

Abstract
A simple and sensitive liquid chromatography/tandem mass spectrometry (LC-MS/MS) method using an atmospheric pressure chemical ionization source (APCI) for the quantification of fenretinide (4-HPR) in mouse plasma was developed and validated. After a simple protein precipitation of plasma sample by acetonitrile, 4-HPR was analyzed by LC-APCI-MS/MS. High performance liquid chromatography (HPLC) separation was conducted on a Hypurity C18 column (50 × 2.1 mm, 5μ) with a flow rate 0.60 mL/min using a gradient mobile phase comprised of 0.05% formic acid in water (A) and methanol (B), and a run time of 4.5 min. The elimination of a tedious sample preparation process and a shorter run time substantially reduced total analysis time. The method was linear over the range 0.5−100 ng/mL, with r>0.998. The intra- and inter-assay precisions were 1.4−9.2% and 5.1−8.2%, respectively, and the intra- and inter-assay accuracies were 93.9−98.6% and 92.7−95.3%, respectively. The absolute recoveries were 90.3% (1.5 ng/mL), 97.0% (7.5 ng/mL) and 92.1% (75.0 ng/mL) for 4-HPR, and 99.1% for the internal standard (150 ng/mL). The analytical method had excellent sensitivity using a small sample volume (30 μL) with the lower limit of quantification (LLOQ) 0.5 ng/mL. This method is robust and has been successfully employed in a pharmacokinetic study of 4-HPR in a mouse xenograft model of neuroblastoma.
Keywords: Fenretinide (4-HPR), N-(4-methoxyphenyl) retinamide (4-MPR), LC-APCI-MS/MS, Plasma
1. Introduction
N-(4-hydroxyphenyl) retinamide or fenretinide (4-HPR), a synthetic amide of alltrans-retinoic acid (ATRA), has emerged as a promising chemopreventive and antiproliferative agent, which is used against various tumor types [1-4,11,13]. It continues to be studied in cancer clinical trials for the treatment of breast, bladder, renal, and neuroblastoma [4–10,12,14].
Since the introduction of 4-HPR in 1985, quantitative analysis of 4-HPR has been performed using high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection [15-22]. Although this technique is well established, most of the published methods require labor-intensive, time-consuming liquid–liquid extraction steps for sample clean up and have long analysis times (retention times ranging between 3.7−9 min). In addition, the lack sensitivity of UV detection method, ranging from 20 ng/mL to 500 ng/mL [15,20,22], requires the use of a large volume (≥ 0.5 ml) of biological sample.
In recent years, the combination of the separation power of HPLC with the selective mass spectrometry (MS) detection has become an important technique in bioanalytical research areas. To our knowledge, LC with MS detection has not yet been reported for the quantitative analysis of 4-HPR. The objective of the investigation was to develop and validate a simple, selective and sensitive LC-MS/MS method for the quantification of 4-HPR in mouse plasma to support a pharmacokinetic study of 4-HPR in a xenograft model of neuroblastoma.
2. Experimental
2.1. Reagents and chemicals
N-(4-hydroxyphenyl) retinamide or fenretinide (4-HPR) and N-(4-methoxyphenyl) retinamide (4-MPR) were provided by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Bethesda, MD, USA), and all-trans-retinoic acid (ATRA) was obtained from Spectrum Inc. (New Brunswick, NJ, USA). The different lots of drug-free (blank) heparinized mouse plasma were obtained from Bioreclamation Inc. (Hicksville, NY, USA). HPLC-grade methanol and acetonitrile were purchased from Fisher-Scientific (Pittsburgh, PA, USA) and reagent-grade formic acid (∼96%) and ammonium acetate (98%) were purchased from Sigma–Aldrich Co. (St. Louis, MO, USA). De-ionized water was prepared using a Milli-Q water purifying system from Millipore Corp. (Bedford, MA, USA).
2.2. Liquid chromatography (LC)
The Shimadzu HPLC system consisted of two LC-20AD delivery pumps, a DGU-20A5 Shimadzu vacuum degasser, a SIL-20AC Shimadzu autosampler and a CBM-20A system controller (Shimadzu Scientific Instruments; Columbia, MD, USA). HPLC separations were performed on a Hypurity C18 analytical column (50 × 2.1 mm, i.d., 5 μ) (Thermo Electron Corp, Waltham, MA, USA), protected by a C8 guard column (2.0 mm × 4.0 mm, i.d.) (Phenomenex Corp., Torrance, CA, USA). Mobile phase A consisted of water with 0.05% formic acid (A) and mobile phase B was 100% methanol. The gradient was as follows: 0−3.00 min, solvent B linear gradient from 70 to 100% B; 3.01−3.50 min, maintain at 100% B; 3.51−4.50 min, maintain at 70% B. The flow rate was 0.60 mL/min and 40 μL was injected for each analysis. The column and autosampler were maintained at room temperature and 4 °C, respectively. An electronic valve actuator with a Rheodyne selector valve was used to divert LC flow to waste, at the first 1.5 min, when no data acquisition was taking place.
2.3. Mass spectrometry analysis
Samples were analyzed with an API 4000 tandem mass spectrometer (MDS Sciex; Toronto, Canada) equipped with an atmospheric pressure chemical ionization (APCI) interface. Software for controlling this equipment, acquiring and processing data was Analyst version 1.4.1 software (MDS Sciex; Toronto, Canada). APCI was performed in the positive ion mode with nitrogen as the nebulizer, auxiliary, collision and curtain gases. Analytes were detected by tandem mass spectrometry using multiple reaction monitoring (MRM) with a dwell time of 200 ms. For the determination of the precursor and product ion spectra, a solution of 500 ng/mL 4-HPR or internal standard in mobile phase was infused directly into the ion sources with a Harvard Apparatus syringe pump at a flow rate of 10 μL/min. The most intense precursor-to-fragment transitions using positive APCI were: 4-HPR, m/z 392.4→283.3; 4-MPR, m/z 406.3→283.3; ATRA, m/z 301.3→123.0. The positive APCI product ion spectra of 4-HPR, 4-MPR and ATRA are shown in Fig. 1.
Figure 1.
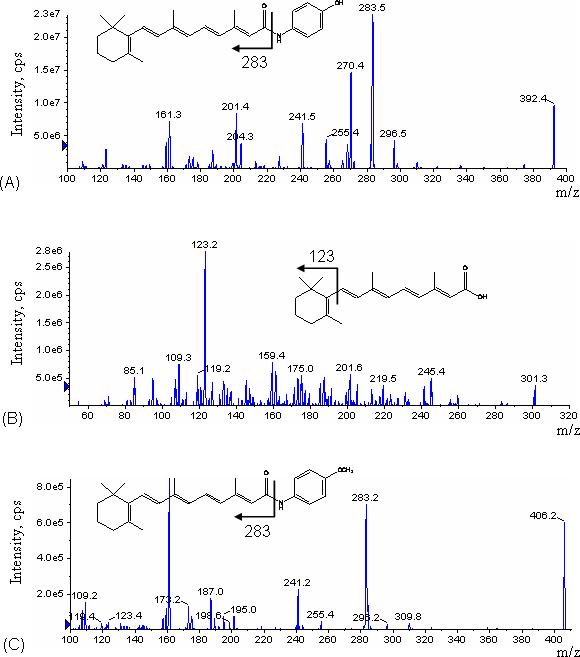
Product ion mass spectra for (A) 4-HPR, (B) ATRA (internal standard), and (C) 4-MPR under the APCI-MS/MS conditions used in MRM mode.
The conditions for ionization of 4-HPR, 4-MPR and internal standard were optimized using individual standard solutions, each at 500 ng/mL, which were infused by a syringe pump through a Tee device at a flow rate of 10 μL/min into the stream of mobile phase eluting from the LC column through a mixing Tee and then into the APCI source, to mimic the LC-MS/MS conditions. The APCI temperature was optimized in the range of 200−500 °C using 25 °C intervals, a linear increase in intensity with increasing APCI temperature observed up to 375 °C, followed by a decrease in intensity with further increase in the APCI temperature, suggesting that 4-HPR may start to decompose at temperatures exceeding 400 °C. The main working parameters of the mass spectrometer were: Collision activate dissociation (CAD) gas 2; Curtain gas, 30; Gas 1 (nebulizer gas) 24; Gas 2 (heater gas) 40; Needle current (NC) 5.00 μA; Source temperature 350 °C. The optimized declustering potential (DP), entrance potential (EP), collision energy (CE), collision cell exit potential (CXP) were set at 72, 8, 19, and 9 for 4-HPR; 70, 8, 18, and 4 for 4-MPR; 61, 8, 21 and 8 for ATRA.
2.4. Preparation of standards and quality control (QC) samples
Two stock solutions were prepared for each analyte from independent preparations. Standard solutions were prepared from one stock solution, and QC samples were prepared from the other. The primary stock solutions of 4-HPR were prepared by dissolving 4-HPR in dimethyl sulfoxide producing a concentration of 1.0 mg/mL and were stored at −20 °C. Two stock solutions of concentration 10 μg/mL were freshly prepared by diluting each primary stock solution with acetonitrile. Working solutions of 4-HPR were freshly prepared by appropriately diluting the respective stock solution with plasma at concentrations of 25, 50 and 100 ng/mL. Eight standards containing 4-HPR concentrations of 0.5, 1.0, 2.5, 5.0, 10.0, 25.0, 50.0 and 100 ng/mL was prepared by adding the appropriate volumes of working solution into 2.0 mL microcentrifuge tubes containing plasma. Three QC levels were prepared in the same manner by adding appropriate volumes of working solution to obtain concentrations of three QC levels were prepared in the same manner by diluting the QC stock solution of 1.5, 7.5 and 75.0 ng/ml, representing low, medium, and high QCs, respectively. The internal standard stock solution was prepared by dissolving 1 mg ATRA per mL of dimethyl sulfoxide and diluting into acetonitrile to a concentration of 10 μg/mL. Internal standard working solution was prepared by diluting the internal standard stock solutions with acetonitrile into a single working solution with a final concentration of 200 ng/mL. Amber glass vials for storing stock solutions and amber plastic vials for processing sample extraction were used, with aluminum foil covering to minimize exposure of the solutions to light to minimize photodegradation.
2.5. Sample preparation
To 30 μL of mouse plasma sample, 90 μL of acetonitrile containing 200 ng/mL of internal standard was added. The sample was vortex-mixed for 1 min and centrifuged at 13,000 rpm for 5 min in an amber plastic vial. A volume of 40 μL of the supernatant was injected into the LC-MS/MS system.
2.6. Method Validation
Method validation and documentation were performed according to guidelines set by the United States Food and Drug Administration (FDA) for bioanalytical method validation [23]. This method was validated in terms of linearity, specificity, lowest limit of quantitation (LLOQ), recovery, intra- and inter-day accuracy and precision, and stability of analyte during the sample storage and processing procedures. Each analytical run included a double blank sample (without internal standard), a blank sample (with internal standard), eight standard concentrations for calibration, and replicate sets (n = 6) of QC samples: low QC (LQC) 1.5 ng/mL, medium QC (MQC) 7.5 ng/mL, and high QC (HQC) 75.0 ng/mL.
2.6.1. Linearity and sensitivity
For the evaluation of the linearity of the standard calibration curve, the analyses of 4-HPR in plasma samples were performed on three independent days using fresh preparations. The calibration curves were prepared over a linear range of 0.5−100 ng/mL at eight concentrations: 0.5, 1.0, 2.5, 5.0, 10.0, 25.0, 50.0 and 100 ng/mL. Each calibration curve consisted of a double blank sample, a blank sample and eight calibrator concentrations. Another double blank sample was analyzed immediately following the highest concentration standard in each run to monitor the carry-over of 4-HPR or the internal standard.
The calibration curve was developed using the following criteria: 1) the mean value should be within ±15% of the theoretical value, except at LLOQ, where it should not deviate by more than ± 20%; 2) the precision around the mean value should not exceed a 15% coefficient of variation (CV), except for LLOQ, where it should not exceed a 20% CV; 3) at least 75% of the non-zero standards of each nominal concentration should meet the above criteria; and 4) the correlation coefficient (r) should be greater than or equal to 0.98.
Each calibration curve was constructed by plotting the analyte to internal standard peak area ratio (y) against analyte concentrations (x). The calibration curves were fitted using a least-square linear regression model y = ax + b, weighted by 1/x2 using the Analyst® software. The resulting a, b, and c parameters were used to determine back-calculated concentrations, which were then statistically evaluated.
2.6.2. Specificity
The specificity was defined as non-interference at retention times of 4-HPR from the endogenous plasma components and no cross-interference between 4-HPR and internal standard using the proposed extraction procedure and LC-MS/MS conditions. Six different lots of blank (4-HPR-free plasma) were evaluated with and without internal standard to assess the specificity of the method.
2.6.3. Accuracy and precision
The intra- and inter-assay precisions were determined using the CV (%), and the intra- and inter-assay accuracies were expressed as the percent difference between the measured concentration and the nominal concentration. The % accuracy of the method was expressed by [(Measured concentration)/(Nominal concentration)] × 100
Intra-assay precision and accuracy were calculated using replicate (n=6) determinations for each concentration of the spiked plasma sample during a single analytical run. Inter-assay precision and accuracy were calculated using replicate (n=6) determinations of each concentration made on three separate days.
2.6.4. Recovery (extraction efficiency) and matrix effect
The extraction efficiency of 4-HPR was determined by analyzing six replicates of 4-HPR plasma samples at three QC concentration levels of 1.5, 7.5, 75.0 ng/mL, respectively. Recovery was calculated by comparing the peak areas of 4-HPR added into blank plasma and extracted using the protein precipitation procedure with those obtained from 4-HPR spiked directly into post-protein precipitation solvent at three QC concentration levels (1.5, 7.5, 75.0 ng/mL). The matrix effect was measured by comparing the peak response of the post-extracted spiked sample with those of the pure standards containing equivalent amounts of the 4-HPR prepared in mobile phase.
2.6.5. Stability study
The stability of 4-HPR in mouse plasma was assessed by analyzing replicates (n=6) of QC samples at concentrations of 1.5, 7.5 and 75.0 ng/mL, during the sample storage and processing procedures. For all stability studies, freshly prepared and stability testing QC samples were evaluated by using freshly prepared standard curve for the measurement. The short-term stability was assessed after exposure of the plasma samples to room temperature prevent from light for 8 hours. The long-term stability was assessed after storage of the plasma samples at −20 °C for 30 days. The freeze/thaw stability was determined after three freeze/thaw cycles (room temperature to −20 °C). The sample stability in the autosampler tray was evaluated by comparing QC samples at 0 and 8 hours in the autosampler tray at 4 °C. This sample stability evaluation mimics the residence time of the samples in the autosampler for each analytic run. The concentrations obtained from all stability studies were compared with the freshly prepared QC samples, and the percentage concentration deviation was calculated. The analytes were considered stable in mouse plasma when the concentration difference was less than 15% between the freshly prepared samples and the stability testing samples.
2.7. Pharmacokinetic study
As component of ongoing studies of retinoids in neuroblastoma xenograft models, 10 female CB17-SCID mice (Taconic Corp., Germantown, NY, USA) received 10 mg/kg fenretinide (5 mg fenretinide dissolved in 0.25 mL ethanol and 8.25 mg bovine serum albumin and made up to 5 mL with 0.9% NaCl) via i.p. administration. The i.p. route was selected for drug administration, as the oral bioavailability of 4-HPR in mouse models is low and variable. One group of 5 mice had blood sampled at 0.5, 2 and 6 hours post-dose, and the other group at 1, 4 and 8 hours post-dose. For each sample, 100 μL of blood was collected via a retro-orbital bleed into lithium heparin tubes. After collection, blood samples were seated for approximately 30 min and centrifuged at 13,000 rpm for 20 min to separate the plasma supernatant. Specimens were processed in the dark, covered with aluminum foil, and plasma stored at −20 °C until analysis
3. Results
3.1. Method validation
3.1.1. Linearity and sensitivity
The method was validated using the above criteria and found to be linear from the concentrations 0.5−100 ng/mL. A representative calibration curve for 4-HPR is shown in Fig. 2. The correlation coefficient (r) from inter-day analysis was found to be greater than 0.998 in all cases. The LLOQ was 0.5 ng/mL, demonstrating a % CV of less than 20% (precision) and an accuracy greater than 80%, with a signal to noise (S/N) ratio of greater than 10. As shown in Table 1 and 2, the intra- and inter-day precision values were 0.1 (n=18) and 3.9% (n=3), respectively, and the intra- and inter-day accuracy values were 100.8 (n=18) and 100.7% (n=3), respectively. A representative chromatogram of double blank, blank, LLOQ and the upper limit of quantification (ULOQ) samples are shown in Fig. 3. The limit of detection (LOD) for the method was 0.07 ng/mL based on a signal to noise ratio of 3. No carry-over peaks were observed at the retention times and the ion channels of either 4-HPR or internal standard.
Figure 2.
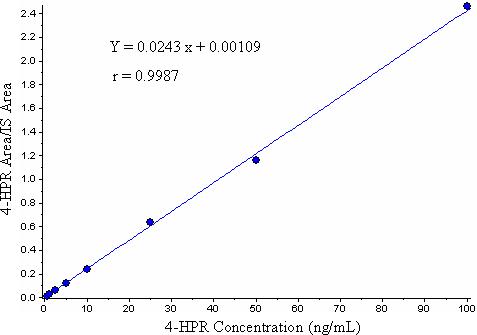
A representative calibration curve for 4-HPR.
Table 1.
Specificity and limit of quantitation of 4-HPR in mouse plasma: lot-to-lot variation
| Nominal conc. LLOQ (0.50 ng/mL) |
|
|
|
|
|---|---|---|---|---|
| Lot # | Measured | STDEV | % CV | % Accuracy |
| Lot 1 (n=3) | 0.44 | 0.05 | 12.20 | 87.87 |
| Lot 2 (n=3) | 0.46 | 0.06 | 13.09 | 91.40 |
| Lot 3 (n=3) | 0.52 | 0.06 | 11.55 | 103.13 |
| Lot 4 (n=3) | 0.58 | 0.02 | 3.34 | 115.67 |
| Lot 5 (n=3) | 0.52 | 0.04 | 8.09 | 103.53 |
| Lot 6 (n=3) |
0.52 |
0.03 |
5.48 |
103.27 |
| n | 18 | |||
| Mean | 0.50 | |||
| STDEV | 0.05 | |||
| % CV | 0.10 | |||
| % Accuracy | 100.81 |
Table 2.
Inter-day accuracy and precision of 4-HPR calibration standards (n = 3)
| Nominal concentration (ng/ml) | Mean | STDEV | CV (%) | Accuracy (%) |
|---|---|---|---|---|
| 0.5 | 0.50 | 0.02 | 3.90 | 100.73 |
| 1 | 0.99 | 0.09 | 8.73 | 99.03 |
| 2.5 | 2.49 | 0.06 | 2.21 | 99.47 |
| 5 | 4.91 | 0.08 | 1.71 | 98.13 |
| 10 | 10.15 | 0.28 | 2.80 | 101.47 |
| 25 | 25.13 | 0.85 | 3.38 | 100.53 |
| 50 | 49.00 | 1.51 | 3.09 | 98.00 |
| 100 | 102.00 | 1.73 | 1.70 | 102.00 |
Figure 3.
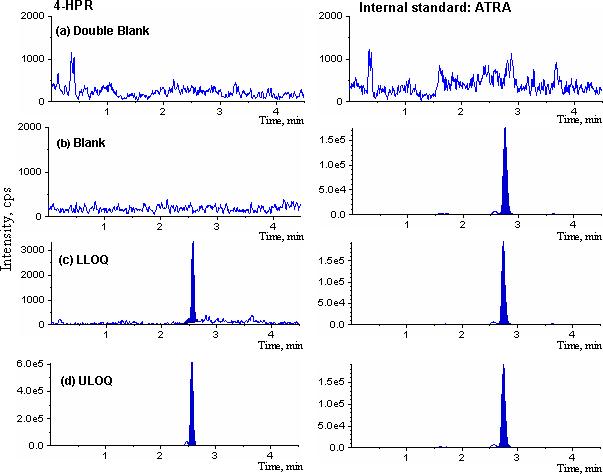
Representative MRM chromatograms of 4-HPR in mouse plasma: (a) double blank plasma; (b) blank plasma; (c) LLOQ, 0.5 ng/mL; and (d) ULOQ, 100 ng/mL. 4-HPR (left panels, a–d) and its internal standard (right panels).
3.1.2. Precision and accuracy
At the eight calibration standards, the inter-day precision ranged from 1.7−8.7% and the accuracy ranged from 98.0−102.0% (Table 2). These data confirm that the present method has a satisfactory accuracy, precision and reproducibility for the quantification of 4-HPR throughout a wide dynamic range. The intra-day and inter-day precision and accuracy of QC samples is summarized in Table 3. The intra-day precision ranged from 1.4−9.2% with the accuracy ranging from 93.9−98.6%. The inter-day precision ranged from 5.1−8.2% and the accuracy ranged from 92.7−95.3%.
Table 3.
Accuracy and precision of 4-HPR QC samples in mouse plasma
| Nominal concentration (ng/ml) | Mean | STDEV | CV (%) | Accuracy (%) |
|---|---|---|---|---|
| Intra-day | ||||
| 1.5 | 1.48 | 0.14 | 9.17 | 98.56 |
| 7.5 | 7.04 | 0.10 | 1.42 | 93.91 |
| 75 | 70.55 | 2.58 | 3.65 | 94.07 |
| n | 6 | 6 | 6 | 6 |
| Inter-day (3 days) | ||||
| 1.5 | 1.43 | 0.12 | 8.24 | 95.33 |
| 7.5 | 7.04 | 0.54 | 7.68 | 93.89 |
| 75 | 69.54 | 3.53 | 5.07 | 92.73 |
| n | 18 | 18 | 18 | 18 |
3.1.3. Recovery and ionization suppression (matrix effect)
Recovery study of protein precipitation was performed on three concentration levels as shown in Table 4. At 1.5, 7.5, and 75.0 ng/mL concentration levels, mean extraction recoveries after 6 replicates were 90.3, 97.0 and 92.1%, respectively. The % CV for all recoveries was less than 4.1%. Data indicated that the extraction efficiency for 4-HPR and internal standard using protein precipitation was sufficient and was not concentration-dependent.
Table 4.
Recovery of 4-HPR in mouse plasma
| Nominal conc. (ng/mL) | |||
|---|---|---|---|
| QC Low 1.5 | QC Mid 7.5 | QC High 75 | |
| % Recovery | 90.29 | 96.98 | 92.05 |
| STDEV | 2.97 | 3.95 | 3.56 |
| % CV | 3.30 | 4.05 | 3.87 |
| n | 6 | 6 | 6 |
Matrix effect can affect on the reproducibility from the analyte or the internal standard of the assay [24-27]. The matrix effect, i.e., the intensity of ion suppression or enhancement is caused by co-eluting matrix components. The matrix effects of 4-HPR and the internal standard were calculated using the following formula: % matrix effect = (A/B) × 100%. A represents the corresponding peak areas of the analytes in spiked plasma post-precipitation and B peak responses of the pure standards prepared in mobile phase. A value of >100% indicated ionization enhancement, and a value of <100% indicated ionization suppression. The matrix effect was tested on the three QCs levels and six individual lots of blank plasma were evaluated. As shown in Table 5, no difference was observed between the pure standards and the post-extracted spiked samples, which means that the HPLC separation conditions had little or no affected by any background signal of plasma after simple protein precipitation clean up step. Matrix effect from dilution of plasma sample was also examined to demonstrate that plasma with concentration greater than the upper limit of the standard curve could be analyzed with acceptable results (data not shown).
Table 5.
Matrix effect of 4-HPR in mouse plasma
| Nominal conc. (ng/mL) | |||
|---|---|---|---|
| QC Low 1.5 | QC Mid 7.5 | QC High 75 | |
| % Matrix effect | 108.67 | 103.54 | 100.84 |
| STDEV | 5.05 | 2.52 | 2.03 |
| % CV | 4.65 | 2.44 | 2.01 |
| n | 6 | 6 | 6 |
3.1.4. Assay specificity
The assay specificity of the method was assessed by the analysis of double blank, blank and LLOQ samples prepared in six different batches of mouse plasma. As demonstrated in Fig. 3 and Table 1, none of the control plasma samples in any of the plasma lots evaluated had any interfering peak from endogenous plasma components at the retention time corresponding to the 4-HPR or its internal standard.
3.1.5. Analyte stability
The stability of 4-HPR was investigated to cover expected conditions during all of the sample storage and process periods, which included the stability data from freeze/thaw, bench-top, autosampler and long-term stability tests. These data are summarized in Table 6. The precision for freeze/thaw samples ranged from 3.8−5.7% and the accuracy ranged from 99.6−107.3%. The results indicated that the analyte was stable in plasma for three cycles when stored at −20 °C and thawed to room temperature. The precision for bench-top stability ranged from 3.3−8.5% and the accuracy ranged from 90.7−97.4%. This indicates reliable stability under the experimental conditions of the analytical runs. The precision and accuracy for long-term stability samples ranged from 3.4−6.7% and 87.7−92.2%, respectively. The results of long-term storage stability data indicated that the plasma samples were stable at −20 °C over 1 month. Further long-term stability study is in progress. The precision ranged from 3.2−8.4% and accuracy ranged from 107.4−110.0% for autosampler stability study. The result suggested that 4-HPR could be analyzed over 8 h in the autosampler tray at 4 °C with acceptable precision and accuracy. The results of stability experiments showed that no stability-related problems occurred during sample storage, extraction and chromatography processes for 4-HPR in plasma samples.
Table 6.
Stability data for 4-HPR under various conditions (n=6)
| Storage period and storage condition | Nominal conc. (ng/mL) | Mean | STDEV | % CV | % Accuracy |
|---|---|---|---|---|---|
| 3 Freeze/thaw cycles |
|||||
| −20 °C | 1.5 | 1.61 | 0.09 | 5.65 | 107.33 |
| 7.5 | 7.64 | 0.35 | 4.62 | 101.84 | |
| |
75 |
74.68 |
2.85 |
3.81 |
99.58 |
| Process sample stability |
|||||
| RT; 8 h | 1.5 | 1.46 | 0.12 | 8.54 | 97.44 |
| 7.5 | 7.29 | 0.52 | 7.15 | 97.18 | |
| |
75 |
68.0 |
2.26 |
3.32 |
90.67 |
| Long term stability |
|||||
| −20 °C; One month | 1.5 | 1.37 | 0.05 | 3.37 | 91.44 |
| 7.5 | 6.92 | 0.46 | 6.58 | 92.22 | |
| |
75 |
65.80 |
4.44 |
6.74 |
87.73 |
| Autosampler stability |
|||||
| 4 °C; 8 h | 1.5 | 1.65 | 0.08 | 4.63 | 110.00 |
| 7.5 | 8.06 | 0.25 | 3.15 | 107.42 | |
| 75 | 81.72 | 6.88 | 8.42 | 108.96 |
3.2. Application to pharmacokinetic study
The method described above was successfully applied to the murine pharmacokinetic study, readily allowing for drug quantitation up to 8 hours following i.p. administration of 10 mg/kg of 4-HPR (Fig. 4A). Figure 4B shows the chromatogram of the separation of 4-HPR, 4-MPR (metabolite) and ATRA (internal standard) from a plasma sample obtained 1 hour after completion of the drug dose.
Figure 4.
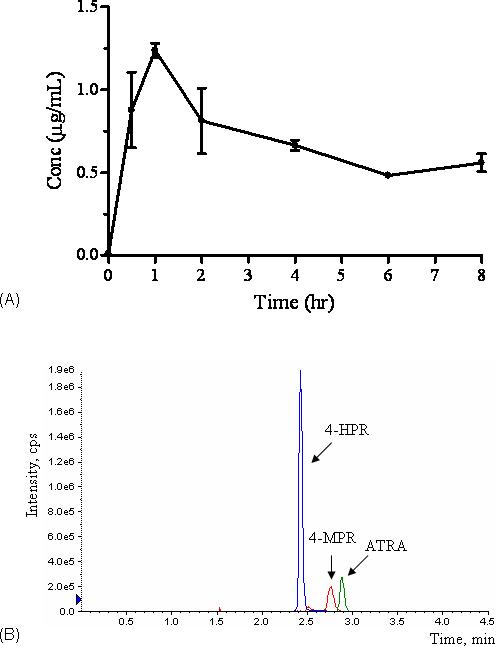
A. The plasma concentration (mean±SD) vs. time profile for 4-HPR following i.p. administration of 10 mg/kg 4-HPR in mice. B. Chromatogram of 4-HPR, 4-MPR (metabolite) and ATRA obtained 1 hour post dosing. Analytes are labeled.
4. Discussion
We developed a fenretinide assay that has a simple and rapid sample clean up step and a rapid, selective and sensitive LC-MS/MS method capable of analyzing a large numbers of light sensitive plasma samples. In addition, the microvolume of sample required allowed for a mouse pharmacokinetic study. A protein precipitation procedure with methanol or acetonitrile was initially evaluated for plasma sample clean up. Although the extraction efficiency for both methods was similar, the methanol precipitation procedure ultimately resulted in high backpressure of LC column. The problem was overcome by using acetonitrile as the protein precipitant.
In this study, ATRA was selected as internal standard, as previously reported internal standards were no longer available. In humans, the fasting plasma ATRA concentration is in the range of 1.5−3.0 ng/mL [28]. In our study using mouse plasma, a small peak eluting with a retention time close to ATRA was observed (Fig. 4 a, b). The ratio of the peak height to that of internal standard used was less than 0.5%, and thus did not impact upon the quantitation of 4-HPR. For pharmacokinetic specimens in which pharmacologic doses of ATRA have been administered, this assay would require modification using an alternate internal standard (e.g. stable 4-HPR isotope).
The organic modifier plays an important role in the resolution of LC and in the ionization efficiency of MS. Various combinations of either methanol or acetonitrile as an organic solvent with and without addition of different content of ammonium acetate or formic acid were evaluated and compared to identify the optimal mobile phase that produced the best sensitivity and peak shape. Methanol was selected based on the best chromatographic separation generated from LC and the highest signal-to-noise (S/N) ratio produced from APCI source. An acidic modifier (formic acid) in the mobile phase enhanced sensitivity approximately two-fold as compared to solvent with no additive while maintaining the separation pattern. A mobile phase consisting of methanol/water containing 0.05% formic acid and the elution gradient profile as described in experimental section were selected.
After the optimal mobile phase was selected, the influence of the LC flow rate (0.2−1.0 mL/min) on the intensity (peak area) of 4-HPR in APCI was investigated. There was little effect on APCI ionization efficiency with larger amounts of eluent sprayed into the APCI source. However, the retention time of 4-HPR was shortened significantly when the flow rate of the mobile phase was greater than 0.8 mL/min, which then caused endogenous substances to interfere with the detection of 4-HPR. The flow rate 0.6 mL/min and the injection volume 40 μL were selected for the optimal chromatographic separation of 4-HPR. Under optimized LC and MS conditions, 4-HPR and ATRA were separated with retention times of 2.6 and 2.8 min, respectively.
5. Conclusion
The LC-APCI-MS/MS method we developed overcomes sample volume limitations encountered with previously described HPLC methods. Our method is accurate, validated and does not require tedious and time-consuming liquid-liquid sample extraction procedures. The method satisfied the requirements of high sensitivity, specificity and rapid sample throughput. Plasma concentrations of 4-HPR can be quantified from 0.5 to 100 ng/ml, making it possible to analyze samples up to 8 hours or longer following an i.p. dose of 10 mg/kg of 4-HPR in mice. This simple and rapid method is suitable for the analysis of microvolume plasma samples for pharmacokinetic studies.
Acknowledgements
This work was supported by a National Cancer Institute grant P01-CA97323.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abou-Issa H, Webb TE, Minton JP, Moeschberger M. J. Natl. Cancer Inst. 1989;81:1820. doi: 10.1093/jnci/81.23.1820. [DOI] [PubMed] [Google Scholar]
- 2.Meyskens F, Jr, Alberts DS, Salmon SE. Int. J. Cancer. 1983;32:295. doi: 10.1002/ijc.2910320306. [DOI] [PubMed] [Google Scholar]
- 3.Moon RC, Metha RG, Rao KVN. Retinoids and cancer in experimental animals. In: Sporn MB, Roberts AB, Goodmans DS, editors. The Retinoids: Biology, Chemistry, and Medicine; 2nd Ed. Raven Press Ltd.; New York, NY: 1994. pp. 573–596. [Google Scholar]
- 4.Pienta KJ, Nguyen NM, Lehr JE. Cancer Res. 1993;53:224. [PubMed] [Google Scholar]
- 5.Decensi A, Fontana V, Fioretto M, Rondanina G, Torrisi R, Orengo MA, Costa A. Eur. J. Cancer. 1997;33:80. doi: 10.1016/s0959-8049(96)00351-6. [DOI] [PubMed] [Google Scholar]
- 6.Decensi A, Bruno S, Costantini M, Torrisi R, Curotto A, Gatteschi B, Nicolo G, Polizzi A, Perloff M, Malone WF, Bruzzi P. J. Natl. Cancer Inst. 1994;86:138. doi: 10.1093/jnci/86.2.138. [DOI] [PubMed] [Google Scholar]
- 7.Rotmensz N, De Palo G, Formelli F, Costa A, Marubini E, Campa T, Crippa A, Danesini GM, Delle Grottaglie M, Di Mauro MG, Filiberti A, Gallazzi M, Guzzon A, Magni A, Malone W, Mariani L, Palvarini M, Perloff M, Pizzichetta M, Veronesi U. Eur. J. Cancer. 1991;27:1127. doi: 10.1016/0277-5379(91)90309-2. [DOI] [PubMed] [Google Scholar]
- 8.Reynolds CP. Curr. Oncol. Rep. 2000;2:511. doi: 10.1007/s11912-000-0104-y. [DOI] [PubMed] [Google Scholar]
- 9.Veronesi U, De Palo G, Costa A, Formelli F, Decensi A. IARC Sci. Publ. 1996;136:87. [PubMed] [Google Scholar]
- 10.Vaishampayan U, Heilbrun LK, Parchment RE, Jain V, Zwiebel J, Boinpally RR, LoRusso P, Hussain M. Invest. New Drugs. 23(225):179. doi: 10.1007/s10637-005-5864-7. [DOI] [PubMed] [Google Scholar]
- 11.Ponthan F, Lindskog M, Karnehed N, Castro J, Kogner P. Oncology Rep. 2003;10:1587. [PubMed] [Google Scholar]
- 12.Gopal AK, Pagel JM, Hedin N, Press OW. Blood. 2004;103:3516. doi: 10.1182/blood-2003-08-2795. [DOI] [PubMed] [Google Scholar]
- 13.Formeli F, Cleris L. European Journal of Cancer. 2000;36:2411. doi: 10.1016/s0959-8049(00)00335-x. [DOI] [PubMed] [Google Scholar]
- 14.Sabici AL, Modiano MR, Lee JJ, Peng Y-M, Xu M-J, Villar H, Dalton WS, Lippman SM. Clin. Cancer Res. 2003;9:2400. [PubMed] [Google Scholar]
- 15.Peng YM, Dalton WS, Alberts DS, Xu MJ, Lim H, Meyskens FL. Int. J. Cancer. 1989;43:22. doi: 10.1002/ijc.2910430106. [DOI] [PubMed] [Google Scholar]
- 16.Lewis KC, Zech LA, Phang JM. Cancer Res. 1994;54:4112. [PubMed] [Google Scholar]
- 17.Conaway CC, Jiao D, Kelloff GJ, Steele VE, Rivensona A, Chung F. Cancer Lett. 1998;124:85. doi: 10.1016/s0304-3835(97)00454-0. [DOI] [PubMed] [Google Scholar]
- 18.Lewis KC, Hochadel JF. Cancer Res. 1999;59:5947. [PubMed] [Google Scholar]
- 19.Kurie JM, Lee JS, Khuri FR, Mao L, Morice RC, Lee JK, Walsh GL, Broxson A, Lippman SM, Ro JY, Kemp BL, Liu D, Fritsche HA, Xu X, Lotan R, Hong WK. Clin. Cancer Res. 2000;6:2973. [PubMed] [Google Scholar]
- 20.Doze FL, Debruyne D, Albessard F, Barre L, Defer GL. Drug Matab. Dispos. 2000;28:205. [PubMed] [Google Scholar]
- 21.Sabichi AL, Modiano MR, Lee JK, Peng Y, Xu M, Villar H, Dalton WS, Lippman SM. Clin. Cancer Res. 2003;9:2400. [PubMed] [Google Scholar]
- 22.Vratilova J, Frgala T, Maurer BJ, Reynolds CP. J. Chromatogr. B. 2004;808:125. doi: 10.1016/j.jchromb.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 23.United States Health and Human Services Guidance for Industry: Bioanalytical Method Validation. 2001 http://www.fda.gov/cder/guidance/index.htm# Biopharmaceutics.
- 24.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal. Chem. 1998;70:882. doi: 10.1021/ac971078+. [DOI] [PubMed] [Google Scholar]
- 25.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal. Chem. 2003;75:3019. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 26.Dams R, Huestis MA, Lambert WE, Murphy CM. J. Am. Soc. Mass Spectrom. 2003;14:1290. doi: 10.1016/S1044-0305(03)00574-9. [DOI] [PubMed] [Google Scholar]
- 27.Taylor PJ. Clin. Biochem. 2005;38:328. doi: 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Sacchi S, Russo D, Avvisati G, Dastoli G, Lazzarino M, Pelicci PG, Bonora MR, Visani G, Grassi C, Iacona I, Luzzi L, Vanzanelli P. Haematologica. 1997;82:106. [PubMed] [Google Scholar]