

Summary
The recruitment of chromatin-modifying coregulator complexes by transcription factors to specific sites of the genome constitutes an important step in many eukaryotic transcriptional regulatory pathways. The histone deacetylase-associated Sin3 corepressor complex is recruited by a large and diverse array of transcription factors through direct interactions with the N-terminal PAH domains of Sin3. Here we describe the solution structures of the mSin3A PAH1 domain in the apo-form and when bound to SAP25, a component of the corepressor complex. Unlike the apo-mSin3A PAH2 domain, the apo-PAH1 domain is conformationally pure and is largely, but not completely, folded. Portions of the interacting segments of both mSin3A PAH1 and SAP25 undergo folding upon complex formation. SAP25 binds through an amphipathic helix to a predominantly hydrophobic cleft on the surface of PAH1. Remarkably, the orientation of the helix is reversed compared to that adopted by NRSF, a transcription factor unrelated to SAP25, upon binding to the mSin3B PAH1 domain. The reversal in helical orientations is correlated with a reversal in the underlying PAH1-interaction motifs, echoing a theme previously described for the mSin3A PAH2 domain. The definition of these so-called type I and type II PAH1-interaction motifs has allowed us to predict the precise location of these motifs within previously experimentally-characterized PAH1 binders. Finally, we explore the specificity determinants of protein-protein interactions involving the PAH1 and PAH2 domains. These studies reveal that even conservative replacements of PAH2 residues with equivalent PAH1 residues are sufficient to dramatically alter the affinity and specificity of these protein-protein interactions.
Keywords: Coregulator recruitment, eukaryotic transcription regulation, PAH domain, protein-protein interactions, Sin3 corepressor, NMR
Introduction
The transcriptional status of a eukaryotic gene is determined, in part, by the pattern of post-translational modifications of histones in the promoter regions.1 Chromatin-modifying activities such as histone deacetylases (HDACs) play an important role in this process erasing the marks established by acetyltransferases and setting the stage for further alterations of the histone code. In mammalian cells, the nuclear HDACs including HDAC1, HDAC2, and HDAC3 are typically found in large multi-subunit complexes with coregulator proteins that are in turn recruited by sequence-specific DNA binding transcription factors and chromatin-binding proteins to specific regions of the genome.2-7 The Sin3 corepressor complex is one of the better-characterized HDAC-associated coregulator complexes and is found in a broad range of species from yeast to human.5 Besides its well-documented role in negatively regulating promoter-dependent transcription, the Sin3 complex is also implicated in heterochromatin silencing,7-10 certain types of DNA repair,11,12 and in the suppression of spurious intragenic transcription initiation.13-16 Underscoring the Sin3 protein’s fundamental role in cellular physiology, sin3 knockouts are characterized by embryonic lethality or defects in development caused by deregulation of genes that control proliferation, homeostasis, and apoptosis.17,18
The mammalian Sin3 corepressor complex consists of at least nine subunits besides Sin3 including SAP30, SAP45/Sds3, SAP130, SAP180/BCAA, RBP1, HDAC1, HDAC2, RbAp46, and RbAp48.19-25 In mammals, Sin3 exists as two closely related polypeptides designated mSin3A and mSin3B each serving not only a scaffolding function for the assembly of the corepressor complex but also being available for direct recruitment by a surprisingly large and diverse array of transcription factors. The Sin3 proteins harbor six conserved regions, four of which are thought to be related to each other and were originally predicted to form a pair of amphipathic helices (PAH), a histone deacetylase interaction domain (HID), and a highly conserved C-terminal region (HCR). In the emerging view, the PAH1 and PAH2 domains are broadly available for recruitment by and interactions with diverse transcription factors,26 whereas the segment spanning the PAH3, HID and PAH4 regions is involved in the scaffolding function of Sin3 by serving as interaction sites for other subunits of the corepressor complex.5 The sequence and structural requirements for recruitment of the mammalian Sin3 proteins via the PAH2 domain are well-defined.27-31 By contrast, less is known about the mode of interaction with and recruitment of the Sin3 proteins via the PAH1 domain. Although the PAH2 domain is the closest relative of the PAH1 domain, the domains exhibit distinct patterns of sequence conservation, suggesting dissimilar modes of engaging targets (Figure 1). Like the PAH2 domain, the PAH1 domain is targeted by numerous factors including the promyelocytic leukemia-associated zinc finger (PLZF),32 the neuronal restrictive silencer factor (NRSF),33 the PHD-containing factor 1 (Pf1),34 a negative regulator of phospholipid biosynthesis Opi1,35 the myelin transcription factor 1 (Myt1),36 the nuclear receptor corepressor NCoR,37 the host cell-proliferation factor 1 (HCF1),38 and the 25 kDa Sin3 associated polypeptide (SAP25).39 Although the structure of the NRSF Sin3 interaction domain (SID) in complex with mSin3B PAH1 has been reported,33 the rules governing PAH1 interactions are not well understood, as the aforementioned PAH1-interacting proteins share little similarity with each other at the sequence level. We present below the solution structure of the mSin3A PAH1 domain in the apo-form and when bound to SAP25 SID, a recently identified component of the corepressor complex.39 We identify the conformational transitions associated with the binding event, compare the interaction modes of SAP25 and NRSF SIDs with the PAH1 domains, attempt to derive the rules for PAH1 associations, and based on this knowledge, predict the precise locations of the SIDs for various PAH1 interactors along with their binding modes. Finally, we explore the determinants of specificity of protein-protein interactions mediated by the closely related mSin3 PAH1 and PAH2 domains.
Figure 1.
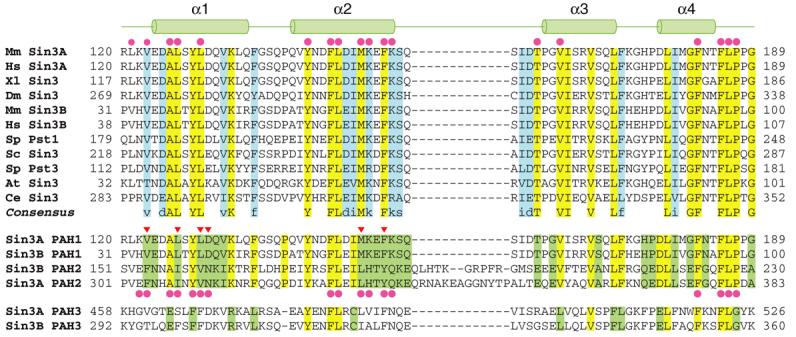
The Sin3 PAH1 domain is highly conserved and distinct from the other PAH domains. CLUSTAL W-guided multiple sequence alignment of Sin3 PAH1 domains from a variety of species (top panel; species abbreviations and GenBank accession codes: Mm: Mus musculus (Sin3A: Q60520; Sin3B: Q62141); Hs: Homo sapiens (Sin3A: Q96ST3; Sin3B: O75182); Xl: Xenopus laevis (AAD34644); Dm: Drosophila melanogaster (CAA07550); Sp: Saccharomyces pombe (Pst1: Q09750; Pst3: O74755); Sc: Saccharomyces cerevisiae (P22579); At: Arabidopsis thaliana (O48686); Ce: Caenorhabditis elegans (CAN86581)). Invariant and conserved residues are highlighted in yellow and blue, respectively. The cartoon on top depicts the location of various helices in the NMR structure of the SAP25-bound mSin3A PAH1. Residues forming the SAP25 SID-binding surface are indicated by solid circles. Multiple sequence alignment of the mouse Sin3A and Sin3B PAH1 and PAH2 domains (middle panel). Residues that are simultaneously invariant in the respective PAH1 and PAH2 domains are highlighted in green. Residues forming the Mad1/Mxd1 SID-binding surface of mSin3A PAH2 are indicated by solid circles below the alignment. Putative specificity determining residues that were mutated in our studies are indicated by inverted triangles. Alignment of the mSin3A and mSin3B PAH3 domain with other PAH1 and PAH2 domains (bottom panel). Identical residues across all three PAH domains are highlighted in yellow. Putative specificity determining residues of mSin3 PAH3 are highlighted in green.
Results and Discussion
Low-resolution analysis of mSin3A PAH1-SAP25 interactions
The 1H-15N HSQC spectrum of the apo-mSin3A PAH1 domain, unlike that of the apo-mSin3A PAH2 domain,40 is characterized by excellent chemical shift dispersion and a single set of correlations of uniform intensity indicative of a folded domain sampling a set of closely related conformations (Supplementary Figure S1(a)). A set of minor resonances could also be detected in the spectrum that is attributed to the incomplete conversion of a glutamine residue at the N-terminus to pyroglutamic acid. To test the functionality of the PAH1 construct, we titrated the protein with SAP25. Previous studies had coarsely mapped the Sin3 interaction domain (SID) of SAP25 to an ∼60 amino acid segment at the carboxy-terminus.39 Titration of 15N-labeled mSin3A PAH1 with approximately one equivalent of unlabeled SAP25 SID led to substantial changes in resonance positions for a number of residues characteristic of a specific interaction (Supplementary Figure S1(b)). This titration as well as the reverse titration of 15N-labeled SAP25 SID with unlabeled mSin3A PAH1 were characterized by the disappearance of resonances of the apo-protein and appearance of resonances at new locations indicative of a complex with slow dissociation kinetics on the NMR chemical shift timescale (Supplementary Figure S1 and S2). Isothermal titration calorimetry (ITC) analysis of the interaction between mSin3A PAH1 and SAP25 SID yielded an equilibrium dissociation constant of 134 ± 13 nM, confirming a high-affinity interaction (Supplementary Figure S3(a)).
Structure and conformation of mSin3A PAH1 and SAP25 SID in the apo and complexed states
To gain insights into the mode of the SID-PAH interaction and to characterize the conformational transitions associated with the binding event, we determined the solution structures of the apo-mSin3A PAH1 domain and that of the SAP25 SID-mSin3A PAH1 complex. The backbone and side chain resonances of each polypeptide were almost completely assigned using standard triple-resonance approaches. The structures were determined using distance and torsion angle restraints derived from 1H-1H NOEs and chemical shifts of backbone nuclei. Structures of reasonable precision and in good agreement with input experimental data were obtained (Figure 2(a) and 2(b); Table 1). Since the 1H-15N correlated spectrum of apo-SAP25 SID was characterized by poor amide proton chemical shift dispersion and uniformly intense correlations indicative of a natively unfolded polypeptide (Supplementary Figure S2(a)), conformational transitions accompanying PAH1 binding were inferred from an analysis of the secondary chemical shifts of backbone nuclei in the apo- and PAH1-bound states.
Figure 2.
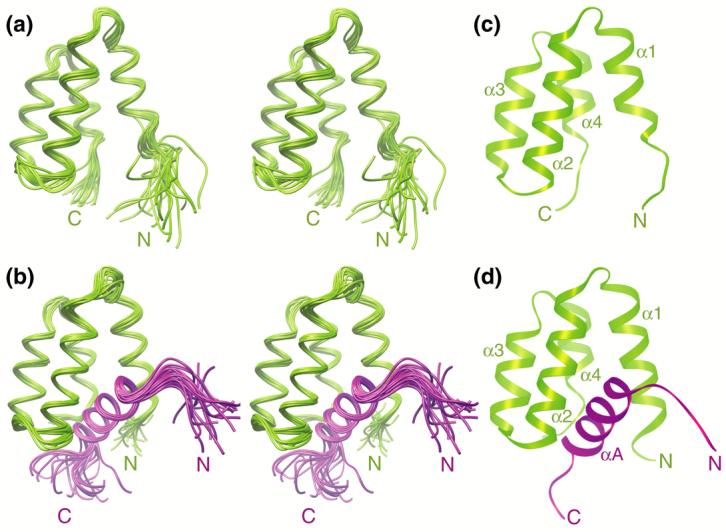
Solution NMR structures of the apo-mSin3A PAH1 domain and the SAP25 SID-mSin3A PAH1 complex. Stereographic views of the ensemble of twenty NMR structures of (a) the apo-mSin3A PAH1 domain and (b) the SAP25 SID-mSin3A PAH1 complex following a best-fit superposition of backbone atoms in structurally ordered regions. Ribbon diagrams of the representative structures of the respective ensembles are shown in panels (c) and (d). The SAP25 SID is depicted in magenta whereas the mSin3A PAH1 domain is colored green. Residues 151-186 of SAP25, although considered for the structure calculations, are essentially unstructured and have been omitted in these views for clarity.
Table 1.
NMR Structure Determination Statistics
| mSin3A PAH1 | SAP25 SID-mSin3A PAH1 complex | |
|---|---|---|
| Restraint Statistics | ||
| NOE-based distance restraints | 1506 | 1913 |
| Unambiguous NOE-based restraints | 1228 | 1420 |
| Intraresidue | 605 | 800 |
| Sequential (|i-j|=1) | 235 | 323 |
| Medium-range (1 <|i-j|≤4) | 189 | 175 |
| Intramolecular long-range (|i-j|>4) | 199 | 122 |
| Intermolecular | - | 173 |
| Ambiguous NOE-based restraints | 278 | 320 |
| Intramolecular NOE-based restraints | 1506 PAH1 | 358 SID, 1382 PAH1 |
| Hydrogen bonding distance restraints | 46 | 80 |
| Torsion angle restraints | 78 (36 ϕ, 36 ψ, 6 χ1) | 127 (58 ϕ, 57 ψ, 12 χ1) |
| Structure Quality of NMR Ensemble | ||
| Restraint satisfaction | ||
| RMS differences for distance restraints | 0.005 ± 0.001 Å | 0.009 ± 0.001 Å |
| RMS differences for torsion angle restraints | 0.054° ± 0.024° | 0.079° ± 0.023° |
| Deviations from ideal covalent geometry | ||
| Bond lengths | 0.001 ± 0.000 Å | 0.001 ± 0.000 Å |
| Bond angles | 0.273° ± 0.005° | 0.306° ± 0.010° |
| Impropers | 0.117° ± 0.007° | 0.182° ± 0.013° |
| Ramachandran plot statistics | ||
| Residues in most favored regions | 83.6% | 71.3 (83.1%)a |
| Residues in allowed regions | 15.8% | 27.1 (16.3%)a |
| Residues in disallowed regions | 0.6% | 1.6 (0.6%)a |
| Average Atomic RMSDs from the Average Structure | ||
| All atoms | 2.24 Å | 8.30 Å |
| All atoms in ordered regionsb | 1.15 Å | 1.09 Å |
| Backbone atoms (N, Cα, C’) | ||
| All residues | 1.50 Å | 8.38 Å |
| Residues in ordered regionsb | 0.44 Å | 0.42 Å |
| Residues in helical regions | 0.31 Å | 0.31 Å |
Statistics provided are for the entire mSin3A PAH1 domain and residues 126-150 of SAP25 SID
Ordered regions in the free mSin3A PAH1 domain encompass residues 127-186; in the binary complex, these include residues 122-188 of mSin3A PAH1 and 132-145 of SAP25 SID
The apo-mSin3A PAH1 domain adopts the canonical left-handed four-helix bundle fold described previously for the two N-terminal mSin3 PAH domains (Figure 2(c)).28-31,33,41 The four helices of apo-mSin3A PAH1 span residues Tyr129-Gln136, Gln142-Lys155, Pro162-Leu171, and Asp177-Phe182. The helical spans are identical to those described previously for the NRSF SID-bound mSin3B PAH1 domain,33 except the α1 helix of apo-mSin3A PAH1 is significantly shorter lacking six residues at the N-terminus and the α4 helix is shorter by two residues at the C-terminus. The backbone segment spanning Tyr129-Phe182 of apo-mSin3A PAH1 superimposes well with the equivalent segment of mSin3B PAH1 with an atomic root-mean-square deviation (RMSD) of 0.89 Å, consistent with the 79% sequence identity shared by these segments. In the SAP25 SID complex, the locations of the mSin3A PAH1 helices are largely unchanged, except the α1 helix is extended by five residues at the N-terminus and is initiated by Glu124 while the α4 helix is extended by two residues at the C-terminus and is terminated by Thr184. This change is accompanied by an enhancement in the precision of the NMR structures both in this region as well as in the segment immediately following the α4 helix extending up to Pro187 (Figure 2(b) and Supplementary Figure S4(a)). Collectively, these changes imply localized folding transitions within PAH1 upon SAP25 SID binding. Indeed, a best-fit superposition of backbone atoms for the segment extending from Val123 to Pro187 of the apo- and SAP25 SID-bound mSin3A PAH1 yields a somewhat high RMSD value of 1.60 Å that reflects these conformational changes. The extension of the first helix by approximately two helical turns was also inferred previously from backbone secondary chemical shifts for the mSin3A PAH2 domain upon complex formation with Mad1/Mxd1 SID.40
The SAP25 SID in the mSin3A PAH1 complex is largely unstructured, except for the segment spanning Ala133-Met144 that adopts a helical conformation (designated the αA helix; Figure 2(b) and 2(d)). The 13Cα secondary chemical shifts, which are exquisitely sensitive to polypeptide backbone conformation,42 suggest that the helix is poorly formed in the apo-form with only seven residues at the N-terminus sampling significant amounts of helical conformations (Supplementary Figure S4(b)). Upon mSin3A PAH1 binding, the helix is stabilized and extended at the C-terminus. These results when considered in light of the folding and conformational transitions occurring in the mSin3A PAH1 domain upon binding indicate that the interacting proteins undergo mutual folding transitions. The N-terminal portion of the PAH1 α1 helix may play a key role in functioning as a gate to lock cognate ligands. Interestingly, the backbone 1H-15N correlations belonging to residues in this region are significantly broadened (Supplementary Figure S1(b)), hinting at a dynamic process, possibly related to the folding transition, on the microsecond-millisecond timescale. Indeed, severe resonance broadening was also noted for equivalent residues in the mSin3A PAH2 domain when bound to HBP1 SID.31
The protein-protein interface in the SAP25 SID-mSin3A PAH1 complex extends over an area averaging 644(±41) Å2 in the NMR ensemble. As in the case of previously described SID-PAH complexes,28-31,33 the interaction involves a helix in the SID on the one hand with a hydrophobic cleft in the PAH domain on the other (Figure 3(a)). The cleft is cavernous and is dominated by hydrophobic residues and a few lysine residues at the periphery (Figure 3(b) and 3(c)). The complex appears to be stabilized by numerous intermolecular interactions that are almost exclusively hydrophobic (Figure 3(c)). In particular, the side chains of Leu138, Leu141, and Leu142 within the ‘LXXLL’ motif and that of Met144 in SAP25 SID make extensive contacts with the cleft. Consistent with these observations, the Leu138Ala mutant and the Leu141Ala,Leu142Ala double mutant exhibited diminished and complete loss of binding, respectively, to mSin3A PAH1.39 In addition to the aforementioned affinity determining interactions made by SAP25 residues, Ala137 and Gly140 appear to play a role in stabilizing the complex, by eliminating or diminishing unfavorable interactions involving their side chains, and may thus serve as key specificity determinants of the interaction. Finally, in half of the structures of the ensemble, a sole intermolecular hydrogen bonding interaction is detected between the hydroxyl and carboxylate groups of mSin3A PAH1 Tyr144 and SAP30 SID Glu134, respectively.
Figure 3.
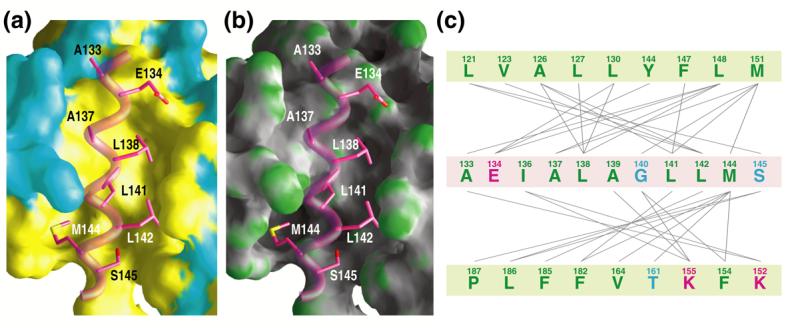
The SAP25 SID binds to a deep hydrophobic cleft in the mSin3A PAH1 domain. Molecular surface views of the cleft color-coded according to (a) residue type (hydrophobic: yellow; polar: cyan) and (b) curvature (concave: gray; planar: white; convex: green). The backbone of the SAP25 SID helix and the PAH1-interacting side chains are shown in worm and stick representations, respectively. (c) A catalogue of intermolecular interactions in the SAP25 SID-mSin3A PAH1 complex detected in ≥60% of conformers in the NMR ensemble.64 SAP25 and mSin3A residues are presented in pale magenta and pale green backgrounds, respectively. The lines connect interacting residues. Line colors indicate the type of interaction (green: electrostatic; red: hydrogen bonding; purple: salt bridge; gray: hydrophobic) whereas font colors indicate the type of residue (green: hydrophobic; blue: polar; magenta: charged).
Elucidation of PAH1-interaction motifs
A comparison of the structure of the SAP25 SID-mSin3A PAH1 complex with that of the NRSF SID-mSin3B PAH1 complex33 yields additional insights into mSin3 PAH1 recognition. In spite of the dissimilar nature of the SIDs at the sequence level, the polypeptide backbones of the PAH domains (corresponding to the Val123-Pro187 and Val34-Pro98 segments of mSin3A and mSin3B, respectively) in these complexes superimpose rather well with an RMSD of 1.10 Å, indicative of closely related conformations (Figure 4(a) and 4(b)). The SIDs engage the cleft through helical structures, but surprisingly, the relative orientations of the helices in the two complexes are almost exactly reversed. Furthermore, the helices engage a highly overlapping surface of the PAH1 domains that is essentially invariant in the two complexes (Figure 4(c) and 4(d)). A close examination of the non-covalent interactions in the SAP25 complex leads us to define a core PAH1-interaction motif of the form s-Φ-x-s-Φ-Φ-x-Φ (where s: residue with short side chain; Φ: bulky hydrophobic; x: any non-proline), which is exactly opposite in chain direction to that deduced for the NRSF complex, and correlates well with the opposite helical orientations. Key equivalent residues in the two PAH1-interaction motifs target equivalent binding ‘pockets’ on the surface of the PAH1 domain (Figure 4(c) and 4(d)). These observations are similar to those reported previously for the mSin3A PAH2 domain,31 implying that these properties are shared by the Sin3 PAH1 and PAH2 domains. Whereas the NRSF SID helix binds PAH1 with an orientation akin to that used by the Mad1/Mxd1 SID helix to bind PAH2,28,29,33 the SAP25 SID binds PAH1 via a helical orientation similar to that used by the HBP1 SID helix to PAH2.31 We propose the terms type I and type II PAH binders to distinguish SIDs belonging to the former and latter categories, respectively. Although PAH binders of each type share overt similarity vis-à-vis helical orientations, the sequence motifs employed in targets recognition are quite distinct (e.g. Φ-x-Φ-Φ-s-x-Φ-s versus Φ-x-x-Φ-Φ-x-A-A used by type I PAH1- and PAH2-interactors, respectively).28,29,33
Figure 4.
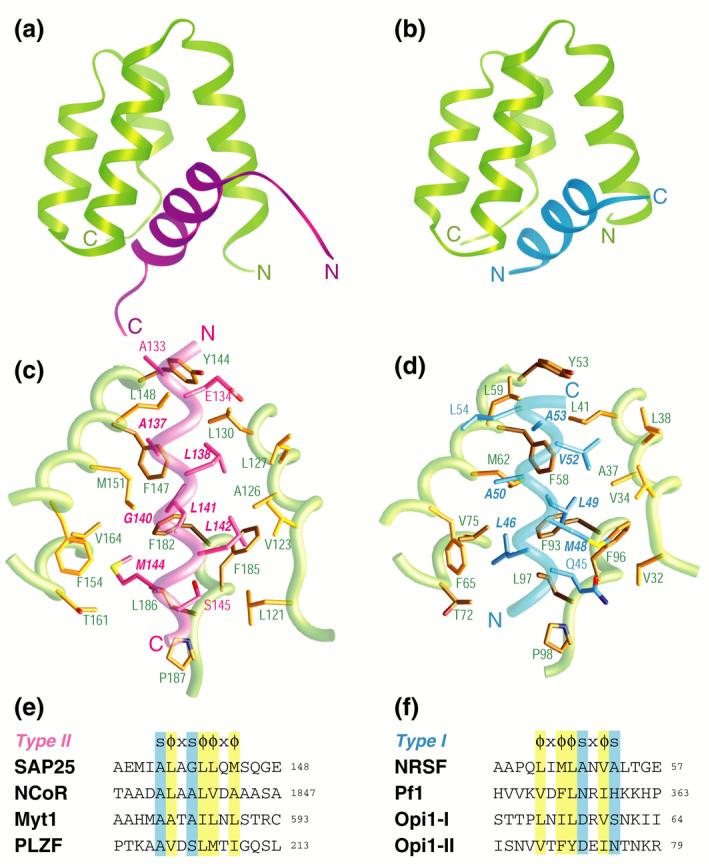
A comparative analysis of intermolecular interactions involving the Sin3 PAH1 domain with diverse targets. A side-by-side comparison of the (a) SAP25 SID-mSin3A PAH1 complex (colored in magenta and green, respectively) and (b) the NRSF SID-mSin3B PAH1 complex (colored in blue and green; PDB code: 2CZY33). Note the contrasting chain directions of the two SID helices. The respective intermolecular interfaces in the two complexes with the backbones rendered as worms and the side chains as sticks (panels (c) and (d)). Residues in the respective SIDs deemed important for the interaction are annotated in bold and italicized. Sequence motifs deduced from structural analysis of the respective complexes (panels (e) and (f); abbreviations: s = any amino acid with a short side chain; x = any non-proline residue; Φ = any bulky hydrophobic residue). The predicted binding sites and modes of a subset of proteins previously shown to target the Sin3 PAH1 domain are also shown. With the exception of yeast Opi1, the proteins are of human or mouse origin (GenBank accession codes: SAP25: NP_001075431; NCoR: AAB17125; Myt1: AAC53456; PLZF: AAD03619; NRSF: NP_005603; Pf1: NP_001028733; Opi1: AAB65073)
The definition of two distinct PAH1-interaction motifs permits predictions to be made regarding the precise location of the SIDs in various PAH1 interacting proteins, as for most of these proteins the SIDs have been mapped only coarsely (we have refrained from using these motifs to discover novel Sin3 interactors as the information content is far too low to allow reliable predictions). In arriving at these predictions, we applied the following criteria: (i) the sequence must have the potential to form a helix and (ii) the motif-defining residues must be solvent accessible and must not engage in any domain-stabilizing tertiary interactions. Criterion i can be assessed by secondary structure prediction, whereas criterion ii requires a known three-dimensional structure. Given the limitations in the accuracy of protein secondary structure prediction and our insufficient knowledge of protein three-dimensional structures, the conclusions from such an analysis must be treated with caution. These concerns notwithstanding, some interesting insights emerge from our analysis. In the case of PLZF, two putative SIDs were previously identified experimentally, each within residues 1-120 and 121-364.32 Our analysis revealed multiple PAH1-interacting motifs in the former segment, but as the region corresponds to the BTB-POZ domain for which a structure is known and since most of the motif-defining residues were not solvent accessible, these were excluded from further consideration. However, PLZF residues 202−209 was found to harbor a type II motif and meets the aforementioned criteria, hinting that it might be a bona fide SID (Figure 4(e)). Similar considerations led us to identify type II motifs for Myt1 (residues 582-589) and NCoR (residues 1836-1843). The PAH1-interacting regions in these proteins were mapped previously using genetic and biochemical approaches to aa 502−623 and 1829−1940, respectively.36,37 We note in passing that the physiological relevance of the interaction between NCoR and Sin3 is unclear as NCoR has been consistently detected in multi-protein complexes containing SMRT and HDAC3.6 The Pf1 corepressor was previously shown to harbor multiple SIDs.34 Pf1 targets both the PAH1 and PAH2 domains of mSin3 via separate SIDs and whereas the PAH2-interacting SID was mapped precisely to the segment spanning aa 209-219,28,34 the PAH1-interacting SID was found to reside within residues 300-743. 34 Our analysis reveals a potential PAH1-interaction motif at aa 352-359 (Figure 4(f)). A similar analysis of the Opi1 SID, previously mapped to residues 45-106,35 reveals two non-overlapping type I PAH1-interaction motifs spanning aa 53-60 and 68-75. We note that the inclusion of the yeast Opi1 protein in this analysis is justified as the S. cerevisiae Sin3 PAH1 domain shares 73% identity and 85% similarity at the sequence level with the mSin3A PAH1 domain; furthermore, residues comprising the SID binding surface are invariant in these domains (Figure 1).
Specificity of mSin3 PAH1 and PAH2 interactions
The close sequence and structural similarity of the mSin3 PAH1 and PAH2 domains (30% sequence identity, 54% sequence similarity) raises questions relating to the specificity of PAH-SID interactions. These questions assume great significance, as out of approximately twenty PAH residues forming the protein-protein interface, half of these are identical in the PAH1 and PAH2 domains, whereas a quarter of them involve conservative substitutions (Figure 1 and Figure 5(a)). However, the non-conservative changes map to the periphery of these protein-protein interfaces suggesting an important role for residues undergoing conservative substitutions in dictating the specificity of these interactions (Figure 5(a)). To test this idea, a panel of mSin3A PAH2/PAH1 chimeras was generated in which one or more PAH1 residues (up to a maximum of six) in the hydrophobic cleft were introduced in a PAH2 background and the affinities for a specific SID was measured. The residues selected included Val311 (corresponding to Leu130 in PAH1; Figure 5(a)), Leu332 (Met151 in PAH1), Phe304 (Val123), Ile308 (Leu 127), Asn312 (Asp131), and Tyr335 (Phe154). Using a fluoresceinated Mad1/Mxd1 SID peptide and employing steady-state fluorescence anisotropy assays, we evaluated the ability of the peptide to bind to single-, double- and multiple-site PAH2 mutants that we refer to as PAH2/PAH1 chimeras. As expected, the wild-type PAH2 protein bound to Mad1/Mxd1 SID with high affinity (Supplementary Figure S3(b); Table 2).28,43 With the exception of the Val311Leu mutant, which exhibited three-fold higher affinity for Mad1/Mxd1 SID relative to wild-type, the single- and double-site mutants exhibited between two- to seven-fold reduction in binding affinities (Table 2). The Leu332→Met mutation had the most profound effect on Mad1/Mxd1 SID binding, indicating that this residue is an important affinity determinant for Mad1/Mxd1 binding. As more mutations were introduced, the binding affinity was correspondingly diminished with quadruple-site mutants exhibiting between ten- and over twenty-fold reductions in binding while the sextuple-site mutant was the most strongly affected showing a hundred-fold reduction in Mad1/Mxd1 SID binding affinity (Table 2).
Figure 5.
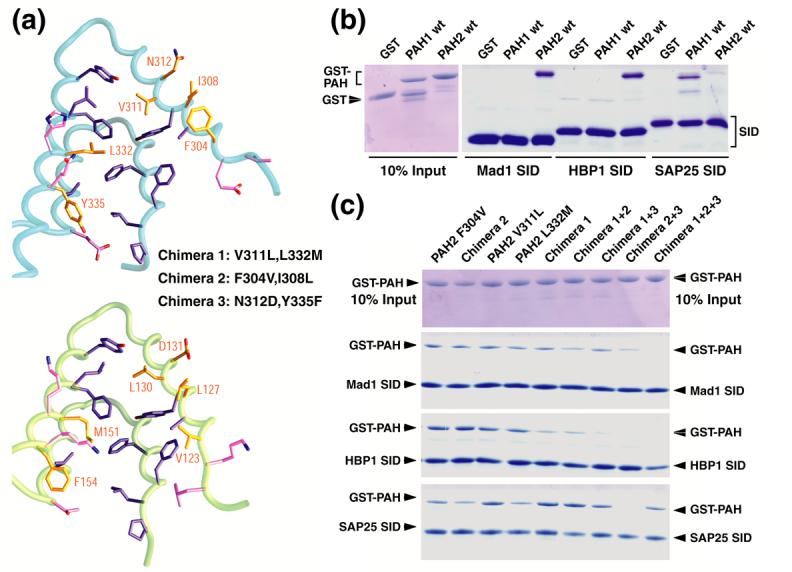
Specificity of protein-protein interactions involving the mSin3A PAH1 and PAH2 domains. (a) The mSin3A PAH2 (top) and PAH1 (bottom) residues involved in binding the SIDs of Mad1/Mxd1 and SAP25, respectively, are depicted. The residues are colored according to the sequence conservation patterns across the Sin3 PAH1 and PAH2 domains (invariant residues: purple; conservative substitutions: gold; non-conservative replacements: magenta). Note the essentially unperturbed side chain conformations of invariant residues. mSin3A PAH2 residues that were mutated to the equivalent PAH1 residue to yield various PAH2/PAH1 chimeras are annotated. (b) Pulldown assays conducted using His6-tagged Mad1/Mxd1 SID, HBP1 SID and SAP25 SID immobilized on a Ni2+ resin with GST or GST-tagged wild-type mSin3A PAH1 and PAH2 proteins. Ten percent of the input proteins used in these assays are shown after resolving them by SDS-PAGE and staining the gels with Coomassie Blue (left panel). Proteins bound to the resin following the pulldown experiments were resolved using SDS-PAGE and visualized by Coomassie Blue staining (right panel). (c) Analogous pulldown assays conducted using the same SID polypeptides with various GST-PAH2/PAH1 chimera proteins. The amounts of the chimera proteins used in these assays are shown in the top panel.
Table 2.
Fluorescence Anisotropy Analysis of Mad1/Mxd1 SID6-21 Binding by GST-PAH2/PAH1 Chimeras
| Protein | Kd (nM) | A0 | A∞ |
|---|---|---|---|
| Wild-type | 51 ± 4 | 0.034 | 0.210 |
| V311L | 16 ± 2 | 0.033 | 0.187 |
| L332M | 369 ± 82 | 0.053 | 0.199 |
| V311L, L332M (Chimera 1) | 104 ± 13 | 0.040 | 0.183 |
| F304V | 163 ± 22 | 0.038 | 0.171 |
| F304V, I308L (Chimera 2) | 187 ± 19 | 0.034 | 0.168 |
| Chimera 1+2 | 1223 ± 72 | 0.037 | 0.163 |
| N312D, Y335F (Chimera 3)+Chimera 1 | 510 ± 87 | 0.035 | 0.190 |
| Chimera 2+3 | 947 ± 146 | 0.038 | 0.159 |
| Chimera 1+2+3 | 5524 ± 810 | 0.037 | 0.167 |
If any or all six residues played a role in determining the specificity of PAH-SID interactions, not only should the PAH2 domain exhibit diminished binding as it starts to increasingly resemble PAH1, but it should also acquire the ability to bind PAH1-specific targets. To test this idea, we conducted binding assays in pulldown format with the wild-type proteins as well as the PAH2/PAH1 chimeras for three different SIDs, including Mad1/Mxd1 SID, HBP1 SID, and SAP25 SID. As expected, the Mad1/Mxd1and HBP1 SIDs bound efficiently to wild-type PAH2, but not to wild-type PAH1 (Figure 5(b)). Similarly, SAP25 SID bound well to wild-type PAH1, but not to wild-type PAH2 (the affinity of the interaction was difficult to measure reliably but from competition assays conducted using fluorescence anisotropy we estimate the Kd >> 2.5 μM; ITC measurements placed the affinity in the >100 μM range). In addition, our aforementioned findings with regard to the binding affinities of various PAH2/PAH1 chimeras and Mad1/Mxd1SID matched qualitatively with those obtained in the pulldown assays (Figure 5(c)). Unlike Mad1/Mxd1SID, HBP1 SID showed greater sensitivity towards PAH1 substitutions in the PAH2 domain. For example, the quadruple-site mutants exhibited strongly diminished binding for HBP1 than for Mad1/Mxd1(Figure 5(c)). Importantly, both Mad1/Mxd1and HBP1 exhibited severe reduction in binding for the sextuple-site mutant. On the other hand, the SAP25 SID bound to almost all PAH2/PAH1 chimeras tested with varying efficiencies. The Val311Leu mutation appears to be sufficient for SAP25 to bind efficiently. Indeed, all multiple-site mutants harboring this mutation also bound SAP25 SID efficiently (Figure 5(c)). Similarly, the Phe304Val mutation also bound SAP25 SID but comparatively with slightly lower efficiency than the Val311Leu mutant. We note that the phenylalanine residue was previously implicated as an important specificity determinant for Mad1/Mxd1interactions by another study.44 Intriguingly, the quadruple-site mutant lacking the Val311Leu and Leu332Met mutations did not exhibit significant binding to SAP25 SID, implying that the contributions made by individual residues towards binding affinity are not necessarily additive or even approximately additive (Figure 5(c)). More detailed structural and functional studies are required to clarify the basis for these findings.
Comparisons with other transcription factor-coregulator complexes
Short amphipathic helices harboring Φ-x-x-Φ-Φ sequence motifs (where Φ and x denote bulky hydrophobic and non-proline residues, respectively) is a recurring theme in transcription factor-coregulator interactions.45 The ubiquity of this feature in transcription factors on the one hand, and the seeming promiscuity of coregulators as reflected in their ability to interact with disparate transcription factors on the other hand, raises questions regarding how such interactions are facilitated at the molecular level while retaining some degree of specificity. The Sin3 PAH1 and PAH2 domains seem to have solved this problem by accommodating ‘reversible’ sequence and helical motifs within a common binding site. It is unclear whether this property is shared with the presently poorly characterized PAH3 domain, which is more distantly related to the PAH1 and PAH2 domains (Figure 1). Interestingly, this property is also exhibited by ubiquitin as evidenced by its interactions with the helical UIM and MIU motifs found in endocytic proteins.46,47 The ligand-binding domains of nuclear receptors engage distinct sequence motifs of coactivators and corepressors through overlapping but somewhat altered binding surfaces.48,49 The KIX domain of the CBP transcriptional coactivator interacts with distinct sequence motifs of the CREB and c-Myb transcription factors through an overlapping surface.50,51 However, the domain despite its relatively small size exhibits considerable versatility in engaging the MLL transcription factor through a completely different surface.52
Conclusions
In conclusion, our studies show that the mSin3A PAH1 domain shares many structural and functional features with the well-characterized mSin3A and mSin3B PAH2 domains including the formation of a four-helix bundle structure, interaction with helical targets through a comparable and an almost exclusively hydrophobic surface, and recognition of chain-reversible sequence motifs. The sequence motifs recognized by the Sin3 PAH1 domains differ significantly from those described for the Sin3 PAH2 domains. Both PAH1 and its target become more ordered and helical in the complex with the extended helix in PAH1 serving as a gate or barrier to prevent target escape. Finally, the PAH1 and PAH2 domains exhibit a high degree of specificity in their interactions with cognate targets and conservative replacements of PAH2 residues with equivalent PAH1 residues are sufficient to dramatically alter interaction affinity and specificity.
Materials and Methods
Production of the mSin3A PAH1 and SAP25 SID domains
The coding sequences for the mSin3A PAH1 (residues 119-189) and murine SAP25 SID (residues 126-186) domains were amplified by PCR and cloned into the pET-30 Xa/LIC expression vector (Novagen). Escherichia coli BL21(DE3) cells harboring these vectors were grown at 37 °C in M9 minimal medium and shifted to 20 °C 15 minutes before induction. Protein expression was induced using 1 mM isopropyl-1-thio-β-D-galactopyranoside when A600nm was ∼0.7 and cells were harvested 7-8 hours thereafter. Cell pellets were suspended in 20 mM sodium phosphate buffer, pH 7.2, containing 0.5 M NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 μM leupeptin, 1 mM pepstatin and 0.1% Triton X-100, lysed via sonication, centrifuged and the supernatant loaded onto a HiTrap Chelating column (GE Healthcare) charged with Zn2+. The His6-tagged protein was eluted using 20 mM sodium phosphate buffer, pH 7.2, containing 0.5 M NaCl and 50 mM EDTA. Protein-containing fractions were assayed by SDS-PAGE and these were further purified via reversed-phase high-pressure liquid chromatography (HPLC) using a C18 column (Vydac) and a mobile aqueous phase containing 80% acetonitrile and 0.1% trifluoroacetic acid. Fractions containing the desired polypeptide were pooled, lyophilized, dissolved in aqueous buffer, and treated with Factor Xa (Novagen) following the vendor’s recommendations. The resulting polypeptide, sans the His6-tag, was purified using reversed-phase HPLC. The identity of the purified protein was confirmed by electrospray ionization-mass spectrometry (ESI-MS). Polypeptides uniformly labeled with 15N and/or 13C isotopes were produced by following the same procedure as above, except that the cells were grown in M9 minimal media containing 15N-(NH4)2SO4 (Martek) and/or 13C6-D-glucose (Isotec). The integrity of the purified proteins and the extent of isotope enrichment (typically >98% for 15N and >96% 13C) were confirmed by ESI-MS.
NMR sample generation
Lyophilized protein samples of mSin3A PAH1 and SAP25 SID were dissolved in 20 mM sodium phosphate buffer, pH 6.0, containing 2 mM DTT-d11 and 0.2% NaN3 in 90% H2O/10% D2O. Approximately equimolar complexes of SAP25 SID and mSin3A PAH1 were generated for NMR studies by titrating increasing amounts of unlabeled SAP25 SID with 15N/15N,13C-mSin3A PAH1 and separately, unlabeled mSin3A PAH1 with 15N/15N,13C-labeled SAP25 SID. The progress of the titration was monitored by recording two-dimensional (2D) 1H-15N correlated spectra and the titration was terminated when no changes in the NMR spectrum were detected. NMR sample concentrations were in the 0.7-1 mM range. All protein concentrations were measured spectrophotometrically.53 For experiments conducted in D2O, the samples of the respective proteins or protein complexes were lyophilized and redissolved in 99.996% D2O (Isotec).
NMR spectroscopy and structure determination
All NMR experiments were performed on a Varian Inova 600 MHz spectrometer equipped with a room temperature pulsed-field-gradient triple-resonance probe. NMR data were acquired either at 15 °C (free mSin3A PAH1 domain) or at 30 °C (free SAP25 SID domain and SAP25 SID-mSin3A PAH1 complex). Data processing and spectral analyses were conducted using Felix 98.0 software (Accelrys) modified in-house to facilitate accelerated resonance and NOE assignments.54 Backbone resonance assignments were made by analyzing three-dimensional (3D) HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, HNCO, and HCACO spectra whereas side chain resonances were assigned using 3D 15N-edited TOCSY, HCCH-COSY, HCCH-TOCSY, C(CO)NH-TOCSY spectra.55,56 Aromatic resonances were assigned by analyzing 2D (HB)CB(CDCGCE)HE and 3D (HB)CBCG(CD)HD and (HC)C(C)CH-TOCSY spectra.57,58
For the determining the structures of the free mSin3A PAH1 domain and the SAP25 SID-mSin3A PAH1 complex, the Hα, Cα, Cβ, C’ and backbone 15N chemical shifts served as the source for backbone ϕ and ψ torsion angle restraints using TALOS.59 These restraints were imposed only for those residues with reliability scores of 10. Side chain χ1 restraints for isoleucine, threonine, and valine residues were derived from measured 3JNCγ and 3JC’Cγ values. Inter-proton distance restraints were derived from 3D 15N-edited NOESY recorded in H2O (τm = 100 ms), 3D aliphatic 13C-edited NOESY (τm = 80 ms), aromatic 13C-edited NOESY (τm = 60 ms), and 3D 13C-filtered, 13C-edited NOESY (τm = 120 ms) recorded in D2O. Intermolecular NOEs were assigned manually and were calibrated indirectly by calculating a scaling factor for the intensities of well-resolved peaks in the 13C-edited NOESY and 13C-filtered, 13C-edited NOESY spectra. These NOEs were assigned upper bounds of 3, 4, 5, and 6 Å. All other NOEs were calibrated and assigned iteratively and automatically by ARIA (version 1.2)60,61 and were checked manually between successive cycles of refinement. Structures were calculated from extended conformations as starting models using ARIA in conjunction with CNS.62 Altogether 80 structures were computed in the final iteration and the 20 structures with the lowest restraint energies, restraint violations, and RMS deviations from ideal covalent geometry were considered for structural analysis. The final structures were analyzed using PROCHECK,63 MONSTER,64 DeepView,65 and CNS.62 Molecular images were generated using RIBBONS66 and GRASP.67
Production of mSin3A PAH2-PAH1 chimeras
Single- and multiple-site mutants of GST-mSin3A PAH2 were generated using the QuikChange site-directed mutagenesis kit (Stratagene). All mutations were confirmed by DNA sequencing. E. coli harboring the wild-type and mutant GST-PAH2 vectors were grown in Luria-Bertani medium at 37 °C. The proteins were expressed for approximately 16 h at 20 °C and prepared for purification using analogous approaches used for producing mSin3A PAH1 and SAP25 SID described above. The proteins were purified using glutathione-sepharose beads (GE Healthcare) following the manufacturer’s recommendations. To preclude adventitious protease-mediated degradation, the proteins were stored at 4 °C in 20 mM sodium phosphate buffer (pH 8.0) containing 25 mM EDTA.
Binding assays
Isothermal titration calorimetry experiments were performed on a MicroCal MCS calorimeter. Experiments were performed at 30 °C in 20 mM sodium phosphate buffer (pH 6.0). Binding isotherms were analyzed using Origin 5.0 software provided by the manufacturer.
GST-PAH binding assays were performed by immobilizing His6-SAP25 SID, His6-Mad1/Mxd1SID (corresponding to residues 1-35 of human Mad1) and His6-HBP1 SID (corresponding to residues 342-398 of human HBP1) domain constructs to a Co2+ resin (Clonetech). The His-tagged proteins were incubated with 1 μM solutions of the various GST-tagged proteins in a binding buffer comprising 50 mM sodium phosphate (pH 7), 5 mM imidazole, and 0.1% Triton for a final reaction volume of 1 ml. Reactions were incubated for 30 min at room temperature. After washing the beads twice with 1 ml of binding buffer, the bound proteins were eluted by boiling in the SDS-PAGE sample-loading buffer containing 250 mM imidazole, resolved by gel electrophoresis, and analyzed by Coomassie Blue staining.
Equilibrium binding assays in free solution were performed using fluorescence spectroscopy. A 16-residue peptide corresponding to residues 6-21 of human Mad1/Mxd1and incorporating a fluorescein moiety at the N-terminus was synthesized using automated procedures (University of Utah DNA/Peptide Facility) and purified by reversed-phase HPLC. The peptide was dissolved in fluorescence buffer comprising 20 mM sodium phosphate buffer (pH 8) and 0.5 mM EDTA. Stock solutions of the various GST-PAH proteins were exchanged into the fluorescence buffer and were diluted to appropriate concentrations, ranging from 0.5 nM to 35 μM. The final volume of each sample was 1 ml while the concentration of the fluoresceinated peptide was maintained at 2 nM.
Steady-state fluorescence experiments were performed at room temperature on an ISS PC-1 fluorimeter configured in the L-format. The excitation wavelength was set to 492 nm while an excitation interference filter (Coherent/Ealing #35-3482; wavelength center: 490 nm, bandpass: 7.3 nm) and an emission cut-off filter (Omega optical #535AF45 multilayer) were employed to attenuate contributions from the excitation bandpass and scattering effects, respectively.68 Since we employed these optical filters, we chose not to use the emission monochromator in order to increase the sensitivity of the instrument. Data collection and processing were performed using the program Vinci (ISS). Ten measurements were recorded for each sample, each measurement being the average of 10 readings of 100 μs-long intervals. The reported anisotropy values are the average and standard deviations from three independent samples. The average anisotropy was graphed as a function of the GST-PAH2 protein concentration and the resulting curves were fit to a dose-response function using the program Origin 5.0 (Microcal). In the fitting procedure, the EC50 value and the limiting anisotropies at zero and infinite protein concentration were allowed to vary while the Hill’s coefficient was fixed at 1. The EC50 values are reported as the equilibrium dissociation constants (Kd) in Table 2 and the associated errors are the fitted errors.
Supplementary Material
Acknowledgements
We are grateful to Bob Eisenman and Yuzuru Shiio for generously providing reagents including the SAP25 cDNA, sharing pre-publication results, and many useful discussions. We also thank Stephanie Wang and Nahir Vélez Avilés for their contributions towards the development of this work. This work was supported by a grant from the NIH to I.R. (R01 GM64715). I.R. is a Scholar of the Leukemia and Lymphoma Society. Access to instrumentation in the Keck Biophysics Facility and the WCAS Structural Biology NMR Facility as well as support for structural biology research from the Robert H. Lurie Comprehensive Cancer Center at Northwestern is gratefully acknowledged.
Abbreviations
- SID
Sin3 interaction domain
- PAH
paired amphipathic helix
- HDAC
histone deacetylase
- SAP
Sin3 associated polypeptide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession codes. The RCSB PDB accession codes for the atomic coordinates of apo-mSin3A PAH1 and the SAP25 SID-mSin3A PAH1 complex are 2RMR and 2RMS, respectively.
References
- 1.Jenuwen T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Roguev A, Krogan NJ. SIN-fully silent: HDAC complexes in fission yeast. Nat Struct Mol Biol. 2007;14:358–9. doi: 10.1038/nsmb0507-358. [DOI] [PubMed] [Google Scholar]
- 3.Ahringer J. NuRD and SIN3 histone deacetylase complexes in development. Trends Genet. 2000;16:351–6. doi: 10.1016/s0168-9525(00)02066-7. [DOI] [PubMed] [Google Scholar]
- 4.Ayer DE. Histone deacetylases: transcriptional repression with SINers and NuRDs. Trends Cell Biol. 1999;9:193–8. doi: 10.1016/s0962-8924(99)01536-6. [DOI] [PubMed] [Google Scholar]
- 5.Silverstein RA, Ekwall K. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet. 2005;47:1–17. doi: 10.1007/s00294-004-0541-5. [DOI] [PubMed] [Google Scholar]
- 6.Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26:5439–49. doi: 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- 7.Knoepfler PS, Eisenman RN. Sin meets NuRD and other tails of repression. Cell. 1999;99:447–50. doi: 10.1016/s0092-8674(00)81531-7. [DOI] [PubMed] [Google Scholar]
- 8.David G, Turner GM, Yao Y, Protopopov A, DePinho RA. mSin3-associated protein, mSds3, is essential for pericentric heterochromatin formation and chromosome segregation in mammalian cells. Genes Dev. 2003;17:2396–405. doi: 10.1101/gad.1109403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xin H, Yoon HG, Singh PB, Wong J, Qin J. Components of a pathway maintaining histone modification and heterochromatin protein 1 binding at the pericentric heterochromatin in Mammalian cells. J Biol Chem. 2004;279:9539–46. doi: 10.1074/jbc.M311587200. [DOI] [PubMed] [Google Scholar]
- 10.Silverstein RA, Richardson W, Levin H, Allshire R, Ekwall K. A new role for the transcriptional corepressor SIN3; regulation of centromeres. Curr Biol. 2003;13:68–72. doi: 10.1016/s0960-9822(02)01401-x. [DOI] [PubMed] [Google Scholar]
- 11.Scott KL, Plon SE. Loss of Sin3/Rpd3 histone deacetylase restores the DNA damage response in checkpoint-deficient strains of Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:4522–31. doi: 10.1128/MCB.23.13.4522-4531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jazayeri A, McAinsh AD, Jackson SP. Saccharomyces cerevisiae Sin3p facilitates DNA double-strand break repair. Proc Natl Acad Sci U S A. 2004;101:1644–9. doi: 10.1073/pnas.0304797101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–92. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 14.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, Boone C, Emili A, Weissman JS, Hughes TR, Strahl BD, Grunstein M, Greenblatt JF, Buratowski S, Krogan NJ. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 15.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–8. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, Grewal SI. Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol. 2007;14:372–80. doi: 10.1038/nsmb1239. [DOI] [PubMed] [Google Scholar]
- 17.Cowley SM, Iritani BM, Mendrysa SM, Xu T, Cheng PF, Yada J, Liggitt HD, Eisenman RN. The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol Cell Biol. 2005;25:6990–7004. doi: 10.1128/MCB.25.16.6990-7004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–95. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alland L, David G, Shen-Li H, Potes J, Muhle R, Lee HC, Hou H, Jr., Chen K, DePinho RA. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol Cell Biol. 2002;22:2743–50. doi: 10.1128/MCB.22.8.2743-2750.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleischer TC, Yun UJ, Ayer DE. Identification and characterization of three new components of the mSin3A corepressor complex. Mol Cell Biol. 2003;23:3456–67. doi: 10.1128/MCB.23.10.3456-3467.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997;89:341–7. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- 22.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression. Cell. 1997;89:349–56. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 23.Laherty CD, Billin AN, Lavinsky RM, Yochum GS, Bush AC, Sun JM, Mullen TM, Davie JR, Rose DW, Glass CK, Rosenfeld MG, Ayer DE, Eisenman RN. SAP30, a component of the mSin3 corepressor complex involved in N-CoR-mediated repression by specific transcription factors. Mol. Cell. 1998;2:33–42. doi: 10.1016/s1097-2765(00)80111-2. [DOI] [PubMed] [Google Scholar]
- 24.Lai A, Kennedy BK, Barbie DA, Bertos NR, Yang XJ, Theberge MC, Tsai SC, Seto E, Zhang Y, Kuzmichev A, Lane WS, Reinberg D, Harlow E, Branton PE. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol Cell Biol. 2001;21:2918–32. doi: 10.1128/MCB.21.8.2918-2932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Sun ZW, Iratni R, Erdjument-Bromage H, Tempst P, Hampsey M, Reinberg D. SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol Cell. 1998;1:1021–31. doi: 10.1016/s1097-2765(00)80102-1. [DOI] [PubMed] [Google Scholar]
- 26.He Y, Radhakrishnan I. Solution NMR studies of apo-mSin3A and mSin3B reveal that the PAH1 and PAH2 domains are structurally independent. Protein Sci. 2008 doi: 10.1110/ps.073097308. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eilers AL, Billin AN, Liu J, Ayer DE. A 13-amino acid amphipathic alpha-helix is required for the functional interaction between the transcriptional repressor Mad1 and mSin3A. J. Biol. Chem. 1999;274:32750–6. doi: 10.1074/jbc.274.46.32750. [DOI] [PubMed] [Google Scholar]
- 28.Brubaker K, Cowley SM, Huang K, Loo L, Yochum GS, Ayer DE, Eisenman RN, Radhakrishnan I. Solution structure of the interacting domains of the Mad-Sin3 complex: implications for recruitment of a chromatin-modifying complex. Cell. 2000;103:655–65. doi: 10.1016/s0092-8674(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 29.Spronk CA, Tessari M, Kaan AM, Jansen JF, Vermeulen M, Stunnenberg HG, Vuister GW. The Mad1-Sin3B interaction involves a novel helical fold. Nat. Struct. Biol. 2000;7:1100–4. doi: 10.1038/81944. [DOI] [PubMed] [Google Scholar]
- 30.van Ingen H, Lasonder E, Jansen JF, Kaan AM, Spronk CA, Stunnenberg HG, Vuister GW. Extension of the binding motif of the Sin3 interacting domain of the Mad family proteins. Biochemistry. 2004;43:46–54. doi: 10.1021/bi0355645. [DOI] [PubMed] [Google Scholar]
- 31.Swanson KA, Knoepfler PS, Huang K, Kang RS, Cowley SM, Laherty CD, Eisenman RN, Radhakrishnan I. HBP1 and Mad1 repressors bind the Sin3 corepressor PAH2 domain with opposite helical orientations. Nat Struct Mol Biol. 2004;11:738–46. doi: 10.1038/nsmb798. [DOI] [PubMed] [Google Scholar]
- 32.David G, Alland L, Hong SH, Wong CW, DePinho RA, Dejean A. Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein. Oncogene. 1998;16:2549–56. doi: 10.1038/sj.onc.1202043. [DOI] [PubMed] [Google Scholar]
- 33.Nomura M, Uda-Tochio H, Murai K, Mori N, Nishimura Y. The neural repressor NRSF/REST binds the PAH1 domain of the Sin3 corepressor by using its distinct short hydrophobic helix. J Mol Biol. 2005;354:903–15. doi: 10.1016/j.jmb.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Yochum GS, Ayer DE. Pf1, a novel PHD zinc finger protein that links the TLE corepressor to the mSin3A-histone deacetylase complex. Mol. Cell. Biol. 2001;21:4110–8. doi: 10.1128/MCB.21.13.4110-4118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner C, Dietz M, Wittmann J, Albrecht A, Schuller HJ. The negative regulator Opi1 of phospholipid biosynthesis in yeast contacts the pleiotropic repressor Sin3 and the transcriptional activator Ino2. Mol Microbiol. 2001;41:155–66. doi: 10.1046/j.1365-2958.2001.02495.x. [DOI] [PubMed] [Google Scholar]
- 36.Romm E, Nielsen JA, Kim JG, Hudson LD. Myt1 family recruits histone deacetylase to regulate neural transcription. J Neurochem. 2005;93:1444–53. doi: 10.1111/j.1471-4159.2005.03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–8. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 38.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003;17:896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiio Y, Rose DW, Aur R, Donohoe S, Aebersold R, Eisenman RN. Identification and characterization of SAP25, a novel component of the mSin3 corepressor complex. Mol Cell Biol. 2006;26:1386–97. doi: 10.1128/MCB.26.4.1386-1397.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Zhang Z, Demeler B, Radhakrishnan I. Coupled unfolding and dimerization by the PAH2 domain of the mammalian Sin3A corepressor. J Mol Biol. 2006;360:7–14. doi: 10.1016/j.jmb.2006.04.069. [DOI] [PubMed] [Google Scholar]
- 41.van Ingen H, Baltussen MA, Aelen J, Vuister GW. Role of Structural and Dynamical Plasticity in Sin3: The Free PAH2 Domain is a Folded Module in mSin3B. J Mol Biol. 2006 doi: 10.1016/j.jmb.2006.01.100. [DOI] [PubMed] [Google Scholar]
- 42.Eliezer D, Yao J, Dyson HJ, Wright PE. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat Struct Biol. 1998;5:148–55. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 43.Cowley SM, Kang RS, Frangioni JV, Yada JJ, DeGrand AM, Radhakrishnan I, Eisenman RN. Functional analysis of the Mad1-mSin3A repressor-corepressor interaction reveals determinants of specificity, affinity, and transcriptional response. Mol. Cell. Biol. 2004;24:2698–709. doi: 10.1128/MCB.24.7.2698-2709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Le Guezennec X, Vriend G, Stunnenberg HG. Molecular determinants of the interaction of Mad with the PAH2 domain of mSin3. J Biol Chem. 2004;279:25823–9. doi: 10.1074/jbc.M313860200. [DOI] [PubMed] [Google Scholar]
- 45.Plevin MJ, Mills MM, Ikura M. The LxxLL motif: a multifunctional binding sequence in transcriptional regulation. Trends Biochem Sci. 2005;30:66–9. doi: 10.1016/j.tibs.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Swanson KA, Kang RS, Stamenova SD, Hicke L, Radhakrishnan I. Solution structure of Vps27 UIM-ubiquitin complex important for endosomal sorting and receptor downregulation. EMBO J. 2003;22:4597–4606. doi: 10.1093/emboj/cdg471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, Schneider TR. Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell. 2006;124:1183–95. doi: 10.1016/j.cell.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 48.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–43. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 49.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, Parks DJ, Moore JT, Kliewer SA, Willson TM, Stimmel JB. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–7. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 50.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–52. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 51.Zor T, De Guzman RN, Dyson HJ, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of c-Myb. J Mol Biol. 2004;337:521–34. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 52.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol. 2006;355:1005–13. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 53.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–26. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 54.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Structural analyses of CREB-CBP transcriptional activator-coactivator complexes by NMR spectroscopy: implications for mapping the boundaries of structural domains. J. Mol. Biol. 1999;287:859–65. doi: 10.1006/jmbi.1999.2658. [DOI] [PubMed] [Google Scholar]
- 55.Grzesiek S, Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J Biomol NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 56.Ferentz AE, Wagner G. NMR spectroscopy: a multifaceted approach to macromolecular structure. Q. Rev. Biophys. 2000;33:29–65. doi: 10.1017/s0033583500003589. [DOI] [PubMed] [Google Scholar]
- 57.Yamazaki T, Forman-Kay JD, Kay LE. 2-Dimensional NMR experiments for correlating 13Cβ and 1Hδ/ε chemical-shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J. Am. Chem. Soc. 1993;115:11054–11055. [Google Scholar]
- 58.Lohr F, Ruterjans H. Novel Pulse Sequences for the Resonance Assignment of Aromatic Side Chains in 13C-Labeled Proteins. J Magn Reson B. 1996;112:259–68. doi: 10.1006/jmrb.1996.0140. [DOI] [PubMed] [Google Scholar]
- 59.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 60.Linge JP, Habeck M, Rieping W, Nilges M. ARIA: automated NOE assignment and NMR structure calculation. Bioinformatics. 2003;19:315–6. doi: 10.1093/bioinformatics/19.2.315. [DOI] [PubMed] [Google Scholar]
- 61.Linge JP, Habeck M, Rieping W, Nilges M. Correction of spin diffusion during iterative automated NOE assignment. J Magn Reson. 2004;167:334–42. doi: 10.1016/j.jmr.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 62.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta. Crystallogr. D Biol. Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 63.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 64.Salerno WJ, Seaver SM, Armstrong BR, Radhakrishnan I. MONSTER: inferring non-covalent interactions in macromolecular structures from atomic coordinate data. Nucleic Acids Res. 2004;32:W566–8. doi: 10.1093/nar/gkh434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 66.Carson M. Macromolecular Crystallography, Pt B. Vol. 277. Academic Press Inc; San Diego: 1997. Ribbons; pp. 493–505. [Google Scholar]
- 67.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–96. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 68.Chin J, Langst G, Becker PB, Widom J. Fluorescence anisotropy assays for analysis of ISWI-DNA and ISWI-nucleosome interactions. Methods Enzymol. 2004;376:3–16. doi: 10.1016/S0076-6879(03)76001-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.