

Abstract
Background and purpose:
Systemic administration of N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB), an antagonist of nicotinic acetylcholine receptors (nAChRs) attenuated the nicotine-induced increase in dopamine levels in nucleus accumbens (NAcc).
Experimental approach:
Using in vivo microdialysis, we investigated the effects of local perfusion of the novel nAChR antagonist bPiDDB into the NAcc or ventral tegmental area (VTA) on increased extracellular dopamine in NAcc, induced by systemic nicotine. We also examined the concentration-dependent effects of bPiDDB on the acetylcholine (ACh)-evoked response of specific recombinant neuronal nAChR subtypes expressed in Xenopus oocytes, using electrophysiological methods.
Key results:
Nicotine (0.4 mg kg−1, s.c.) increased extracellular dopamine in NAcc, which was attenuated by intra-VTA perfusion of mecamylamine (100 μM). Intra-VTA perfusion of bPiDDB (1 and 10 μM) reduced nicotine-induced increases in extracellular dopamine in NAcc. In contrast, intra-NAcc perfusion of bPiDDB (1 or 10 μM) failed to alter the nicotine-induced increase in dopamine in NAcc. Intra-VTA perfusion of bPiDDB alone did not alter basal dopamine levels, compared to control, nor the increased dopamine in NAcc following amphetamine (0.5 mg kg−1, s.c.). Using Xenopus oocytes, bPiDDB (0.01–100 μM) inhibited the response to ACh on specific combinations of rat neuronal nAChR subunits, with highest potency at α3β4β3 and lowest potency at α6/3β2β3.
Conclusions and implications:
bPiDDB-Sensitive nAChRs involved in regulating nicotine-induced dopamine release are located in the VTA, rather than in the NAcc. As bPiDDB has properties different from the prototypical nAChR antagonist mecamylamine, further development may lead to novel nAChR antagonists for the treatment of tobacco dependence.
Keywords: nicotine, acetylcholine receptor, dopamine, nucleus accumbens, ventral tegmental area, in vivo microdialysis, HPLC, electrophysiology, Xenopus oocytes
Introduction
The mesolimbic dopamine system, which originates in the ventral tegmental area (VTA) and projects to the nucleus accumbens (NAcc), is a critical component of the reward circuitry activated by drugs of abuse, including nicotine (Corrigall et al., 1992; Picciotto and Corrigall, 2002; Dani, 2003; Balfour, 2004; Di Chiara et al., 2004; Wonnacott et al., 2005; Lecca et al., 2006). Nicotine, the principal reward-relevant alkaloid in tobacco smoke, exerts its effects by stimulating a heterogeneous family of nicotinic acetylcholine receptor (nAChR; Alexander et al., 2007) subtypes in various regions of the CNS, including the NAcc and VTA (Klink et al., 2001; Wooltorton et al., 2003; Gotti et al., 2006). Evidence suggests that these nAChRs are formed by the pentameric assembly of either homomeric or heteromeric subunits that constitute nAChRs with different functional and pharmacological properties, expressed in distinct localization patterns in the CNS (Quik et al., 2000; Le Novere et al., 2002; Nashmi and Lester, 2006). Among the diverse population of nAChRs, high-affinity α4β2* and α6β2* nAChRs, as well as low-affinity α7* nAChRs, have been identified in the VTA and NAcc (Clarke and Pert, 1985; Champtiaux et al., 2003; McCallum et al., 2006). Although both high- and low-affinity nAChR subtypes found in the mesolimbic dopamine system may play a role in mediating the rewarding effect of nicotine (Corrigall et al., 1994; Markou and Patterson, 2001), it is generally believed that nicotine, at a concentration relevant to smoking, activates dopaminergic neurons by stimulating predominantly high-affinity β2-containing nAChRs in the VTA (Picciotto et al., 1998; Mansvelder et al., 2002; Tapper et al., 2004). In addition, nicotine indirectly stimulates VTA dopaminergic neurons through α7 nAChRs located on glutamatergic presynaptic terminals leading to glutamate release (Mansvelder and McGehee, 2000; Wooltorton et al., 2003; Dani and Harris, 2005). Thus, differential effects of nicotine are determined by the location and functional status of nAChR subtypes in the mesolimbic system (Wooltorton et al., 2003; Gaimarri et al., 2007).
Neurochemical studies in vivo employing microdialysis techniques demonstrate that extracellular dopamine in NAcc is increased by nicotine administered either systemically or directly into the NAcc or VTA (Imperato et al., 1986; Benwell and Balfour, 1992; Nisell et al., 1994; Marshall et al., 1997; Rahman et al., 2004). nAChRs are known to be involved in the reward-relevant release of dopamine following systemic nicotine, as this effect is inhibited by the non-selective nAChR antagonist mecamylamine administered either systemically (Imperato et al., 1986; Brazell et al., 1990; Benwell et al., 1995) or directly into the VTA (Nisell et al., 1994; Fu et al., 2000; Sziraki et al., 2002). Although the exact nAChR subtypes involved in regulating mesolimbic dopamine release are not known with certainty, as mentioned previously, β2-containing receptors in the VTA are generally thought to be critically involved (Picciotto et al., 1998; Mansvelder et al., 2002; Tapper et al., 2004). However, as local perfusion of mecamylamine or methyllycaconitine (an α7 nAChR antagonist) into the NAcc reduces the dopamine-releasing effect of nicotine applied directly into the NAcc (Marshall et al., 1997), nAChR subtypes within the terminal fields may also play a role.
Currently available treatments for tobacco dependence provide therapeutic benefit predominantly by activating reward-relevant dopamine circuitry either directly (nicotine replacement) or indirectly by monoamine reuptake blockade (bupropion). Recently, varenicline, a partial agonist at α4β2 nAChRs and a full agonist at α7 nAChRs, has been added to the armamentarium for treating tobacco dependence (Coe et al., 2005; Mihalak et al., 2006). However, all of these potentially dopamine-enhancing pharmacotherapies appear to be limited in efficacy, as relapse rates among smokers attempting to quit continues to be high. As VTA dopaminergic neurons play a crucial role for nicotine reward and, given the heterogeneity of nAChR subtypes in the region (Klink et al., 2001; Wooltorton et al., 2003; Gotti et al., 2006), it is possible that the development of nAChR antagonists that specifically block nAChR subtypes modulating the effect of nicotine on mesolimbic dopamine function may offer a novel therapeutic strategy for the treatment of nicotine dependence. In support of this concept, clinical studies show that mecamylamine may be useful in a smoking cessation therapy (Rose et al., 1994; Lundahl et al., 2000). However, the therapeutic utility of mecamylamine is limited due to its lack of selectivity for these nAChRs regulating dopamine release, which leads to adverse peripheral effects such as constipation and hypotension. Therefore, development and characterization of selective novel pharmacological antagonists that target these brain nAChR subtypes regulating dopamine, specifically involved in nicotine reward, may offer therapeutic benefit over existing treatments.
Recently, our laboratory has synthesized a number of bis-aza-aromatic quaternary ammonium analogues, which have been shown to act as nAChR antagonists (Ayers et al., 2002). The novel nAChR antagonist N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB; Figure 1) was found to potently inhibit nicotine-evoked striatal [3H]dopamine release in vitro, with an IC50 of 2 nM (Dwoskin et al., 2003, 2004), probably by selective blockade of dopamine release mediated via nAChRs of the α4α6β2β3* subtypes (Salminen et al., 2004; Gotti et al., 2006). bPiDDB has also been shown to reduce nicotine-induced hyperactivity and intravenous nicotine self-administration in rats (Neugebauer et al., 2006). Consistent with its antagonist action at CNS nAChRs, we have also demonstrated that bPiDDB readily enters the CNS compartment via the blood–brain barrier choline transporter (Geldenhuys et al., 2005; Lockman et al., 2006). Using in vivo microdialysis, we have shown recently that systemic injection of bPiDDB dose-dependently reduced the nicotine-induced increase in extracellular dopamine levels in NAcc (Rahman et al., 2007), possibly by targeting brain α6β2β3* or α4α6β2β3* nAChRs. However, the location and regional specificity of bPiDDB-sensitive nAChR subtypes within the NAcc and/or VTA that mediate the bPiDDB-induced inhibition of the nicotine-induced increase in dopamine levels in the NAcc remain unknown.
Figure 1.
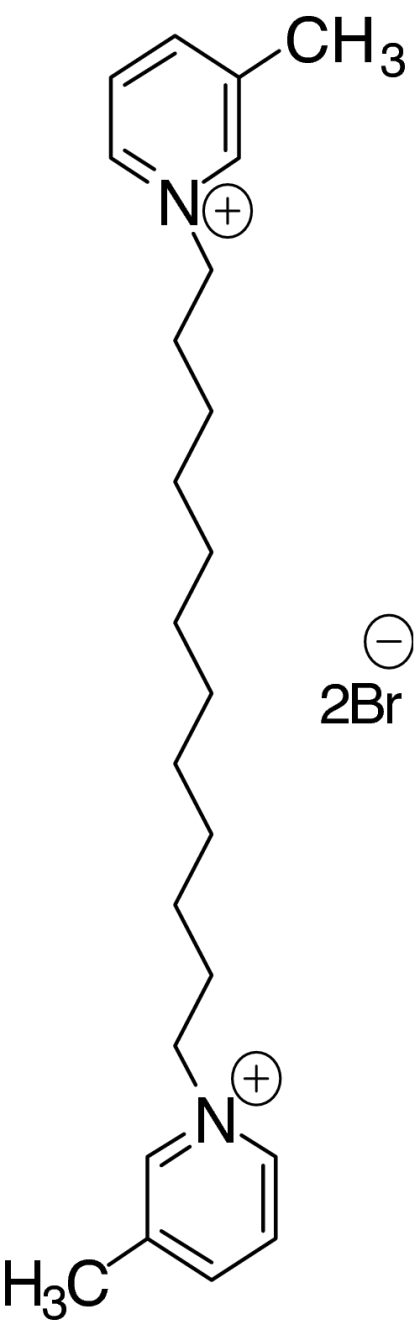
Structure of N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB).
In the current study, we investigated the effects of bPiDDB administered directly into the NAcc or VTA to alter nicotine-induced increases in extracellular dopamine in the NAcc. Using in vivo microdialysis in freely moving rats, coupled with high-performance liquid chromatography and electrochemical detection, we measured extracellular dopamine levels in rat NAcc dialysate. For these experiments, nicotine was given acutely by systemic injection, while bPiDDB or mecamylamine was perfused directly into either the VTA or NAcc through a dialysis probe. We also examined the concentration-dependent effects of bPiDDB on the acetylcholine (ACh)-evoked response of specific recombinant neuronal nAChR subtypes expressed in Xenopus oocytes using electrophysiological methods.
Methods
In vivo microdialysis
Animals
Experimental protocols were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky.
Male Sprague–Dawley rats (250–275 g) from Harlan Industries (Indianapolis, IN, USA) were used. Rats had ad libitum access to food and water and were maintained on a 12:12-h light/dark cycle (lights on at 0700 hours) in standard individual cages. Rats were acclimatized to the animal colony for at least 1 week and were handled briefly on 3–5 consecutive days prior to the start of the experiment.
Treatment regimen
To examine the effect of intra-VTA perfusion of bPiDDB on extracellular dopamine levels in NAcc following systemic nicotine injection, rats were assigned randomly to one of the six groups (n=6 per group): (1) artificial cerebral spinal fluid (ACSF)+saline; (2) ACSF+nicotine (0.4 mg kg−1); (3) mecamylamine (100 μM)+nicotine (0.4 mg kg−1); (4) bPiDDB (0.1 μM)+nicotine (0.4 mg kg−1); (5) bPiDDB (1 μM)+nicotine (0.4 mg kg−1); and (6) bPiDDB (10 μM)+nicotine (0.4 mg kg−1). In a separate experiment, rats were also given intra-VTA perfusions of bPiDDB (1–10 μM) alone, and extracellular dopamine levels determined in NAcc. The nicotine dose (0.4 mg kg−1, s.c.) used in this study was similar to the doses used in previous reports (Benwell and Balfour, 1992; Rahman et al., 2007). However, since the diffusion characteristics of bPiDDB using reverse dialysis are not known currently, the concentrations of bPiDDB used in the present study were selected from the high end of the range of concentrations used previously in an in vitro study showing that bPiDDB inhibits dopamine release in rat striatal slices (Dwoskin et al., 2003).
To determine the effect of intra-NAcc perfusion of bPiDDB on extracellular dopamine levels in NAcc following systemic nicotine injection, rats were assigned randomly to one of the six groups (n=6 per group): (1) ACSF+saline; (2) ACSF+nicotine (0.4 mg kg−1); (3) mecamylamine (100 μM)+nicotine (0.4 mg kg−1); (4) bPiDDB (0.1 μM)+nicotine (0.4 mg kg−1); (5) bPiDDB (1 μM)+nicotine (0.4 mg kg−1); and (6) bPiDDB (10 μM)+nicotine (0.4 mg kg−1). In a separate experiment, rats were also given intra-NAcc perfusions of bPiDDB (1 or 10 μM) alone and extracellular dopamine levels determined in NAcc.
In an additional experiment, to determine the effect of intra-VTA perfusion of bPiDDB on extracellular dopamine levels in NAcc following systemic amphetamine, rats were assigned randomly to one of the three groups (n=6 per group): (1) ACSF+saline; (2) ACSF+amphetamine (0.5 mg kg−1, s.c.); and (3) bPiDDB (10 μM)+amphetamine (0.5 mg kg−1, s.c.). This dose of amphetamine has been shown to reliably increase extracellular dopamine levels in NAcc (Rahman et al., 2007).
Stereotaxic surgery
Surgeries for microdialysis were performed as described previously (Rahman et al., 2007), using anaesthesia induced by xylazine (8 mg kg−1, i.p.) and ketamine hydrochloride (60 mg kg−1, i.p.). Rats were placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). For intra-VTA perfusion experiments, two microdialysis guide cannulae (18 gauge; Plastics One, Roanoke, VA, USA) were implanted ipsilaterally into the NAcc and VTA, according to the atlas of Paxinos and Watson (1986). For experiments with NAcc, one microdialysis guide cannula was implanted unilaterally into the NAcc. The cannulae were implanted at a 10° angle from the midline using the following coordinates with the incisor bar set at −3.3 mm: VTA coordinates were AP −5.2 mm from bregma, L +2.0 mm and DV −7.6 mm; and NAcc coordinates were AP +1.7 mm from bregma, L +2.1 mm and DV −6.2 mm. Rats were given carpofen (5 mg kg−1, s.c.), a non-opioid analgesic, for 3 days after the surgical procedure. Rats were allowed to recover for 3–4 days in their home cages following surgery, during which time they were allowed free access to food and water.
Microdialysis probes and procedure
Loop-style probes were made as described previously (Rahman and McBride, 2002). Loop-style probes were used instead of concentric probes, because they sample a large portion of the target area and provide higher basal levels of dopamine. Comparable loop-style probes have been used previously to measure dopamine levels in the NAcc (Benwell and Balfour, 1992; Rahman et al., 2007). Probes were constructed with dialysis membrane (Spectra/Por regenerated cellulose dialysis membrane, molecular weight cutoff of 13 000; Medical Industries, Los Angeles, CA, USA), with an outside membrane diameter of 220 μm. The probe had 2 mm of active dialysis membrane for the NAcc placement and 1 mm active membrane for the VTA. All microdialysis probes were inserted 18–24 h before initiating the microdialysis procedure (see below).
Microdialysis experiments were performed in clear Plexiglas chambers (25 × 44 × 38 cm). ACSF (in mM: 145 NaCl, 2.7 KCl, 1.0 MgCl2, 1.2 CaCl2; pH adjusted to 7.3–7.4 with 2 mM sodium phosphate buffer; filtered through a 0.2 μm sterile filter) was perfused through the probe at a flow rate of 1 μl min−1 for 120 min prior to collection of the baseline samples. After equilibration, baseline samples were collected into polyethylene microfuge tubes containing 5 μl of 0.1 N perchloric acid every 20 min for an additional 60 min before inclusion of antagonist in the ACSF. Stable baseline values for extracellular levels of dopamine in NAcc usually occurred within 60 min, as reported previously (Kohl et al., 1998; Rahman and McBride, 2002). Antagonists were dissolved in ACSF and perfused through the microdialysis probe into either VTA (intra-VTA perfusion) or NAcc (intra-NAcc perfusion). For antagonist–nicotine groups, perfusion with antagonist alone began 40 min prior to the systemic injection of nicotine and continued for an additional 100 min before switching to ACSF in the absence of antagonist. Concentrations of nAChR antagonists used for reverse dialysis were within the range of those used in previous neurochemical studies (for mecamylamine see Nisell et al., 1994; Rahman et al., 2007; for bPiDDB see Dwoskin et al., 2003). Although the actual brain tissue concentrations of the drugs delivered by reverse dialysis are not known, the concentrations are likely to be significantly lower than the amount in the probe (Kohl et al., 1998). Samples were analysed either immediately or were frozen on dry ice and stored at −70 °C until HPLC analysis. At the end of the experiments, a 1% bromphenol blue solution was perfused through the probes to verify probe placement. Rats were anaesthetized deeply with pentobarbital and brains were removed and fixed for subsequent sectioning to determine the location of the microdialysis probe. Probe placements were evaluated according to the atlas of Paxinos and Watson (1986). Only data from rats with correct probe placements in VTA or NAcc were used in the data analyses.
HPLC analysis of extracellular dopamine
Samples were analysed for dopamine using high-performance liquid chromatography and electrochemical detection (ESA Inc., Chelmsford, MA, USA). The system consisted of a solvent delivery unit, a Coulochem III electrochemical detector equipped with a 5014B analytical cell and 5020 guard cell. The guard cell was set at +350 mV, electrode 1 at −150 mV and electrode 2 at +200 mV, with the gain at 10 nA. The mobile phase was composed of 75 mM NaH2PO4, 1.7 mM 1-octanesulphonic acid, 25 μM EDTA, 100 μl l−1 triethylamine and 10% acetonitrile (pH 3.0 adjusted with phosphoric acid), and the flow rate was 0.5 ml min−1. Samples were loaded into a 20-μl sample loop and injected onto a reverse phase analytical column (BetaBasic-18 column, 150 × 3 mm; Thermo Hypersil-Keystone, Bellefonte, PA, USA). Retention times of dopamine standards were used to identify respective peaks. Chromatograms were integrated, compared with the standards and analysed using an ESA chromatography data system (EZChrom Elite, Chelmsford, MA, USA).
Data analysis of in vivo experiments
Values were not corrected for in vitro probe recovery efficiency, which was ∼15% and in close agreement with published values (Thielen et al., 2004; Rahman et al., 2007). Baseline was defined as the average dopamine release in three samples prior to drug administration. Normalized (% of basal) data were analysed by two-way repeated measures ANOVA (treatment × time) using SPSS (version 12.0; Chicago, IL, USA), followed by a post hoc Tukey HSD test for multiple comparisons, unless otherwise stated. Effects of pharmacological treatments on extracellular dopamine levels were assessed by determining measurements of area under the curve (AUC; Reid et al., 1999; Rahman et al., 2007). Increases in extracellular dopamine above the baseline (that is, percentage change from baseline value) starting from the time of drug application until the dopamine levels returned to baseline, were used to calculate AUC using the trapezoidal rule (GraphPad Prism Software Inc., San Diego, CA, USA). AUC values were analysed by one-way ANOVA, followed by post hoc analysis. In all cases, statistical significance was set at P<0.05.
Xenopus oocyte electrophysiology
nAChR expression in Xenopus oocytes
Mature (>9 cm) female Xenopus laevis African frogs (NASCO, Fort Atkinson, WI, USA) were used as a source of oocytes. Prior to surgery, anaesthesia was induced by placing the frog in a 1.5 g l−1 solution of MS222 (3-aminobenzoic acid ethyl ester) for 30 min. Oocytes were removed from an incision made in the abdomen. To remove the follicular cell layer, harvested oocytes were treated with 1.25 mg ml−1 type 1 collagenase (Worthington Biochemical Corporation, Freehold, NJ, USA) for 2 h at room temperature in calcium-free Barth's solution (in mM: 88 NaCl, 1 KCl, 0.8 MgS04, 2.4 NaHCO3, 15 HEPES (pH 7.6), 12 mg l−1 tetracycline). Subsequently, stage 5 oocytes were isolated and injected with 50 nl (5–20 ng) each of the appropriate subunit cRNAs. Recordings were made 2–15 days after injection. All procedures involving frogs were approved by the IACUC, University of Florida.
Preparation of RNA
Rat neuronal nAChR clones and mouse muscle nAChR cDNA clones were used. The wild-type clones were obtained from Dr J Boulter (University of California, Los Angeles). The rat α6/3 clone (Dowell et al., 2003) was obtained from Dr M McIntosh and modified (that is, sequence corrected; Papke et al., 2005). After linearization and purification of cloned cDNAs, RNA transcripts were prepared in vitro using the appropriate mMessage mMachine kit from Ambion Inc. (Austin, TX, USA).
Electrophysiology
Experiments were conducted using an OpusXpress 6000A unit (Molecular Devices, Union City, CA, USA). OpusXpress is an integrated system that provides automated impalement and voltage clamp of up to eight oocytes in parallel. Cells were perfused automatically with bath solution, and ACh solutions were delivered from a 96-well plate. Both voltage and current electrodes were filled with 3 M KCl. The ACh solutions (with or without bPiDDB) were applied via disposable tips, which eliminated any possibility of cross-contamination. Antagonist applications alternated between ACh controls and experimental coapplications of ACh and bPiDDB. Flow rates were set at 2 ml min−1 for experiments with α7 receptors and 4 ml min−1 for all other nAChR subtypes. Cells were voltage-clamped at a holding potential of −60 mV. Data were collected at 50 Hz and filtered at 20 Hz. ACh applications were 12 s in duration followed by 181 s washout periods for α7 receptors, and 8 s with 241 s washout periods for other nAChR subtypes.
Experimental protocols in oocytes
Each oocyte received two initial control applications of ACh, an experimental coapplication of ACh and antagonist, and then a follow-up control application of ACh. The control ACh concentrations for α4β2, α3β4, α3β2β3, α6/3β2β3, α3β4β3, α6β4β3, α7 and α1β1ɛδ receptors were 10, 100, 100, 100, 100, 100, 300 and 30 μM, respectively. These concentrations were selected, because they gave large responses with little desensitization to repeated applications, so that the same oocyte could be stimulated repeatedly with minimal decline in the amplitude of the ACh responses. This allowed us to separate the inhibitory effects of the antagonist from possible cumulative desensitization.
Data analysis of in vitro experiments
Responses to antagonist coapplications with ACh were calculated relative to the preceding ACh control responses to normalize the data, compensating for the varying levels of channel expression among the oocytes. Responses were characterized based on net charge (Papke and Papke, 2002), which is a more sensitive measure of antagonist activity than peak currents (Papke et al., 2005). For net charge measurement, a 90-s segment of data beginning 2 s prior to drug application was analysed from each response. Data were first adjusted to account for any baseline offset by subtracting the average value of the 5-s baseline period prior to drug application from all succeeding data points. Following baseline adjustment, net charge was then calculated by taking the sum of all the adjusted points. The normalized net charge values were calculated by dividing the net charge value of the experimental response by the net charge value calculated for the preceding ACh control response. The mean and standard error of the mean (s.e.mean) were calculated from the normalized responses of at least four oocytes for each antagonist concentration. To determine if there were residual inhibitory effects, the subsequent control response was compared to the pre-application control ACh response.
For concentration–response relationships, data derived from net charge analyses were plotted using Kaleidagraph 3.0.2 (Abelbeck Software, Reading, PA, USA), and curves were generated from a modified form of the Hill equation:
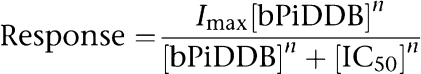 |
where Imax denotes the control response for a particular ACh/subunit combination, and n represents the Hill coefficient. On the basis of our normalization procedure, Imax was by definition equal to 1 (the response to ACh alone), whereas n and IC50 were unconstrained for the fitting procedures, and Hill slopes were seeded with the value of −1.
Drugs
S(–)-Nicotine ditartrate (Sigma-Aldrich, St Louis, MO, USA) was prepared in a 0.9% NaCl (saline) solution, to which NaOH was added to obtain a pH of 7.4. bPiDDB was synthesized according to previously established methods (Ayers et al., 2002). Mecamylamine HCl and D-amphetamine sulphate were purchased from Sigma-Aldrich. Nicotine and amphetamine solutions were prepared in saline and administered subcutaneously (s.c.) in a volume of 1 ml kg−1 body weight. Mecamylamine and bPiDDB were dissolved in ACSF (see below) and perfused through the microdialysis probe into the NAcc or VTA. The dose of nicotine represents the free base weight, whereas the doses of all other drugs represent the salt weights.
Results
Representative probe placements for the rats used in this study are shown in Figure 2. Only rats with verified probe placements in the VTA and NAcc were included. Overlapping probe placements are not shown for clarity of presentation.
Figure 2.

Representative locations of microdialysis probe placements in the nucleus accumbens (NAcc; left) and ventral tegmental area (VTA; right) as indicated in black. Numbers to the right indicate distance in millimetres from bregma according to Paxinos and Watson (1986). The active probe protruded 2 mm below the distal end of the guide cannulae for NAcc and 1 mm for the VTA as described in the Methods.
Effects of intra-VTA perfusion of bPiDDB on the nicotine-induced increase in extracellular dopamine in NAcc
Local perfusion of ACSF (n=4) through the microdialysis probe into the VTA did not alter extracellular dopamine levels in NAcc (102±3% of baseline), consistent with results obtained previously (Kohl et al., 1998; Rahman and McBride, 2002; Rahman et al., 2004). However, systemic nicotine (0.4 mg kg−1, s.c.) significantly increased extracellular levels of dopamine in NAcc (P<0.05; Figure 3). Nicotine increased extracellular dopamine to a peak ∼265% of basal and dopamine levels returned to baseline over the 100-min post-injection collection period (Figure 3a). Basal levels of dopamine did not differ significantly between control (1.1±0.2 nM) and nicotine (1.2±0.3 nM) groups.
Figure 3.

(a) Time course of the effects of acute nicotine (NIC; 0.4 mg kg−1, s.c.) alone or in combination with intra-VTA bPiDDB (0.1–10 μM) or mecamylamine (MEC; 100 μM) on extracellular dopamine levels in nucleus accumbens (NAcc). On the test day, after collection of stable basal samples, rats were perfused with bPiDDB or MEC for 40 min before NIC injection as indicated by arrow. Perfusion of bPiDDB or MEC continued for an additional 100 min. Dopamine levels represent the mean±s.e.mean of four–six rats. The average basal extracellular level of dopamine in these groups was 1.2±0.3 nM. (b) Area under the curve (AUC) for the effect of NIC alone or in combination with bPiDDB (0.1–10 μM) or MEC (100 μM). AUC values represent arbitrary units (mean ± s.e.mean) of four–six rats. *Different from control (artificial cerebral spinal fluid (ACSF)), P<0.05. #Different from NIC alone, P<0.05. (c) The effect of intra-VTA bPiDDB (1 or 10 μM) alone on extracellular dopamine levels in NAcc. After collection of basal samples, rats were perfused with ACSF or bPiDDB in the VTA, and dopamine levels were measured from NAcc. Data are the mean±s.e.mean of four–five rats. VTA, ventral tegmental area.
To examine the role of nAChRs in the VTA, systemic nicotine was combined with perfusion of mecamylamine (100 μM) or bPiDDB (0.1, 1 or 10 μM) in the VTA through the dialysis probe starting 40 min before nicotine administration and continuing for an additional 100 min (Figure 3a). A two-way mixed factor ANOVA revealed significant main effects of treatment (F(5, 27)=9.29, P<0.05) and time (F(4, 101)=13.7, P<0.05) as well as a significant treatment × time interaction (F(19, 101)=5.29, P<0.05). Post hoc comparisons revealed that mecamylamine significantly decreased the nicotine-induced enhancement of dopamine levels in NAcc (P<0.05). bPiDDB also attenuated the nicotine-induced enhancement of extracellular dopamine levels in NAcc in a concentration-dependent manner, with both 1 and 10 μM bPiDDB significantly inhibiting the time-dependent effect of nicotine (P<0.05). A one-way ANOVA of AUC data showed a significant effect of treatment (F(5, 12)=18.6, P<0.05; Figure 3b). Post hoc comparisons revealed that nicotine produced a significant increase in AUC over ACSF control, while the nicotine-induced increase in AUC was significantly inhibited by 1 and 10 μM bPiDDB (P<0.01). Local perfusion of bPiDDB (1 or 10 μM) alone in the VTA did not significantly alter extracellular dopamine levels in NAcc compared to ACSF control (Figure 3c).
Effects of intra-NAcc perfusion of bPiDDB on the nicotine-induced increase in extracellular dopamine in NAcc
To determine the role of nAChRs in NAcc, systemic nicotine was combined with perfusion of mecamylamine (100 μM) or bPiDDB (0.1, 1 or 10 μM) in the NAcc through the dialysis probe starting 40 min before nicotine and continuing for an additional 100 min (Figure 4a). A two-way mixed factor ANOVA revealed significant main effects of treatment (F(5, 21)=6.01, P<0.05) and time (F(4, 84)=49.69, P<0.05) as well as a significant treatment × time interaction (F(20, 84)=4.14, P<0.05). In contrast to the effects observed with VTA perfusion, mecamylamine produced only a modest inhibition of nicotine-induced enhancement of dopamine levels in the NAcc that was not statistically significant. Similarly, at the concentrations tested, bPiDDB failed to attenuate the nicotine-induced enhancement of extracellular dopamine in NAcc. A one-way ANOVA of AUC data showed a significant effect of treatment (F(5, 30)=3.95, P<0.05; Figure 4b). Post hoc tests revealed that, while nicotine significantly increased AUC compared to ACSF control, there was no significant inhibition of this effect of nicotine with either mecamylamine (10 μM) or bPiDDB (0.1, 1 or 10 μM). Local perfusion of bPiDDB (1–10 μM) alone into NAcc did not significantly alter extracellular dopamine in NAcc compared to the ACSF control (Figure 4c).
Figure 4.

(a) Time course of the effects of acute nicotine (NIC; 0.4 mg kg−1, s.c.) alone or in combination with intra-NAcc bPiDDB (0.1–10 μM) or mecamylamine (MEC; 100 μM) on extracellular dopamine levels in NAcc. On the test day, after collection of stable basal samples, the rats were perfused with bPiDDB or MEC for 40 min before NIC injection as indicated by arrow. Perfusion of bPiDDB or MEC continued for an additional 100 min. Data represent the mean±s.e.mean of four–six rats. (b) Area under the curve (AUC) for the effect of NIC alone or in combination with bPiDDB (0.1–10 μM) or MEC (100 μM). AUC values represent arbitrary units (mean±s.e.mean) of four–six rats. *Different from control (artificial cerebral spinal fluid (ACSF)), P<0.05. (c) The effect of intra-NAcc bPiDDB (1 or 10 μM) alone on extracellular dopamine levels in NAcc. After collection of basal samples, rats were perfused with ACSF or bPiDDB in the NAcc, and dopamine levels were measured from NAcc. Data are the mean±s.e.mean of four–five rats. NAcc, nucleus accumbens; VTA, ventral tegmental area.
Effects of intra-VTA perfusion of bPiDDB on the amphetamine-induced increase in extracellular dopamine in NAcc
We have shown previously that amphetamine (0.5 mg kg−1, s.c.) increases extracellular dopamine in NAcc, with dopamine levels returning to baseline over a 100-min post-injection collection period (Rahman et al., 2007). To determine if the ability of bPiDDB to inhibit the effect of systemic nicotine was specific to nicotine, systemic amphetamine was combined with perfusion of bPiDDB (10 μM) into the VTA through the dialysis probe starting 40 min before amphetamine and continuing for an additional 100 min (Figure 5a). A two-way mixed factor ANOVA showed significant main effects of treatment (F(2, 13)=22.38, P<0.05) and time (F(3, 5)=20.1 3, P<0.05) as well as a significant treatment × time interaction (F(3, 34)=9.83, P<0.05). Post hoc comparisons revealed that amphetamine produced a time-dependent increase in extracellular dopamine in NAcc, and that this increase was not significantly inhibited by bPiDDB (10 μM). A one-way ANOVA of AUC data showed a significant effect of treatment, F(5, 30)=3.95, P<0.05 (Figure 5b). Post hoc tests revealed that, while amphetamine significantly increased AUC compared to ACSF control, no significant inhibition of this amphetamine effect was obtained with bPiDDB (10 μM).
Figure 5.

(a) Time course effect of acute amphetamine (AMPH; 0.5 mg kg−1, s.c.) alone or in combination with intra-VTA bPiDDB (10 μM) on extracellular dopamine levels in nucleus accumbens (NAcc). On the test day, after collection of stable basal samples, rats were perfused with bPiDDB for 40 min before AMPH injection as indicated by arrow. Perfusion of bPiDDB continued for an additional 100 min. Data represent the mean±s.e.mean of four–six rats. The average basal extracellular level of dopamine in these groups was 0.9±0.2 nM. (b) Area under the curve (AUC) for the effect of AMPH alone or in combination with bPiDDB (10 μM). AUC values represent arbitrary units (mean±s.e.mean) of four–six rats. *Different from control (artificial cerebral spinal fluid (ACSF)), P<0.05.
Concentration–response relationships for bPiDDB inhibition of recombinant nAChR subtypes expressed in oocytes
After measuring control responses to ACh applications from oocytes expressing nAChR subunit combinations, ACh was coapplied with bPiDDB. Results are shown in Figure 6, and the IC50 values derived from the curve fits are given in Table 1. Of the neuronal nAChRs assessed, nAChRs containing the combination of α3 and β4 subunits (α3β4 and α3β4β3) were most sensitive to inhibition by bPiDDB with IC50 values of less than 1 μM. Other recombinant nAChR subunit combinations were sensitive to bPiDDB at higher concentrations, within the range of 5–35 μM. In all cases, the bPiDDB-induced inhibition was readily reversed with a 5-min washout (results not shown).
Figure 6.

Inhibition of acetylcholine (ACh)-evoked responses obtained from oocytes expressing rat α4β2, α3β4, α3β2β3, α6/3β2β3, α3β4β3, α6β4β3, α7 and α1β1ɛδ nicotinic acetylcholine receptor (nAChR) subunits, by bPiDDB at varying concentrations. The mean net charges of coapplication responses (±s.e.mean, n⩾4) normalized to the net charge of the ACh controls obtained from the same cells are shown. Data obtained from oocytes expressing rat subunits are represented as follows: α4β2, α3β4, α3β2β3, α6/3β2β3, α3β4β3, α6β4β3, α7 and α1β1ɛδ.
Table 1. bPiDDB inhibition of nAChR subunit combinations in Xenopus oocytes.
| Subunits | IC50 (μM) |
|---|---|
| α4β2 | 8.2±1.3 |
| α3β4 | 0.17±0.02 |
| α3β2β3 | 20±2.5 |
| α6/3β2β3 | 34±10 |
| α3β4β3 | 0.4±0.06 |
| α6β4β3 | 4.8±1.6 |
| α7 | 6.5±1.3 |
| α1β1ɛδ | 0.25±0.065 |
Abbreviations: bPiDDB, N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide; nAChR, nicotinic acetylcholine receptor.
The values shown are means±s.e.mean derived from the dose–response curves shown in Figure 6.
Discussion and conclusions
The major finding from this study is that intra-VTA perfusion of bPiDDB attenuated the nicotine-induced increase in dopamine levels in NAcc in a concentration-dependent manner. In contrast, intra-NAcc perfusion of bPiDDB failed to reduce the nicotine-induced increase in dopamine levels in NAcc. In addition, intra-VTA perfusion of bPiDDB alone did not significantly alter basal dopamine levels when compared to control. Furthermore, intra-VTA perfusion of bPiDDB failed to prevent the amphetamine-induced increase in dopamine levels in NAcc. Electrophysiological results also revealed that bPiDDB (>1 μM) inhibited ACh-induced currents in a concentration-dependent manner at multiple neuronal nAChR subtypes expressed in Xenopus oocytes.
The inhibitory effects on nicotine-induced response produced by intra-VTA bPiDDB are consistent with our previous neurochemical results employing systemic bPiDDB (Rahman et al., 2007). Within the VTA, both dopaminergic neurons and non-dopaminergic neurons, which influence nicotine-induced activity express multiple nAChR subtypes. For example, nicotine activates and rapidly desensitizes nAChRs on dopaminergic neurons in the VTA, and these nAChRs are thought to be predominantly high-affinity α4β2* nAChRs (Pidoplichko et al., 1997; Picciotto et al., 1998; Mansvelder et al., 2002; Tapper et al., 2004). Additionally, dopaminergic neurons in the VTA are activated indirectly through stimulation of α7* nAChRs located on glutamatergic presynaptic terminals leading to glutamate release and subsequent excitation of VTA dopaminergic neurons (Mansvelder and McGehee, 2000; Wooltorton et al., 2003; Dani and Harris, 2005). Electrophysiological studies demonstrate that nicotine enhances excitation of dopaminergic neurons by interacting with α7* nAChRs on excitatory glutamatergic neurons that are not readily desensitized; nicotine also activates, but more importantly, desensitizes α4β2* nAChRs located on GABAergic terminals, which impinge on the dopaminergic neurons, thus leading to disinhibition of dopaminergic neurons (Dani et al., 2001; Klink et al., 2001; Mansvelder et al., 2002; Bonci et al., 2003). Given this diversity of nAChR subtypes in the VTA, it is unclear whether single or multiple nAChR subtypes might be involved in the bPiDDB-sensitive mechanisms regulating nicotine-induced dopamine release in NAcc. However, based on results from an in vitro striatal dopamine release assay (Dwoskin et al., 2004), it is possible that α4α6β2β3* and/or α6β2β3* nAChRs in the cell body region are potential candidate receptor subtypes for those regulating dopamine release in the NAcc. Interestingly, the α6-containing nAChRs expressed in the oocyte preparation were sensitive to the inhibitory effects of bPiDDB, and the concentration of bPiDDB (0.1–10 μM) applied by reverse dialysis to the VTA was within the concentration range where inhibitory effects were observed in the oocyte preparation. However, the concentrations of bPiDDB used in these two preparations cannot be compared directly, because bPiDDB was exposed uniformly to nAChRs in the in vitro oocyte electrophysiological experiments, whereas bPiDDB was diffused across a concentration gradient in the in vivo reverse dialysis experiments. Although the exact tissue concentration of drug delivered by the microdialysis probe is not known, this concentration is lower than the drug concentration in the probe (Kohl et al., 1998; Rahman and McBride, 2002). Thus, additional studies using knockout and recombinant receptor techniques are needed to characterize the specific nAChR subtype(s) involved in the inhibitory effect of intra-VTA bPiDDB on nicotine-induced mesoaccumbal dopamine release.
As observed with bPiDDB, intra-VTA perfusion of mecamylamine blocked the acute systemic effect of nicotine on extracellular dopamine levels in NAcc. In addition, similarly to bPiDDB, intra-NAcc perfusion of mecamylamine did not significantly alter the acute systemic effect of nicotine on extracellular dopamine levels in NAcc. These findings with mecamylamine are generally consistent with previous microdialysis studies measuring extracellular dopamine levels in NAcc (Nisell et al., 1994; Fu et al., 2000) or VTA (Nisell et al., 1994; Rahman et al., 2004). The inability of intra-NAcc mecamylamine to decrease the effect of systemic nicotine suggests that nAChRs in NAcc are not involved in nicotine-induced release of accumbal dopamine. Alternatively, however, it is possible that diffusion of mecamylamine from the dialysis probe may not have been sufficient to antagonize nAChRs within the entire NAcc region that was activated by systemically administered nicotine. Consistent with this latter possibility, intra-NAcc mecamylamine has been shown to block the dopamine-releasing effect of nicotine that is perfused through the dialysis probe (Marshall et al., 1997); similar results were obtained with coperfusion of the competitive nicotinic antagonist dihydro-β-erythroidine and nicotine into NAcc (Quarta et al., 2007). Thus, it appears that there is a differential sensitivity of terminal nAChRs in blocking accumbal dopamine release induced by systemic or locally applied nicotine.
In the present study, intra-VTA perfusion of bPiDDB failed to alter the increase in extracellular accumbal dopamine induced by systemic amphetamine. This finding corroborates previous results showing that systemic bPiDDB (3 mg kg−1, s.c.) also does not affect the increase in extracellular dopamine levels in NAcc following systemic amphetamine (Rahman et al., 2007). Taken together, it appears that the neurochemical blockade produced by bPiDDB does not extrapolate from nicotine to other psychostimulant drugs. However, in another line of work, Schoffelmeer et al. (2002) found that systemic administration of mecamylamine or dihydro-β-erythroidine blocked the ability of amphetamine to induce behavioural sensitization in a locomotor assay and blocked the ability of amphetamine to enhance electrically-evoked [3H]dopamine release in an ex vivo assay using NAcc tissue slices. Although these latter findings appear to conflict with the results reported here, several experimental differences exist between the present study and that of Schoffelmeer et al. (2002). In particular, the present study measured endogenous dopamine release in vivo following acute amphetamine, whereas Schoffelmeer et al. (2002) measured electrically evoked [3H]dopamine ex vivo following repeated amphetamine. Additional studies are needed to determine if these discrepant findings reflect methodological differences between studies or are due to some inherent difference between the novel nAChR antagonist, bPiDDB, used here, and the classical nicotinic antagonists, mecamylamine and dihydro-β-erythroidine, used by Schoffelmeer et al. (2002).
Although the reverse dialysis results indicate that bPiDDB-sensitive nAChRs in the VTA are involved critically in the reward-relevant increase in mesolimbic dopamine release following nicotine administration, the electrophysiological results obtained with the oocyte heterologous expression system indicate that bPiDDB also shows activity at peripheral nAChRs. From the oocyte results, bPiDDB, as shown for mecamylamine, is a potent blocker (IC50=170 nM) of α3β4 receptors, although bPiDDB also shows comparable potency (IC50=250 nM) for blocking muscle-type receptors. Some caution is warranted in interpreting the latter results however, as a limitation of the oocyte expression system is that it is unable to express a predominant α3α5β4 nAChR ganglionic receptor subtype. That is, when these subunits are coexpressed in oocytes, the resulting receptors function similarly to α3β4 alone, and thus, there is no certainty to what extent the α5 is incorporated into these receptors.
We were surprised to find a discrepancy between the oocyte results reported here and the potent inhibition produced in the striatal superfusion assay reported earlier (Dwoskin et al., 2003). bPiDDB is potent in inhibiting nicotine-evoked dopamine release from rat striatal slices (IC50=2 nM) and does not interact at the agonist-binding site of α4β2* and α7* nAChRs (IC50=50 and >100 μM, respectively; Dwoskin et al., 2003). More recent studies show that bPiDDB interacts at α-conotoxin-sensitive nAChRs, which contain α6 and β2 subunits (Sumithran et al., unpublished). However, based on the current results, bPiDDB appears to have low affinity when evaluated in oocytes expressing α6/3β2β3 and α6β4β3 (IC50=34 and 4.8 μM, respectively). In this regard, none of the α-conotoxin-sensitive nAChRs contain the β4 subunit, and thus, the insensitivity of bPiDDB at the α6β4β3 subtype may not be surprising. Although the expressed α6β4β3 subtype was used because expression of RNA from cDNA clones does not readily produce functional receptors coexpressing α6 with other subunits of interest, such as α4 and β2, an alternative approach was to also use the α6/3 chimaera, as developed by Dowell et al. (2003). However, similar to the α6β4β3 subtype, relatively weak inhibition of α6/3β2β3 activity was observed with bPiDDB. One possible explanation for the relatively low potency of bPiDDB inhibition of the chimaeric receptors compared to putative α6-containing nAChRs receptors in the striatum is that although bPiDDB may be binding in the vestibule of the receptor, which is in the α6 sequence, the structure of the vestibule itself may be affected by how critical extracellular subdomains (such as the Cys-loop) interface with the transmembrane domains and the extracellular loop between TM2 and TM3 (Bouzat et al., 2004), which is in the α3 sequence. Thus, the topography of binding sites in the vestibule of the chimaera may be compromised relative to their conformation in the native receptors. Alternatively, the apparent discrepancy in potencies evident between the striatal superfusion and oocyte expression assays may reflect an inherent difference in the complexity of the native system compared with the oocyte expression system and/or to methodological differences used in the two assay systems. For example, while the oocyte experiments involved brief simultaneous application of agonist and antagonist, the release assays involve a prolonged (greater than 30 min) preincubation with antagonist prior to agonist treatment.
If, as we expect, bPiDDB potently interacts with native α6β2-containing nAChRs as indicated in the superfusion assay, it would be considered selective, as it was 85-fold and 125-fold more potent at α6β2-containing nAChRs than at ganglionic and muscle peripheral receptors, respectively, based on the current results. Nevertheless, from a clinical perspective, to the extent that activity at the neuromuscular junction would produce unwanted side effects, some insight into this issue can be obtained by looking at the history of the clinical use and pharmacology of mecamylamine, our reference compound in these experiments. Mecamylamine is commonly thought to be selective for neuronal (ganglionic) nAChRs. However, the IC50 value of mecamylamine for blocking muscle receptors is only about a factor of 10 higher than for blocking α3β4 (Papke et al., 2001); and although mecamylamine blocks all of the readily detectable effects of nicotine mediated by receptors in the brain (Damaj et al., 1999, 2005), its potency for inhibiting the predominant brain α4β2 receptors is no greater than its potency for muscle receptors. Mecamylamine was first used clinically as an antihypertensive agent over 50 years ago (Moyer et al., 1955; Freis and Wilson, 1956; Dennis et al., 1957; McQueen and Smirk, 1957), usually with no neuromuscular complications (Corcoran et al., 1956). This would suggest that neuromuscular junctions are perhaps protected by having a large receptor reserve. Using an irreversible antagonist to estimate receptor reserve at the neuromuscular junction, Rochel and Robbins (1987) found that 75% receptor blockade at the rat diaphragm still allowed action potentials to be generated in the muscle. If 25% of the bungarotoxin-binding sites were unbound by toxin, it would mean that only about 6% of the receptors would have had two open binding sites and actually been activatable. This would suggest that there is likely to be at least a 20-fold receptor reserve at the neuromuscular junction. Moreover, it is likely that the ‘effective' receptor reserve would be greater still for a rapidly reversible use-dependent antagonist like bPiDDB, as the brief opening of muscle type receptors would make inhibition unlikely under anything but the near steady-state conditions of the oocyte experiments. Given these considerations at the muscle-type nAChR, bPiDDB appears to represent a promising prototypical antagonist for nAChRs containing the α6 subunit that has potential as a lead compound for the clinical development of related molecules.
In conclusion, a growing body of evidence indicates that nicotine reward depends, at least in part, on activation of nAChRs at the level of the cell body region. For example, destroying dopamine-containing cells in the VTA with 6-hydroxydopamine disrupts the rewarding effect of nicotine in rodents (Corrigall et al., 1992) and local microinjection of nAChR antagonists into the VTA prevents nicotine self-administration (Corrigall et al., 1994). bPiDDB has been shown to decrease nicotine self-administration in the rat (Neugebauer et al., 2006). In vitro data also indicate that bPiDDB exhibits selective and competitive antagonism at nAChR subtypes mediating nicotine-evoked striatal dopamine release (Dwoskin et al., 2003, 2004). Thus, bPiDDB-sensitive somatodendritic nAChRs may serve as a potential target in the development of novel nAChR antagonists that may have clinical utility in the treatment of tobacco dependence.
Acknowledgments
This work was supported by USPHS Grant U19 DA17548. We acknowledge Emily Geary for her assistance during surgery, as well as Lisa Jacobs and Dolan-Abu-Aouf for the collection and analysis of the oocyte data. For purposes of full disclosure, the University of Kentucky holds patents on bPiDDB, and a potential royalty stream to LPD and PAC may occur, consistent with the University of Kentucky policy.
Glossary
- bPiDDB
N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide
- nAChR
nicotinic acetylcholine receptor
- NAcc
nucleus accumbens
- VTA
ventral tegmental area
Footnotes
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA.2007Guide to receptors and channels (GRAC)2nd edn (2007 revision)Br J Pharmacol 150Suppl 1S82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers JT, Dwoskin LP, Deaciuc AG, Grinevich VP, Zhu J, Crooks PA. Bis-Azaaromatic quaternary ammonium analogues: ligands for alpha4beta2* and alpha7* subtypes of neuronal nicotinic receptors. Bioorg Med Chem Lett. 2002;12:3067–3071. doi: 10.1016/s0960-894x(02)00687-x. [DOI] [PubMed] [Google Scholar]
- Balfour DJ. The neurobiology of tobacco dependence: a preclinical perspective on the role of the dopamine projections to the nucleus accumbens. Nicotine Tob Res. 2004;6:899–912. doi: 10.1080/14622200412331324965. [DOI] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ. The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br J Pharmacol. 1992;105:849–856. doi: 10.1111/j.1476-5381.1992.tb09067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benwell ME, Balfour DJ, Birrell CE. Desensitization of the nicotine-induced mesolimbic dopamine responses during constant infusion with nicotine. Br J Pharmacol. 1995;114:454–460. doi: 10.1111/j.1476-5381.1995.tb13248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Bernardi G, Grillner P, Mercuri NB. The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction. Trends Pharmacol Sci. 2003;24:172–177. doi: 10.1016/S0165-6147(03)00068-3. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, et al. Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature. 2004;430:896–900. doi: 10.1038/nature02753. [DOI] [PubMed] [Google Scholar]
- Brazell MP, Mitchell SN, Joseph MH, Gray JA. Acute administration of nicotine increases the in vivo extracellular levels of dopamine, 3,4-dihdroxyphenyl acetic acid and ascorbic acid preferentially in the nucleus accumbens of the rat: comparison with caudate-putamen. Neuropharmacology. 1990;29:1177–1185. doi: 10.1016/0028-3908(90)90042-p. [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Corder-Erausquin M, David DJ, Przybylski C, Lena C, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23:7820–7829. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PB, Pert A. Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res. 1985;348:355–358. doi: 10.1016/0006-8993(85)90456-1. [DOI] [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, et al. Varenicline: an alpha 4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;19:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- Corcoran AC, Dustan HP, Page IH, Schneckloth RE. Mecamylamine in treatment of hypertensive disease; observations on an unusual neuromuscular complication. J Am Med Assoc. 1956;162:868–875. doi: 10.1001/jama.1956.02970260018006. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Adamson K. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Franklin KB, Coen KM, Clarke PB. The mesolimbic dopamine system is implicated in the reinforcing effects of nicotine. Psychopharmacology. 1992;107:285–289. doi: 10.1007/BF02245149. [DOI] [PubMed] [Google Scholar]
- Damaj MI, Glassco W, Dukat M, Martin BR. Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther. 1999;291:1284–1291. [PubMed] [Google Scholar]
- Damaj MI, Wiley JL, Martin BR, Papke RL. In vivo characterization of a novel inhibitor of CNS nicotinic receptors. Eur J Pharmacol. 2005;521:43–48. doi: 10.1016/j.ejphar.2005.06.056. [DOI] [PubMed] [Google Scholar]
- Dani JA. Roles of dopamine signaling in nicotine addiction. Mol Psychiatry. 2003;8:255–256. doi: 10.1038/sj.mp.4001284. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–352. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- Dennis EW, Ford RV, Moyer JH, Hershberger RL. Mecamylamine in the treatment of hypertension. J Med Assoc Ga. 1957;46:427–430. [PubMed] [Google Scholar]
- Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology. 2004;47:227–241. doi: 10.1016/j.neuropharm.2004.06.032. [DOI] [PubMed] [Google Scholar]
- Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A, et al. Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J Neurosci. 2003;23:8445–8452. doi: 10.1523/JNEUROSCI.23-24-08445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwoskin LP, Sumithran SP, Ayers JT, Crooks PA. Development of novel treatments for nicotine addiction: bis-Picolinium, a very potent and selective antagonist at nicotinic receptor subtypes mediating nicotine-evoked dopamine release. Soc Res Nicotine Tob Abst. 2003;9:POS3–POS13. [Google Scholar]
- Dwoskin LP, Sumithran SP, Zhu J, Deaciuc AG, Ayers JT, Crooks PA. Subtype-selective nicotinic receptor antagonists: potential as tobacco use cessation agents. Bioorg Med Chem Lett. 2004;14:1863–1867. doi: 10.1016/j.bmcl.2003.10.073. [DOI] [PubMed] [Google Scholar]
- Freis ED, Wilson IM. Mecamylamine, a new, orally effective, hypotensive agent; experimental and clinical evaluation. AMA Arch Intern Med. 1956;97:551–561. doi: 10.1001/archinte.1956.00250230045005. [DOI] [PubMed] [Google Scholar]
- Fu Y, Matta SG, Gao W, Sharp BM. Local α-bungarotoxin-sensitive nicotinic receptors in the nucleus accumbens modulate nicotine-stimulated dopamine secretion in vivo. Neurosci. 2000;101:369–375. doi: 10.1016/s0306-4522(00)00371-7. [DOI] [PubMed] [Google Scholar]
- Gaimarri A, Moretti M, Riganti L, Zanardi A, Clementi F, Gotti C. Regulation of neuronal nicotinic receptor traffic and expression. Brain Res Rev. 2007;55:134–143. doi: 10.1016/j.brainresrev.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Geldenhuys WJ, Lockman PR, Nguyen TH, Van der Schyf CJ, Crooks PA, Dwoskin LP, et al. 3D-QSAR Study of bis-azaaromatic quaternary ammonium analogs at the blood–brain barrier choline transporter. Biorg Med Chem. 2005;13:4253–4261. doi: 10.1016/j.bmc.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Imperato A, Mulas A, Di Chiara G. Nicotine preferentially stimulates dopamine in the limbic system of freely moving rats. Eur J Pharmacol. 1986;132:337–338. doi: 10.1016/0014-2999(86)90629-1. [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux J-P. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–1463. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl RR, Katner JS, Chernet E, McBride WJ. Ethanol and negative feedback regulation of mesolimbic dopamine release in rats. Psychopharmacology. 1998;139:79–85. doi: 10.1007/s002130050692. [DOI] [PubMed] [Google Scholar]
- Le Novere N, Corringer P-J, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- Lecca D, Cacciapaglia F, Valentini V, Gronli J, Spiga S, Di Chiara G. Preferential increase of extracellular dopamine in the rat nucleus accumbens shell as compared to that in the core during acquisition and maintenance of intravenous nicotine self-administration. Psychopharmacology. 2006;184:435–446. doi: 10.1007/s00213-005-0280-4. [DOI] [PubMed] [Google Scholar]
- Lockman PR, Crooks PA, Manda V, Thomas F, Dwoskin LP, Allen DD. The blood–brain barrier choline transporter facilitates brain entry of the polar nicotinic receptor antagonist, N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide (bPiDDB) AAPS Annu Meeting Abstr. 2006;20:151. [Google Scholar]
- Lundahl LH, Henningfield JE, Lukas SE. Mecamylamine blockade of both positive and negative effects of IV nicotine in human volunteers. Pharmacol Biochem Behav. 2000;66:637–643. doi: 10.1016/s0091-3057(00)00252-5. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Markou A, Patterson NE. The nicotinic antagonist methyllycaconitine has differential effects on nicotine self-administration and nicotine withdrawal in the rat. Nicotine Tob Res. 2001;3:361–373. doi: 10.1080/14622200110073380. [DOI] [PubMed] [Google Scholar]
- Marshall DL, Redfern PH, Wonnacott S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naïve and chronic nicotine-treated rats. J Neurochem. 1997;68:1511–1519. doi: 10.1046/j.1471-4159.1997.68041511.x. [DOI] [PubMed] [Google Scholar]
- McCallum S, Parameswaran N, Brodia T, Fan H, McIntosh M, Quik M. Differential regulation of mesolimbic {alpha}3*/{alpha}6{beta}2* and alpha}4{beta}2* nAChR sites and function after long-term oral nicotine to monkeys. J Pharmacol Exp Ther. 2006;318:381–388. doi: 10.1124/jpet.106.104414. [DOI] [PubMed] [Google Scholar]
- McQueen EG, Smirk FH. Use of mecamylamine in the management of hypertension. Br Med J. 1957;1:422–425. doi: 10.1136/bmj.1.5016.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- Moyer JH, Ford R, Dennis E, Handley CA. Laboratory and clinical observations on mecamylamine as a hypotensive agent. Proc Soc Exp Biol Med. 1955;90:402–408. doi: 10.3181/00379727-90-22047. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Lester H. CNS localization of neuronal nicotinic receptors. J Mol Neurosci. 2006;30:181–184. doi: 10.1385/JMN:30:1:181. [DOI] [PubMed] [Google Scholar]
- Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT. Effect of a novel nicotinic receptor antagonist, N,N′-dodecane-1,12-diyl-bis-3-picolinium dibromide, on nicotine self-administration and hyperactivity in rats. Psychopharmacology. 2006;184:426–434. doi: 10.1007/s00213-005-0163-8. [DOI] [PubMed] [Google Scholar]
- Nisell M, Nomikos GG, Svensson TH. Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse. 1994;16:36–44. doi: 10.1002/syn.890160105. [DOI] [PubMed] [Google Scholar]
- Papke RL, Buhr JD, Francis MM, Choi KI, Thinschmidt JS, Horenstein NA. The effects of subunit composition on the inhibition of nicotinic receptors by the amphipathic blocker 2,2,6,6-tetramethylpiperidin-4-yl heptanoate. Mol Pharmacol. 2005;67:1977–1990. doi: 10.1124/mol.105.011676. [DOI] [PubMed] [Google Scholar]
- Papke RL, Papke JKP. Comparative pharmacology of rat and human alpha7 nAChR conducted with net charge analysis. Br J Pharmacol. 2002;137:49–61. doi: 10.1038/sj.bjp.0704833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press: New York; 1986. [Google Scholar]
- Picciotto MR, Corrigall WA. Neuronal systems underlying behaviors related to nicotine addiction: neural circuits and molecular genetics. J Neurosci. 2002;22:3338–3341. doi: 10.1523/JNEUROSCI.22-09-03338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Quarta D, Ciruela F, Patkar K, Borycz J, Solinas M, Lluis C, et al. Heteromeric nicotinic acetylcholine-dopamine autoreceptor complexes modulate striatal dopamine release. Neuropsychopharmacology. 2007;32:35–42. doi: 10.1038/sj.npp.1301103. [DOI] [PubMed] [Google Scholar]
- Quik M, Polonskaya AY, Gillespie A, Jakowec M, Lloyd GK, Langston JW. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol. 2000;425:58–69. doi: 10.1002/1096-9861(20000911)425:1<58::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rahman S, McBride WJ. Involvement of GABA and cholinergic receptors in the nucleus accumbens on feedback control of somatodendritic dopamine release in the VTA. J Neurochem. 2002;80:646–654. doi: 10.1046/j.0022-3042.2001.00739.x. [DOI] [PubMed] [Google Scholar]
- Rahman S, Neugebauer NM, Zhang Z, Crooks PA, Dwoskin LP, Bardo MT. The effects of novel N,N-dodecane-1,12-diyl-bis-3-picolinium dibromide on acute and repeated nicotine-induced increases in extracellular dopamine response in rat nucleus accumbens. Neuropharmacology. 2007;52:755–763. doi: 10.1016/j.neuropharm.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Rahman S, Zhang J, Corrigall WA. Local perfusion of nicotine differentially modulates somatodendritic dopamine release in the rat ventral tegmental area after nicotine preexposure. Neurochem Res. 2004;29:1687–1693. doi: 10.1023/b:nere.0000035803.64724.17. [DOI] [PubMed] [Google Scholar]
- Reid RT, Lloyd GK, Rao TS. Pharmacological characterization of nicotine-induced acetylcholine release in the rat hippocampus in vivo: evidence for permissive dopamine synapse. Br J Pharmacol. 1999;127:1486–1494. doi: 10.1038/sj.bjp.0702683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochel S, Robbins N. Acetylcholine receptor availability and transmission efficacy. Brain Res. 1987;435:41–47. doi: 10.1016/0006-8993(87)91584-8. [DOI] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman C, Levin D, Stein RM, Ripka GV. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin Pharmacol Ther. 1994;56:86–99. doi: 10.1038/clpt.1994.105. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Schoffelmeer AN, De Vries TJ, Wardeh G, van de Ven HW, Vanderschuren LJ. Psychostimulant-induced behavioral sensitization depends on nicotinic receptor activation. J Neurosci. 2002;22:3269–3276. doi: 10.1523/JNEUROSCI.22-08-03269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumithran SP, Babu KS, Joyce BM, Ayers JT, Deaciuc AG, Crooks PA, et al. Novel N,N′-alkane-diyl-bis-3-picolinium analogs as antagonists at nicotinic receptors: I. Inhibition of nicotine-evoked neurochemical effects(unpublished).
- Sziraki I, Sershen H, Hashim A, Lajtha A. Receptors in the ventral tegmental area mediating nicotine-induced dopamine release in the nucleus accumbens. Neurochem Res. 2002;27:253–261. doi: 10.1023/a:1014844823534. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:983–985. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Thielen RJ, Engleman EA, Rodd ZA, Murphy JM, Lumeng L, Li T-K, et al. Ethanol drinking and deprivation alter dopaminergic and serotonergic function in the nucleus accumbens of alcohol-preferring rats. J Pharmacol Exp Ther. 2004;309:216–225. doi: 10.1124/jpet.103.059790. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Sidhpura N, Balfour DJ. Nicotine: from molecular mechanisms to behaviour. Curr Opin Pharmacol. 2005;5:53–59. doi: 10.1016/j.coph.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]