

Summary
LRH-1 is a nuclear receptor previously known to play distinct functions during mouse development and essential roles in cholesterol homeostasis. Recently, a new role for LRH-1 has been discovered in tumor progression, giving LRH-1 potential transforming functions. In order to identify critical factors stimulating LRH-1 expression leading to deregulated cellular proliferation, we studied its expression and its regulation in several breast cancer cell lines. We observed that LRH-1 expression was increased in Estrogen Receptor (ER) α expressing cell lines, whereas weak to no expression was found in non-expressing ERα cell lines. In MCF7, LRH-1 expression was highly induced after treatment with 17β-estradiol (E2). This transcriptional regulation was the result of a direct binding of the estrogen receptor to the LRH1 promoter, as demonstrated by gelshift and chromatin immunoprecipitation assays. Interestingly, siRNA-mediated inactivation of LRH-1 decreased the E2-dependent proliferation of MCF7 cells. Finally, LRH-1 protein expression was detected by immunohistochemistry in tumor cells of human mammary ductal carcinomas. Altogether, these data demonstrate that LRH-1 is transcriptionally regulated by the estrogen receptor α and reinforce the hypothesis that LRH-1 could exert potential oncogenic effects during breast cancer formation.
Keywords: Binding Sites; Breast Neoplasms; Cell Line, Tumor; DNA-Binding Proteins; genetics; Disease Progression; Estrogen Receptor alpha; physiology; Female; Gene Expression Regulation, Neoplastic; Humans; Promoter Regions (Genetics); RNA, Small Interfering; genetics; Receptors, Cytoplasmic and Nuclear; genetics; Transcription Factors; genetics; Transcription, Genetic
Keywords: Breast Cancer, E2, Estrogen receptor, Ftz-F1, Gene expression, LRH-1, Nuclear receptors, Transcription
Introduction
Liver receptor homolog-1 (LRH-1, NR5A2 (Committee, 1999)) is a monomeric nuclear receptor that belongs to the FTZ-F1 subgroup of nuclear receptors (for review, see Fayard et al., 2004). Recent findings demonstrate that LRH-1 has constitutive transcriptional activity by adopting an active conformation with a large but empty ligand pocket (Sablin et al., 2003), but also identify phosphatidyl inositols as ligands modulating LRH-1 transcriptional activity (Krylova et al., 2005).
Since its molecular cloning, LRH-1 has been linked to a plethora of cellular functions linked to developmental, metabolic and proliferative process. This nuclear receptor plays important developmental functions in mouse, as demonstrated by the genetic dissection of LRH-1 KO mice (Falender et al., 2003; Pare et al., 2004). LRH-1 −/− mice die at e6.5–7.5, due to profound developmental defects, such as impaired node formation and gastrulation (Pare et al., 2004). During adulthood, LRH-1 plays important roles in cholesterol and lipid homeostasis by controlling the expression of genes involved in these metabolic pathways (for review, see Fayard et al., 2004). Several studies reported elevated expression of LRH-1 in granulosa cells and corpus luteum of the ovary, suggesting potential functions for this nuclear receptor in follicular growth and function (Falender et al., 2003; Hinshelwood et al., 2003). By controlling expression of cyclin E1 and D1 through interaction with β-catenin, LRH-1 has recently been shown to promote intestinal cell renewal (Botrugno et al., 2004). However, despite its transcriptional control on G1 phase cyclins, the contribution of LRH-1 to tumorigenesis remains unclear.
17β-estradiol (E2) binds to and activates the estrogen receptor (ER) α (NR3A1) and β (NR3A2) (for review, see Ali & Coombes, 2002). It is well documented that E2 and ERα have promoting effects on cell proliferation and have been involved in breast cancer development. In contrast, ERβ could have a protective effect on breast cancer formation (Bardin et al., 2004). Blocking E2 synthesis by inhibition of aromatase activity or blocking ERα activity using receptor antagonists remain the standard therapy in the treatment of hormone-sensitive breast cancers.
In this study, we analyzed the expression of LRH-1 in several breast cancer cell lines. We observed that LRH-1 is detected in ERα-positive cell lines, whereas no expression is found in ERα-negative cell lines. Moreover, in MCF7 cells, we showed that E2 is a potent activator of LRH-1 expression, through direct binding of ERα to the human LRH-1 promoter in vivo. Interestingly, siRNA-mediated inactivation of LRH-1 decreased the E2-dependent proliferation of MCF7 cells. Finally, we observed, by immunohistochemistry studies, that LRH-1 was expressed in human breast cancers. These findings demonstrate that LRH-1 is an estrogen-responsive gene and represent, to our knowledge, the first direct implication of this nuclear receptor in breast cancer development.
Results
LRH-1 is expressed in breast cancer cell lines
To evaluate a potential implication of LRH-1 in breast cancer development, we first studied LRH-1 mRNA expression in several breast cancer cell lines by real time quantitative PCR (Q-PCR, figure 1). These cell lines are divided in two groups, the first class comprises cell lines that express ERα (ER+), the second class contains cell lines that do not express ERα (ER−). Interestingly, most of the ER+ cell lines express LRH-1 at different levels, with the highest LRH-1 expression in ZR75 cells (figure 1, upper panel). In ER− cell lines, a weak to undetectable expression was observed compared to ER+ cells, suggesting a potential role for ERα in regulating LRH-1 expression (figure 1, lower panel).
Figure 1. LRH-1 is specifically expressed in ER+ breast cancer cell lines.
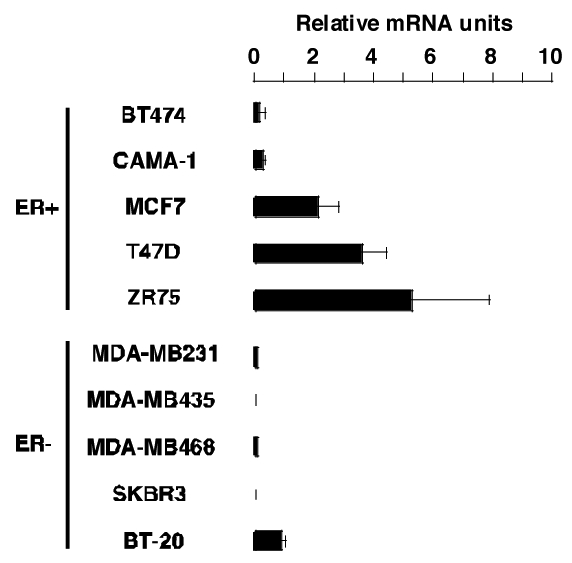
Total mRNA was isolated from several ERα expressing (ER+) and non expressing (ER−) breast cancer cell lines cultured in phenol red free medium containing 10% DCC-FCS. RNA was reverse transcribed as described in the Experimental Procedures. Levels of LRH-1 mRNA were determined by Q-PCR and normalized to RS9.
E2 rapidly induces LRH-1 expression in MCF7 cells
Since LRH-1 is expressed in ER+ breast cancer cell lines, we wanted to address the role of ERα and its natural ligand E2 in the control of LRH-1 mRNA expression. We therefore performed an estradiol time-course treatment (figure 2A). LRH-1 expression was rapidly induced upon E2 addition, with a 4- to 5-fold increase in mRNA levels after 2 hours of treatment, and a maximal induction after 6 hours (8- to 9-fold induction, figure 2A). Increased LRH-1 mRNA levels lasted at least 24 hours after E2 induction (figure 2A). This rapid effect of E2 on LRH-1 mRNA levels suggested that ER could directly regulate the expression of LRH-1. We next wanted to determine the effect of known ERα agonists and antagonists on LRH-1 mRNA expression. Both E2 and the ERα specific agonist PPT increased 4–5 fold LRH-1 mRNA expression (figure 2B). In contrast, the partial ER agonist genistein had no effects on LRH-1 expression (figure 2B). Interestingly, the synthetic antiestrogens OHTam, raloxifene and ICI182780 decreased by 8-, 10- and 4.5-fold, respectively, LRH-1 mRNA expression in MCF7 cells (figure 2B). Finally, to evaluate whether ERα or β could exert isoform specific regulation, we transduced the ER deficient cell line MDA-MB231 with an empty adenovirus (AdCMV), or adenovirus encoding the ERα (AdERα) and β (AdERβ) cDNA as previously described (Lazennec et al., 2001). AdCMV infection had no effect on LRH-1 expression either in the absence or presence of E2, suggesting that ER is required to mediate the effects of E2 on LRH-1 mRNA expression (figure 2C). Supporting this hypothesis, infection of MDA-MB231 cells with an adenovirus encoding hERα resulted in a strong effect of E2 on LRH-1 mRNA expression (figure 2C). Infection of the cells with AdERβ resulted in a much lower induction, suggesting an ERα specific effect (figure 2C). Interestingly, expression of pS2, a known ERα target gene, was similar to what observed for LRH-1 (figure 2C). In summary, these results suggest that LRH-1 is an early target gene of ERα in MCF7 cells. Moreover, re-expression of ERα or β in ER deficient cells followed by E2 treatment leads to LRH-1 mRNA induction, further suggesting the role of ER in this transcriptional regulation.
Figure 2. E2 regulates LRH-1 expression in MCF7 cells.
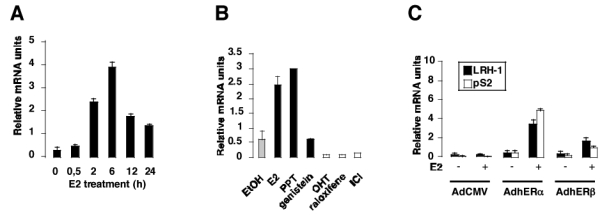
A- MCF7 cells were stripped from endogenous steroids and cultured in phenol red free medium containing 3% DCC-FCS, with E2 at a concentration of 10−8M. Cells were harvested at different time and processed for mRNA extraction. LRH-1 expression was measured by Q-PCR, and data are expressed relative to RS9.
B- Cells were stripped as described in A and incubated with the control vehicle (EtOH), ERα agonists (E2, PPT), ER partial agonist (genistein) or ER antagonists (OHT, raloxifene, ICI) at 10−8M. After 24 hours, RNA was extracted and LRH-1 expression was measured as described in A.
C- The ER- cell line MDA-MB231 was stripped from endogenous steroids as described and infected with non-recombinant adenovirus (AdCMV) or with adenovirus containing the human ERα (AdERα) or β (AdERβ) cDNA. After infection, cells were treated with the vehicle (−) or E2 10−8M (+) for 48 hours. Cells were then harvested and processed for RNA extraction and Q-PCR as described. LRH-1 and pS2 expressions were measured as described in A.
E2 directly regulates LRH-1 transcription
In order to evaluate whether the observed increase in LRH-1 mRNA expression was a direct effect of E2 mediated by ERα, cells were stimulated with E2 in the absence or presence of the protein synthesis inhibitor cycloheximide. Treatment of MCF7 cells with cycloheximide resulted in a strong induction of LRH-1 mRNA expression, suggesting that this transcriptional regulation was not dependent upon de novo synthesis of an intermediate protein (figure 3A). Next, to test if the effect of E2 on LRH-1 mRNA expression was the result of increased LRH1 mRNA stability upon E2 treatment, we performed actinomycin D chase experiments to determine the half-life and stability of LRH-1 mRNA. The apparent half-life of LRH-1 mRNA was approximately 5 hours in the absence of E2, and was not changed upon E2 treatment (figure 3B). These results demonstrated that LRH-1 is directly regulated by E2 and suggested a direct effect of ERα on the human LRH-1 promoter region.
Figure 3. E2 regulation of LRH-1 is not secondary to an intermediate protein or higher mRNA stability.
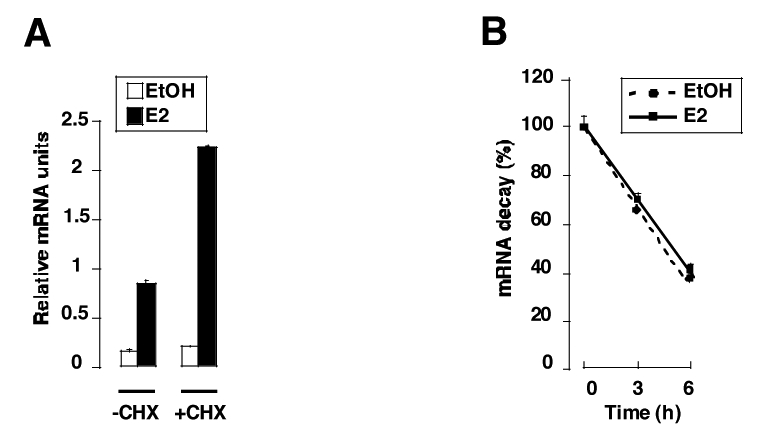
A- MCF7 were treated (+CHX) or not (−CHX) for 1 hour with cycloheximide (25μ/ml) before adding the vehicle (EtOH) or E2 (10−8M). Cells were scrapped 15 hours after E2 treatment and RNA was extracted as described in the Experimental Procedures section. Quantification of LRH-1 expression was performed by Q-PCR and standardized to RS9 levels. The results are means of three independent experiments.
B- MCF7 cells were treated with ethanol (EtOH) or E2 (10−8M) for 24 hours and then incubated for different times (0, 3, 6 hours) with actinomycin D (3μg/ml) before RNA extraction. LRH-1 expression was measured by Q-PCR, normalized to RS9 and the results are expressed as 100% of LRH-1/RS9 RNA levels without actinomycin D treatment (0 hour). The results are means of three independent experiments.
Computational analysis were then performed on the human LRH-1 promoter (figure 4A). In the regulatory region of the human LRH-1 gene, we identified a response element for ER, named ERELRH-1, located at −2338 to −2323 from the transcription initiation site and containing an inverted repeat of the half site GGGTCA separated by 3 nucleotides. This ERELRH-1 sequence is highly homologous to other ERE previously identified, such as the ERE found in the EFP, EBAG9, Cox7A2L and pS2 promoters. To determine whether ER could activate the human LRH-1 promoter, MCF7 cells were then cotransfected with the full-length hLRH-1 promoter reporter construct, containing the ERELRH-1, or a deletion mutant devoid of the ERELRH-1 sequence, with or without an expression vector containing the ERα cDNA (figure 4B). E2 induced LRH-1 promoter activity up to 4-fold in the presence of ERα (figure 4B), whereas a deletion mutant lacking the ERELRH-1 was only slightly induced by E2 treatment, indicating that this sequence is implicated in the transcriptional regulation of LRH-1 promoter by ER. To further demonstrate that the ERELRH-1 was directly implicated in the regulation of LRH-1 expression, we performed cotransfection experiments using a reporter vector consisting of a 210 bp region of the hLRH-1 promoter containing the ERELRH-1 upstream of the minimal Tk promoter driving the expression of a luciferase reporter gene. Cotransfection of this construct and an ERα expression vector resulted in a strong induction of the luciferase activity by E2 in both MCF7 (figure 4C) and MDA-MB231 (figure 4D) cell lines. No effects of E2 in the absence of transfected ERα were observed due to the low levels of expression of ERα in these cells. These results demonstrate that ERα regulates the activity of the LRH-1 promoter through a region containing an ERE.
Figure 4. ERα regulates LRH-1 promoter activity.
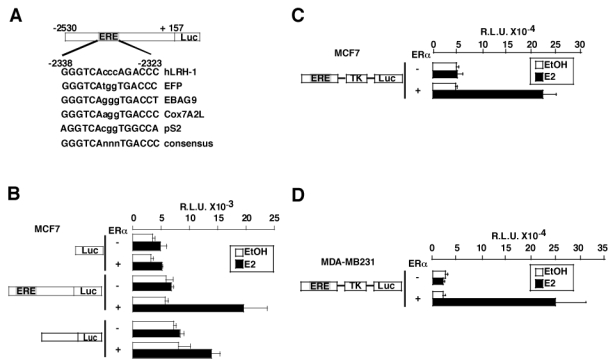
A- Computational analysis of a 2.5 kb fragment of the regulatory region of the human LRH-1 gene demonstrating the presence of a potential binding site for ER homologous to the ERE present in the EFP, ABAG9, Cox7A2L, pS2 genes, except for one base pair. A consensus sequence for the ERE is also represented.
B- MCF7 cells were transfected with (+) or without (−) an expression vector encoding ERα and the pGL3 basic vector, the hLRH-1 reporter vector or a deletion mutant devoid of the ERE. Cells were then treated with the control vehicle ethanol (EtOH) or E2 10−8M for 24 hours. After treatment, cells were lysed and assayed for luciferase activity.
C- The ERELRH-1-Tk-Luc construct was transfected in MCF7 in the presence (+) or absence (−) of ERα. Cells were treated with ethanol (EtOH) or E2 at a concentration of 10−8M, harvested after 24 hours and processed for luciferase activity as described in B.
D- The same construct as in C was cotransfected in the ER- cell line MDA-MB231 with an empty vector or a vector encoding ERα. Cells were stimulated and assayed for luciferase activity as in B.
ERα binds to the human LRH-1 promoter in vitro and in vivo
In order to demonstrate whether this transcriptional regulation occurs through a direct binding of ERα to the ERELRH-1, EMSA were performed using the ERELRH-1 oligonucleotide as a probe. Protein extracts from MDA-MB231 cells infected with an empty adenovirus or an adenovirus encoding ERα were tested for their ability to bind to the ERELRH-1. No binding was observed when proteins from the MDA-MB231 infected with the non-recombinant AdCMV cell line were used (figure 5A, lane 1), whereas a retarded band was observed when proteins from AdERα infected cells were incubated with ERELRH-1 DNA (figure 5A, lane 2). Binding to this site could be competed by adding excess amounts of the respective unlabeled (figure 5A, ERELRH-1, lane 3) or consensus (figure 5A, EREcons, lane 4) ERE oligonucleotides, whereas the mutated ERELRH-1 was not able to efficiently compete with binding (figure 5A, EREmut, lane 5). Addition of an anti-hERα antibody, but not IgG, resulted in a supershifted band, demonstrating the specificity of the binding (figure 5A, lane 6 and 7). Moreover, a radiolabeled double-stranded oligonucleotide containing the mutated ERELRH-1 was unable to bind ERα proteins (figure 5A, lane 8). No binding was observed when radiolabeled EREcons and protein extracts from the non-recombinant AdCMV cell line were used (figure 5A, lane 9), whereas this consensus efficiently bound ERα obtained from AdERα infected cells (figure 5A, lane 10).
Figure 5. ERα binds to the ERE present in the hLRH-1 promoter.
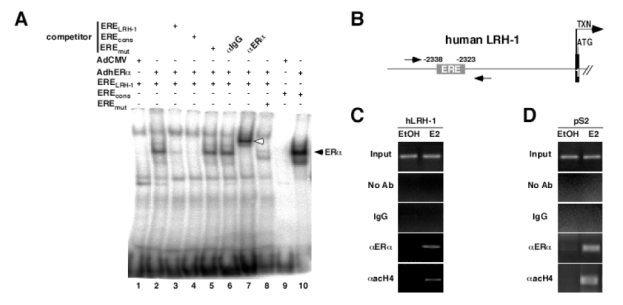
A- EMSA showing binding of ERα to the ERE present in the hLRH-1 promoter. Whole cell nuclear extracts from MDA-MB231 infected with a non recombinant adenovirus (lane 1 and 9) or an adenovirus encoding the human ERα (lane 2 to 8, 10) were incubated with radiolabeled ERELRH-1 (lane 1 to 7), EREmut (lane 8) or EREcons (positive control, lane 9 and 10), in the presence or absence of wild-type (competitor ERELRH-1, lane 3), consensus (competitor EREcons, lane 4) or mutated (competitor EREmut, lane 5) cold probe. Incubation of an anti-ERα antibody resulted in a supershifted band (lane 7, white arrow), whereas no modification in ERα binding was observed with IgG (lane 6).
B- Schematic representation of the 5′ region of the human LRH-1 gene. The ERELRH-1 is indicated. Transcription (arrow, TXN) and translation (ATG) initiation sites are shown. Amplimers are highlighted by the arrows.
C–D ChIP assay demonstrating binding of ERα to the human LRH-1 and pS2 promoters. Cross-linked chromatin from MCF7 treated with the vehicle (EtOH) or E2 10−8M (E2) for 48 hours were incubated without antibody (No Ab), IgG or antibodies against human ERα (aERa) and acetylated histone H4 (aacH4). Immunoprecipitates were analyzed by PCR and positive controls (Input) are shown.
To further prove that ERα binds and activates the LRH-1 promoter, chromatin immunoprecipitation studies of the ERE in the LRH-1 promoter were performed in MCF7 cells treated or not with E2. A 374 bp fragment of the LRH-1 promoter containing the ERE, schematically depicted on figure 6B, was amplified by PCR when anti-ERα or anti acetylated histone H4 were used to immunoprecipitate the chromatin from MCF7 B cells treated with E2 (figure 5C). No amplification was observed in the absence of E2, or when non-specific IgGs were used to immunoprecipitate chromatin (figure 5C). These results suggested that, in the presence of E2, ERα binds to the LRH-1 promoter in vivo. Furthermore, the presence of acetylated histone H4 in this promoter suggested that binding of ERα resulted in promoter activation. As expected, the pS2 promoter, which is a known ER target gene, was immunoprecipitated using the anti-ERα and anti-acetyl H4 antibodies under E2 treatment (figure 5D), validating the observed results for LRH1 promoter.
Figure 6. siRNA-mediated inhibition of LRH-1 expression decreases E2 effect on cell proliferation.
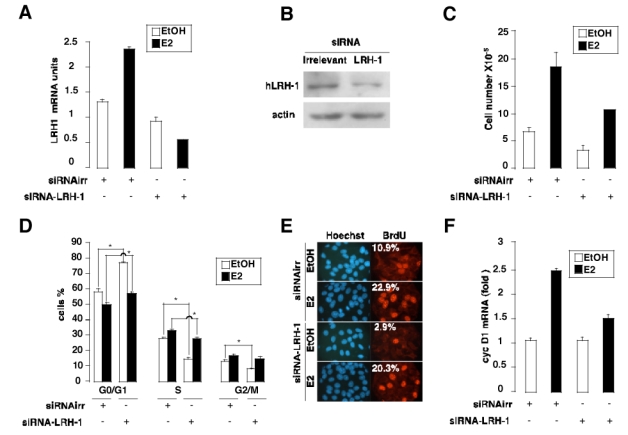
A- RNA levels of LRH-1 are decreased in MCF7 expressing a siRNA-LRH-1 in the absence or presence of E2. Q-PCR results were normalized to RS9.
B- Protein levels of LRH-1 are decreased in MCF7 expressing a siRNA-LRH-1. An anti-β-actin antibody was used as a loading control.
C- Cell proliferation was quantified as described in the Experimental Procedures section. Proliferation of MCF7 cells expressing a LRH-1 siRNA is decreased compared to MCF7 expressing an irrelevant siRNA in the absence (EtOH) or presence of estradiol (E2).
D- FACS analysis of propidium iodide-stained cells comparing the cell cycle profile of MCF7 cells stably expressing an irrelevant or LRH-1 siRNA, in the absence (EtOH) or presence of estradiol (E2).
E- BrdU incorporation in MCF7 siRNAirr and siRNA-LRH-1 cells (Texas red fluorescence, right) compared to the total number of nuclei (Hoechst staining, left). The average percentage of BrdU-labeled cells form five independent field is indicated.
F- siRNA-mediated inhibition of LRH-1 decreases cyclin D1 mRNA expression in MCF7 cells. RNA levels of the Gl cyclin were determined by Q-PCR and normalized to RS9.
LRH-1 inhibition abrogates the proliferative effect of E2 on MCF7 cells
To demonstrate that the regulation of LRH1 expression could explain some of the effects of E2 on breast cancer cell growth, we tested the ability of E2 to trigger proliferation of the ER-expressing breast cancer cell line MCF7 in the absence or in the presence of LRH1, using siRNA technology. The MCF7 cell line stably expressing a siRNA-LRH-1 had lower amounts of LRH-1 mRNA and protein expression, as assayed by Q-PCR analysis and immunoblotting, respectively (figure 6A and B). Cell counting analysis demonstrated that the response to E2 of MCF7 cells was significantly abrogated in cells stably expressing a hairpin RNA that blocked the expression of LRH1 compared to cells expressing a non-relevant siRNA (figure 6C). To determine whether this decrease in cell number was due to decreased cell proliferation and/or a decrease in cell survival, we performed FACS analysis (figure 6D) and BrdU incorporation assays (figure 6E). Interestingly, the cell cycle distribution of cells expressing siRNA-LRH-1 was changed when compared to cells expressing an irrelevant siRNA. A decrease in the proportion of cells in the S phase (28 % for siRNA-irrelevant versus 14,5 % for siRNA-LRH-1) and in the G2/M phase (13,1 % for siRNA-irrelevant versus 8,1 % for siRNA-LRH-1) was observed together with a concomitant increase in the percentage of cells in G0/G1 phase (58,8 % for siRNA-irrelevant versus 77,3 % for siRNA-LRH-1). The effect of the inhibition of LRH-1 expression on cell cycle was less pronounced in the presence of E2, but remains significant for the S phase (33,1 % for siRNA-irrelevant versus 27,8 % for siRNA-LRH-1) and G0/G1 phase (50,1 % for siRNA-irrelevant versus 57,3 % for siRNA-LRH-1, figure 6D). To further prove the effect of inhibition of LRH-1 on cell cycle, BrdU experiments were performed. As observed by FACS analysis, a decreased number of BrdU positive cells was observed when LRH-1 expression was down-regulated by siRNA in the absence of E2 (10.9 % for siRNA-irrelevant versus 2.9 % for siRNA-LRH-1, figure 6E). In the presence of E2, a significant difference in BrdU incorporation was also observed (22.9 % for siRNA-irrelevant versus 20.3 % for siRNA-LRH-1, figure 6E). Furthermore, Q-PCR analysis demonstrated that the expression of cyclin D1, which mediates the effects of E2 on cell proliferation, was down-regulated in MCF7 cells with attenuated expression of LRH1 (figure 6F). These results suggested that LRH1 partially mediates the effects of E2 on the proliferation of MCF7 cells.
LRH-1 is expressed in human breast tumors
Since LRH-1 is transcriptionally regulated by E2 in the breast cancer cell line MCF7, we next wanted to determine whether LRH-1 was expressed in breast cancer biopsies. To validate the specificity of the antibody, LRH-1 protein expression was analyzed in human normal liver and colon adenocarcinoma sections, tissues known to express high level of LRH-1 (Fayard et al., 2004; Schoonjans et al., 2005). In these tissues, LRH-1 immunoreactivity was observed, confirming the specificity of the antibody (figure 7A). To further confirm the specificity of our immunostaining, rabbit IgG were used as primary antibody and incubated with human breast cancer sections (figure 7B). In these conditions, no staining was observed, reinforcing that LRH-1 immunoreactivity using the H2325 anti-LRH-1 antibody is specific. Analysis of LRH-1 expression on human breast cancer sections showed that LRH-1 was expressed in tumor cells of several infiltrating ductal carcinomas, with a nuclear but also cytoplasmatic localization (figure 7B). Moreover, LRH-1 expression was also detected in intraduct carcinomas, suggesting that LRH-1 might be expressed in several types of breast cancers. Interestingly, we observed that the breast tumor region expressing LRH-1 were also expressing ERα and the progesterone receptor (PR, NR3C3), an ERα target gene in breast, suggesting that the in vitro regulation of LRH-1 expression by ERα that we demonstrated in MCF7 cells might be found in situ in ER+ breast carcinomas.
Figure 7. LRH-1 is expressed in tumor cells of mammary infiltrating ductal carcinomas.
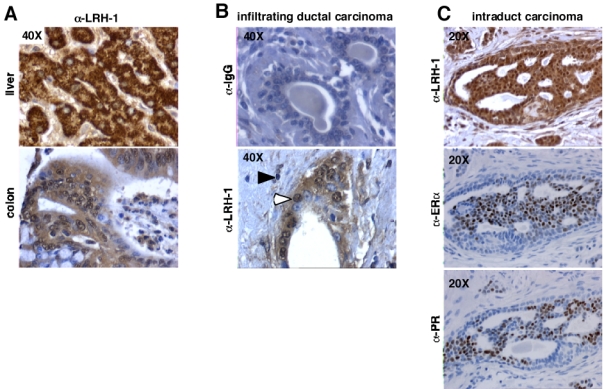
A- We used 5 μm formalin-fixed paraffin-embedded liver and colon tissue sections, as a positive control for immunostaining. A strong immunoreactivity was detected on these tissues.
B- As negative controls, mouse IgGs were used instead of the primary antibody on breast tissue section. No staining was observed in these conditions.
C- LRH-1 immunostaining (open arrowheads) is detected in tumor cells of mammary ductal carcinomas, with a nuclear and cytoplasmatic localization. Inflammatory stromal regions surrounding carcinomas contain some lymphocytes positively stained for LRH-1 (filled arrowheads).
Discussion
Increasing evidence supports a role for LRH1 in the control of proliferative processes. This idea is reinforced by the observation that LRH1 overexpression triggers proliferation of pancreatic and hepatic cancer cell lines (Botrugno et al., 2004). Furthermore, it has been recently demonstrated that LRH-1+/− mice are protected from colon carcinogenesis (Schoonjans et al., 2005). Altogether these data reinforce the idea that LRH-1 could be involved in tumor progression. When overexpressed in pancreatic and hepatic cell lines, LRH-1 promotes cell proliferation, colony formation in soft agar, and tumor progression when LRH-1 overexpressing cells are grafted in athymic mice (Botrugno et al., 2004). The effects of LRH-1 on cell proliferation are mediated through the transcriptional induction of the cyclin E1, D1 and c-myc, in synergy with the complex β-catenin/TCF4. In addition, in two mice models of colon carcinogenesis, i.e. in the genetic model APCMIN/+ and a chemically-induced model using azoxymethane, LRH-1 haploinsufficiency reduces intestinal tumorigenesis (Schoonjans et al., 2005). Interestingly, in this study it was shown that LRH-1 protein was expressed in normal colon and over-expressed in neoplastic lesions with nuclear and cytoplasmatic immunostaining, suggesting a critical role for LRH-1 in intestinal tumorigenesis (Schoonjans et al., 2005). Participation of LRH1 in breast tumor development and progression is supported by our observation that LRH1 inhibition results in decreased proliferation of breast cancer cell lines (Fig. 6), and by the observed overexpression of LRH1 in human breast cancer (Fig. 7 and Zhou et al., 2005).
During breast development and carcinogenesis, E2 and ERα are known to play key roles (Ali & Coombes, 2002). In breast cancers, E2 induces uncontrolled proliferation of ER+ cells (Ali & Coombes, 2002). The switch between a non-proliferating ER+ cell into a proliferating ER+ cell remains, at present, unclear, but is involved in tumorigenesis. One interesting possibility is that upregulation of LRH1 expression could account for the differential effects of E2 in normal epithelial cells compared to breast cancer cells. In this scenario, ERα triggers LRH1 expression, which, in turn, upregulates the expression of cyclins D1 promoting cancer cell proliferation. Interestingly, cyclin D1 is a known target of LRH-1 (Botrugno et al., 2004) and ERα, and is implicated in breast cancer development (Planas-Silva & Weinberg, 1997; Prall et al., 1997). Regulation of cyclin D1 expression in breast cancer involves both genomic (Castoria et al., 2001) and non-genomic (Castro-Rivera et al., 2001; Cicatiello et al., 2004; Sabbah et al., 1999) actions of ERα. ERα regulation of LRH1 expression, which in turn regulates cyclin D1 expression, represents an additional mechanism by which ERα could indirectly increase cyclin D1 levels in breast cancer cells. Furthermore, cyclin D1 directly interacts with ERα and potentiates its transcriptional activity through recruitment of NCOA1 and P/CAF coactivators (McMahon et al., 1999; Neuman et al., 1997; Zwijsen et al., 1998; Zwijsen et al., 1997). This is of particular interest since cyclin D1, through its positive effect on ERα transcriptional activity, could exert a positive feedback loop on LRH-1 expression, finally resulting in an amplification of cell proliferation and tumor progression. Furthermore, it has been shown that local synthesis of aromatase, the cytochrome P450 enzyme responsible for the conversion of C19 adrenal steroids into E2, is under the control of LRH1 (Clyne et al., 2002), resulting in local accumulation of estrogens necessary for tumor progression. Therefore, LRH-1 could be a factor responsible for the local synthesis of E2, promoting epithelial breast tumor progression. This role of LRH1 as a mediator of E2 effects on cell proliferation is supported by our results showing impaired effect of E2 on proliferation of MCF7 cells with attenuated expression of LRH1 (Fig. 7). In summary, we show that LRH1 expression is under the control of ERα in breast cancer cells and mediates the effects of E2 on proliferation of these cells.
Materials and methods
Materials
Propyl pyrazole triol (PPT) was purchased from Tocris Cookson Ltd. (Bristol, UK), estradiol-17β (E2), genistein, cycloheximide and actinomycin D from Sigma (St Louis, MO, USA), the antiestrogens 4-hydroxytamoxifen (OHTam), raloxifen and ICI182780 (ICI) were from AstraZeneca (Rueil Malmaison, France). Anti-ERα antibodies were obtained from Santa Cruz (HC-20, ChIP assay, immunohisotchemistry, Santa Cruz, CA, USA) and from Stressgen (SRA-1000, EMSA, San Diego, CA, USA), anti-acetyl H4 antibody (Lys12) from Cell Signaling (Beverly, MA, USA). The anti-LRH-1 (H-75), anti-PR (C-19) and anti-histone H1 (FL-219) antibodies were purchased from Santa-Cruz.
Cell culture, stable and transient transfections
BT474, CAMA-1, MCF7, T47D, ZR75, MDA-MB231, MDA-MB435, MDA-MB468, SKBR3 and BT-20 breast cancer cell lines were derived from stocks routinely maintained in the laboratory. Monolayer cell cultures were grown in Ham’s F-12/Dulbecco’s modified Eagle’s medium (1:1) (F12/DMEM) or in DMEM supplemented with 10% foetal calf serum (FCS) (InVitrogen, Cergy-Pontoise, France) and antibiotics. Before treatment or transfections, cells were stripped of endogenous estrogens for 5 days using phenol red free medium containing 3% dextran-coated charcoal (DCC) treated FCS (DCC-FCS). Transient transfections were performed in 24 well plates using JetPEI (Qbiogene, Illkirch, France) as described previously (Annicotte et al., 2003). After lysis of the cells, luciferase activity was measured using the centro LB960 luminometer (Berthold technologies, Bad Wildbad, Germany) and measurements were normalized for β-galactosidase activity to correct for differences in transfection efficiency. Graph values represent the mean of three independent experiments.
Recombinant adenovirus construction, propagation and infection
The adenoviruses AdCMV, AdERα and AdERJβ have been described previously (Lazennec et al., 2001). MDA-MB231 were infected for 18 hours at a multiplicity of infection (MOI) of 100 with the different adenoviruses in DMEM/F12 10% DCC-FCS. The next day, the medium was changed and cells were grown for 48 hours before collecting them for RNA or whole cell extract preparation.
RNA extraction, RT-PCR and Q-PCR
RNA extraction and reverse transcription were performed as described (Annicotte et al., 2003). Q-PCR was carried out using a LightCycler and the DNA double strand specific SYBR Green I dye for detection (Roche, Basel, Switzerland). Q-PCR was performed using oligonucleotides specific to hLRH-1 (5′-GGCCCAAACTTATTCCTTCC-3′ and 5′-TGTCCCGTGTGTGGAGATAA-3′), pS2 (5′-TGACTCGGGGTCGCCTTTGGAG-3′ and 5′-GTGAGCCGAGGCACAGCTGCAG-3′), cyclin D1 (5′-CATGGAACACCAGCTCCTGTG-3′ and 5′-GTTCATGGCCAGCGGGAAGAC-3′) and results were then normalized to RS9 levels (5′-AAGGCCGCCCGGGAACTGCTGAC-3′ and 5′-ACCACCTGCTTGCGGACCCTGATA-3′).
Cloning of the human LRH-1 promoter, ERELRH-1 deletion mutant and ERELRH-1-Tk-Luc
The human LRH-1 (hLRH-1) promoter was cloned using BIO-X-ACT DNA polymerase (Bioline GmbH, Luckenwalde, Germany) and human genomic DNA as a template. PCR amplifications were performed according to the manufacturer’s instructions using primers 5′-CGACGCGTCGTGACAGCCAGGATTACCAGTTAT-3′ (M l u I site) and 5′-GAAGATCTTCCAGAAATCATTGAGCAAAAGAAAAGTG-3′ (BglII site) and fragments were subsequently cloned into the pGL3-basic vector (Promega Life Science, Madison, WI) digested by MluI/BglII. A deletion mutant without the ERELRH-1 was obtained using primers 5′-CGACGCGTCGCTGAAAGGCAGTGGACAGCAC-3′ (M l u I site) and 5′-GAAGATCTTCCAGAAATCATTGAGCAAAAGAAA AGTG-3′ (BglII site) and cloned as described previously. A 210 bp PCR fragment containing the ERELRH-1 was cloned in the HindIII site of the pGL3-Tk-Luc vector (a kind gift of P. Balaguer, Montpellier, France) using primers 5′-CCCAAGC TTGGGGATATCCAAATGGGGACATTTCTT-3′ and 5′-CCCAAGCTTGGG CAGTTGAAATGTTGGAATACAGCA-3′. The different pGL3-hLRH-1 promoter constucts were sequenced and used in transient transfections.
Electrophoretic Mobility Shift Assays (EMSA)
Double-stranded oligonucleotides containing the consensus ERE binding site (EREcons 5′-AGCTCTTTGATCAGGTCACTGTGACCTGACTTT-3′) or the ERE present in the hLRH-1 promoter (ERELRH-1 5′-TGACTTCAGGGGTCACCCAGACCCCAAGCCACC-3′) were labeled with T4 polynucleotide kinase and EMSA binding reactions were performed as described previously (Annicotte et al., 2003). For competition experiments, 100-fold molar excess of cold wild type ERELRH-1, EREcons or mutated double-stranded oligonucleotides (EREmut 5′-TGACTTCAGGAATTCCCCAGATCTCAAGCCACC-3′) were included just before adding labeled wild type ERELRH-1 oligonucleotides. Proteins were obtained from whole cell extracts of MDA-MB231 infected with adenoviruses containing the empty vector (AdCMV) or the cDNA of the human ERα (AdERα), as decribed previously (Lazennec et al., 2001). Mouse IgG (negative control) or anti-ERα antibody (SRA-1000) were incubated 20 min at 4°C with whole cell extracts. DNA-protein complexes were separated by electrophoresis on a 4% polyacrylamide gel in 0.25 x TBE buffer at 4°C.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed as described (Annicotte et al., 2003). Briefly, MCF7 cells were cultured for 5 days in phenol red free medium containing 3% DCC-FCS and then treated for 48 hours with EtOH or E2 10−8M. Cells were fixed 10 min in PBS containing 1% formaldehyde and protease inhibitor cocktail (PIC) and subsequently rinsed 5 times in PBS. Cells were collected and centrifuged at 4°C for 5 min at 2000g and resuspended in lysis buffer (50mM HEPES pH 7.5, 140mM NaCl, 1% triton X-100 and PIC). After 30 min of lysis, 3 cycles of sonication (3 pulses of 9 sec, 20% amplitude for cells) were performed to prepare DNA fragments ranging in size from 200 bp to 1000 bp, followed by centrifugation for 10 min. Supernatants were collected and cleared by incubation with protein A-sepharose (2.5 mg), sonicated salmon sperm DNA (2 μg) and 2μg of IgG for 2h at 4°C. Twenty μl of supernatant was collected and used as input. Immunoprecipitation was then carried out overnight at 4°C using no antibody (mock), 2 μg of IgG or 2 μg of antibodies raised against hERa or acetylated histone H4. After centrifugations, washing and elution, the cross-linking was reversed by heating the samples at 65 °C overnight. DNA was then purified using Qiagen PCR purification kit (Qiagen, Courtaboeuf, France) and PCR reaction was performed using primers 5′-TGTGGCCACTTCTGATTCTGACTT-3 ′ and 5′-ACCATGCCCGGCTAATTTTTGTAT-3′, and 5′-ATGGCCACCATGGAGAACAA-3′ and 5′-TAAAACAGTGGCTCCTGGCG-3′ to amplify the human LRH-1 and pS2 promoters, respectively. ChIP assay was performed at least twice for each condition.
RNA interference (RNAi)
RNAi experiment was performed using the pRNAT-U6 RNAi system, following manufacturer’s instructions (GenScript, Piscataway, NJ, USA). Briefly, a double stranded oligonucleotide targeting nucleotides 476 to 494 (5′-AGCGTTGTCCTTACTGTCG-3′) of hLRH-1 mRNA was cloned in the BamHI/HindIII site of the pRNAT-U6-GFP vector. Plasmid constructs were verified by sequencing. Stable transfection was carried out in 100 mm plates using JetPEI and 10 μg of pRNAT-U6 vector containing an irrelevant siRNA (5′-CGTTCTCCGAACGTGTCACGT-3′) or hLRH-1 siRNA. After 48h, cells were selected for 14 days with G418 (1.5 mg/ml). Clones were then amplified for seven days and checked for stable integration by immunofluorescent detection of GFP (data not shown), Q-PCR and immunoblotting as described previously (Annicotte et al., 2003). Positive clones were grown for 5 days using phenol red free medium containing 10% DCC-FCS, plated on cover slips and subsequently treated with EtOH or E2 10−8M for 72 hours. Cells were harvested at 0 and 72 hours and counted as described (Botrugno et al., 2004). RNAi experiments were repeated 3 times.
Flow cytometry analysis and BrdU incorporation assays
MCF-7 stably expressing an irrelevant or hLRH-1 siRNA were grown as described above in phenol red free medium, treated for 48 h with EtOH or E2 10−8M and labelled with propidium iodide. Cells were sorted by FACS analysis (Coulter Electronics, Hialeah, FL, USA) and cell cycle profiles were determined using the ModFit software (Becton Dickinson, San Diego, CA, USA). For BrdU incorporation, cells were plated on coverslips, grown as described above and incubated 2h in the presence of BrdU 100μM. Cells were then processed for BrdU detection as described (Botrugno et al., 2004). At least 500 cells were counted.
Immunohistochemistry (IHC)
IHC was performed on the LandMark™ Tissue MicroArray (Ambion, Austin, TX, USA) containing 5 μm formalin-fixed paraffin-embedded breast tissue sections. After antigen retrieval, sections were incubated with the anti-hLRH-1 antibody (H-75) or the anti-hERα antibody (HC-20) 16 hours at 4°C, and then with a peroxydase-conjugated anti-rabbit secondary antibody (Jackson Immunoresearch, Cambridgeshire, UK). Immunostaining was revealed using the DAB chromogen (DakoCytomation, Glostrup, Denmark) and sections were counterstained with haematoxylin/eosin (DakoCytomation). As a positive control, we used 5 μm formalin-fixed paraffin-embedded human liver and colon carcinoma tissue sections. As negative controls, rabbit IgGs were used instead of the primary antibody on breast tissue section. No specific staining was observed in these conditions.
Acknowledgments
Helpful discussions with Dr M. Esslimani-Sahla and members of the Fajas lab are greatly appreciated. We thank Fanja Rabeolina for excellent technical help and the Vector Core of the University Hospital of Nantes for the production of adenovirus. This work was supported by grants from INSERM, CHU de Montpellier, Association pour la Recherche contre le Cancer, and Fondation pour la Recherche Médicale. J.S.A. is supported by post-doctoral fellowships from the Ligue Nationale Contre le Cancer (2004) and the INSERM (2005). C.C. is supported by a post-doctoral fellowship from the Ligue Nationale Contre le Cancer. This work is dedicated to the memory of the coauthor F. Vignon, deceased while the manuscript was in preparation.
References
- Ali S, Coombes RC. Nat Rev Cancer. 2002;2:101–12. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- Annicotte JS, Fayard E, Swift GH, Selander L, Edlund H, Tanaka T, Kodama T, Schoonjans K, Auwerx J. Mol Cell Biol. 2003;23:6713–24. doi: 10.1128/MCB.23.19.6713-6724.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Endocr Relat Cancer. 2004;11:537–51. doi: 10.1677/erc.1.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, Schoonjans K. Mol Cell. 2004;15:499–509. doi: 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F. Embo J. 2001;20:6050–9. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Rivera E, Samudio I, Safe S. J Biol Chem. 2001;276:30853–61. doi: 10.1074/jbc.M103339200. [DOI] [PubMed] [Google Scholar]
- Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, Cancemi M, Caporali S, Caristi S, Scafoglio C, Teti D, Bresciani F, Perillo B, Weisz A. Mol Cell Biol. 2004;24:7260–74. doi: 10.1128/MCB.24.16.7260-7274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne CD, Speed CJ, Zhou J, Simpson ER. J Biol Chem. 2002;277:20591–7. doi: 10.1074/jbc.M201117200. [DOI] [PubMed] [Google Scholar]
- Committee NRN. Cell. 1999;97:161–3. [Google Scholar]
- Falender AE, Lanz R, Malenfant D, Belanger L, Richards JS. Endocrinology. 2003;144:3598–610. doi: 10.1210/en.2002-0137. [DOI] [PubMed] [Google Scholar]
- Fayard E, Auwerx J, Schoonjans K. Trends Cell Biol. 2004;14:250–60. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Hinshelwood MM, Repa JJ, Shelton JM, Richardson JA, Mangelsdorf DJ, Mendelson CR. Mol Cell Endocrinol. 2003;207:39–45. doi: 10.1016/s0303-7207(03)00257-0. [DOI] [PubMed] [Google Scholar]
- Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, Mackay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA. Cell. 2005;120:343–55. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F. Endocrinology. 2001;142:4120–30. doi: 10.1210/endo.142.9.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C, Suthiphongchai T, DiRenzo J, Ewen ME. Proc Natl Acad Sci U S A. 1999;96:5382–7. doi: 10.1073/pnas.96.10.5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman E, Ladha MH, Lin N, Upton TM, Miller SJ, DiRenzo J, Pestell RG, Hinds PW, Dowdy SF, Brown M, Ewen ME. Mol Cell Biol. 1997;17:5338–47. doi: 10.1128/mcb.17.9.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare JF, Malenfant D, Courtemanche C, Jacob-Wagner M, Roy S, Allard D, Belanger L. J Biol Chem. 2004;279:21206–16. doi: 10.1074/jbc.M401523200. [DOI] [PubMed] [Google Scholar]
- Planas-Silva MD, Weinberg RA. Mol Cell Biol. 1997;17:4059–69. doi: 10.1128/mcb.17.7.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. J Biol Chem. 1997;272:10882–94. doi: 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- Sabbah M, Courilleau D, Mester J, Redeuilh G. Proc Natl Acad Sci U S A. 1999;96:11217–22. doi: 10.1073/pnas.96.20.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablin EP, Krylova IN, Fletterick RJ, Ingraham HA. Mol Cell. 2003;11:1575–85. doi: 10.1016/s1097-2765(03)00236-3. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Dubuquoy L, Mebis J, Fayard E, Wendling O, Haby C, Geboes K, Auwerx J. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0409756102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Suzuki T, Kovacic A, Saito R, Miki Y, Ishida T, Moriya T, Simpson ER, Sasano H, Clyne CD. Cancer Res. 2005;65:657–63. [PubMed] [Google Scholar]
- Zwijsen RM, Buckle RS, Hijmans EM, Loomans CJ, Bernards R. Genes Dev. 1998;12:3488–98. doi: 10.1101/gad.12.22.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. Cell. 1997;88:405–15. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]