1021/bi00097a013. [DOI] [PubMed] [Google Scholar]
- 47.Fisher P J, Prendergast F G, Ehrhardt M R, Urbauer J L, Wand A J, Sedarous S S, McCormick D J, Buckley P J. Nature (London) 1994;368:651–653. doi: 10.1038/368651a0. [DOI] [PubMed] [Google Scholar]
- 48.Jackson M, Mantsch H H. Crit Rev Biochem Mol Biol. 1995;30:95–120. doi: 10.3109/10409239509085140. [DOI] [PubMed] [Google Scholar]
- 49.Martin I, Schaal H, Scheid A, Ruysschaert J M. J Virol. 1996;70:298–304. doi: 10.1128/jvi.70.1.298-304.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajarathnam K, Hochman J, Schindler M, Ferguson M S. Biochemistry. 1989;28:3168–3176. doi: 10.1021/bi00434a009. [DOI] [PubMed] [Google Scholar]
- 51.Pouny Y, Shai Y. Biochemistry. 1992;31:9482–9490. doi: 10.1021/bi00154a022. [DOI] [PubMed] [Google Scholar]
- 52.Simmerman H K B, Kobayashi Y M, Autry J M, Jones L R. J Biol Chem. 1996;271:5941–5946. doi: 10.1074/jbc.271.10.5941. [DOI] [PubMed] [Google Scholar]
- 53.Lemmon M A, Flanagan J M, Hunt J F, Adair B D, Bormann B J, Dempsey C E, Engelman D M. J Biol Chem. 1992;267:7683–7689. [PubMed] [Google Scholar]
- 54.Fung B K, Stryer L. Biochemistry. 1987;17:5241–5248. doi: 10.1021/bi00617a025. [DOI] [PubMed] [Google Scholar]
- 55.Corigliano M M, Xun L A, Ponnamperuma C, Dalzoppo D, Fontana A, Kanmera T, Chaiken I M. Int J Pept Protein Res. 1985;25:225–231. doi: 10.1111/j.1399-3011.1985.tb02168.x. [DOI] [PubMed] [Google Scholar]
- 56.Milton R C, Milton S C, Kent S B. Science. 1992;256:1445–1448. doi: 10.1126/science.1604320. [DOI] [PubMed] [Google Scholar]
- 57.Bohm C, Mohwald H, Leiserowitz L, Als-Nielsen J, Kjaer K. Biophys J. 1993;64:553–559. doi: 10.1016/S0006-3495(93)81386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanein D, Geiger B, Addadi L. Science. 1994;263:1413–1416. doi: 10.1126/science.8128221. [DOI] [PubMed] [Google Scholar]