

Abstract
Vesicoureteral reflux (VUR) is the most common disease of the urinary tract in children. In order to identify gene(s) involved in this complex disorder, we performed a genome-wide search in a selected sample of 31 patients with primary VUR from eight families originating from southern Italy. Sixteen additional families with 41 patients were included in a second stage. Nonparametric, affected-only linkage analysis identified four genomic areas on chromosomes 1, 3, and 4 (p < 0.05); the best result corresponded to the D3S3681-D3S1569 interval on chromosome 3 (nonparametric linkage score, NPL = 2.75, p = 0.008). This region was then saturated with 26 additional markers, tested in the complete group of 72 patients from 24 families (NPL = 2.01, p = 0.01). We identified a genomic area on 3q22.2–23, where 26 patients from six multiplex families shared overlapping haplotypes. However, we did not find evidence for a common ancestral haplotype. The region on chromosome 1 was delimited to 1p36.2–34.3 (D1S228-D1S255, max. NPL = 1.70, p = 0.03), after additional fine typing. Furthermore, on chromosome 22q11.22–12.3, patients from a single family showed excess allele sharing (NPL = 3.35, p = 0.015). Only the chromosome 3q region has been previously reported in the single genome-wide screening available for primary VUR. Our results suggest the presence of several novel loci for primary VUR, giving further evidence for the genetic heterogeneity of this disorder.
Keywords: Primary familial vesicoureteral reflux, VUR, Genetic linkage, Complex disorder
Introduction
Vesicoureteral reflux (VUR) (OMIM 193000) is the most common disease of the urinary tract in children and affects 1–2% of the Caucasian population [1]. VUR may be associated with both acquired postinfectious and congenital parenchymal damage, currently known as reflux nephropathy (RN) [2, 3]. The most serious consequence of RN is chronic renal insufficiency (CRI), leading to end-stage renal failure (ESRF), dialysis, and/or renal transplantation: 25.4% of children affected with CRI have RN [4]. As a consequence, the impact of VUR on public health is considerable and, despite medical and surgical interventions for the past decades, the incidence of VUR-related renal failure has not decreased [5].
The reflux may occur isolated or in association with other congenital abnormalities of kidney/urinary tract (CAKUT) or as part of syndromic entities, such as renal-coloboma or branchio-oto-renal syndromes [6–8]. The exact etiology of primary VUR is not known, but it is probably related to an abnormal morphogenesis of the ureteral bud, leading to a defect of the ureterovesical junction [9]. The initial evidence suggesting a genetic origin of primary VUR came from twin studies, showing an 80–100% concordance for VUR in monozygotic twins vs. a 35–50% concordance in dizygotic twins [10, 11]. Subsequent evidence included familial clustering of VUR [12], ethnic differences between affected and nonaffected individuals [13], and an increased risk (30–50%) of developing VUR in first-degree relatives of an index case [14–16]. From family studies, a range of inheritance patterns was reported, including autosomal dominant with incomplete penetrance [17–20], autosomal recessive [21], polygenic [22], and even X-linked [23].
Previous studies suggest a urinary tract malformation locus on chromosome 6p [24, 25]. Studies of humans with chromosomal abnormalities also suggest candidate loci or genes on chromosomes 10q26 [26], 19q13 (USF2 gene) [27], and 13q33–34 [28]. Because mutations in PAX2 on 10q24 cause renal-coloboma syndrome, a rare autosomal dominant disease with kidney anomalies that include VUR, this gene was also proposed as a candidate [6, 7]. However, none of these loci or genes has been shown causally related to primary VUR [19, 20]. Recently, Lu et al. [29] showed that mutations in the ROBO2 gene contribute to the pathogenesis of VUR/CAKUT in a small proportion of families. In the only genome-wide linkage study reported to date, Feather et al. [18] demonstrated linkage to chromosome 1p13 for primary VUR under a model of autosomal dominant inheritance with reduced penetrance.
Here, we describe the results of the second genome-wide scan for primary VUR. Differently from previous studies and aiming to collect a homogeneous sample set, our patients were ascertained in a single geographic region. Our results suggest the presence of several novel loci for primary VUR, giving further evidence for the genetic heterogeneity of this disorder.
Methods
Patients and families
Fifty-one pedigrees with multiple patients with VUR coming from Campania (southern Italy) were enrolled in the study (Fig. 1). All families were ascertained through an index case, with VUR documented by voiding cystourethrography (VCUG) in males and direct radionuclide cystography (RNC) in females and family members. Three pediatric nephrologists and one radiologist assessed the patients. RN was diagnosed by DMSA scintigraphy (dimercaptosuccinic acid labeled with Technetium-99 m) and defined as focal defects of radionuclide uptake and/or by one-kidney differential uptake below 43% [30]. VUR grading was made according to the International Grading System of Vesicoureteral Reflux [31].
Fig. 1.
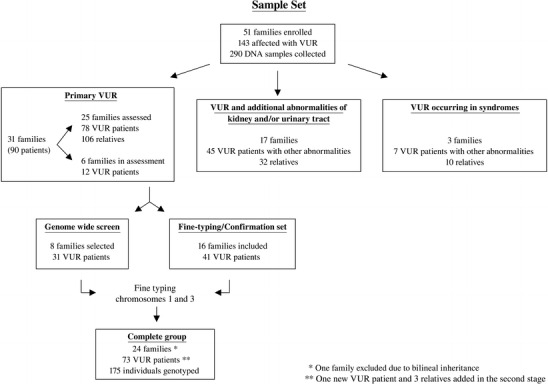
Selection and distribution of families and patients included in the study. VUR vesicoureteral reflux
Additional family members were considered as “affected” based on the presence of reflux documented by VCUG/RNC and/or the diagnosis of RN, or the detection of ESRF/renal replacement in absence of other known causes. As VUR may spontaneously disappear during childhood and adolescence [32], the finding of scintigraphic signs of RN in relatives of VUR patients strongly suggests the previous occurrence of reflux [33]. Individuals with renal symptoms indicative of VUR, such as previous urinary tract infections and/or hypertension and/or proteinuria, not supported by additional findings were classified as “diagnosis unknown”. Patients with secondary VUR, i.e., neurogenic bladder and posterior urethral valves, or other urinary tract abnormalities, i.e., ureterocele and obstructive hydronephrosis, were excluded. The study focused on primary familial VUR. Eight families with 31 patients with VUR were selected for study phase 1 (genome scan) according to the following criteria: diagnosis of primary VUR in absence of any other malformation, two or more affected individuals per family, and a pattern of inheritance compatible with an autosomal dominant model. The second sample (follow-up) consisted of five affected relative pairs (parent–child trios, ten patients) and 11 small families (31 patients) fulfilling the same criteria (Fig. 1).
Informed consent from patients and family members (parents for their children) and approval from the Ethic Committee at Second University of Naples were obtained previously.
Laboratory analysis
Genomic DNA was isolated from peripheral blood leukocytes by standard techniques and was sent from the Paediatrics Department of Second University of Naples to the Department of Clinical Genetics, Erasmus Medical Centre in Rotterdam. A systematic genome scan was performed using the ABI Prism MD-10 set (Applied Biosystems) consisting of 382 short-tandem-repeat polymorphisms markers (STRPs), average spaced 10 cM. Additional markers for further characterization of candidate regions were selected from the gender-average Marshfield genetic map. Information about marker order and distances were obtained from the National Center for Biotechnology Information (NCBI) physical map and Marshfield integrated genetic map. Polymerase chain reaction (PCR) products were resolved on an ABI3100 automated sequencer, and genotypes were analyzed using the GeneMapper software v.2.0 (Applied Biosystems).
Linkage analysis
One thousand simulations were performed (SLINK, MSIM) [34] to investigate the statistical power of our sample set. An autosomal dominant mode of inheritance was assumed with a penetrance of 70%, a mutant allele frequency of 1%, and a phenocopy rate of 1%. The calculations were done under the assumption of genetic heterogeneity. Assuming 35% of unlinked families, a maximum total log of odds (LOD) score of 5.4 was obtained with a low average LOD of 1.58 [standard deviation (SD) = 1.26], after analyzing all eight pedigrees selected previously. Increasing the percentage of unlinked families to 50%, a maximum LOD score of 4.94 was observed (average 0.99, SD = 1.04). Families 1, 4, and 12 had the largest contribution to the total LOD score (LOD = 0.86, 1.04, and 1.62, respectively), followed by families 5, 7, and 13 (LOD = 0.57). Due to their small size, families 2 and 11 had less contribution to the final LOD (LOD = 0.29). Simulations were calculated in the replication group (11 families) after exclusion of five parent–child trios, giving a maximum LOD of 4.57 (average 1.49, SD = 1.07) and a maximum LOD of 4.85 (average 1.14, SD = 1.08), under assumption of 35% and 50% unlinked families, respectively. Family 25 with seven patients was the main contributor to the total LOD score (LOD = 1.47). Finally, simulations were performed in all 19 families (genome scan group and replication group) reaching a maximum LOD of 9.19 (average of 2.76, SD = 1.78) and a maximum LOD of 7.36 (average of 1.71, SD = 1.34), under assumption of 35% and 50% unlinked families, respectively.
Mega2 [35] was used to process the genetic data, whereas the accuracy of allele segregation within the families was confirmed with Pedcheck. The program GENEHUNTER v.2.1 [36] was used to compute multipoint parametric and nonparametric (or model-free) linkage analysis. As VUR may often disappear with age, all individuals in whom VUR, RN, or both were not clinically proven were classified (and analyzed) as “diagnosis unknown”. Thus, a conservative “affected-only” analysis was performed based on these criteria. Due to the uncertain pattern of inheritance, nonparametric analysis was computed. The rationale of a nonparametric analysis is that, among affected relatives, excess sharing of haplotypes identical by descent would be expected, irrespective of the mode of inheritance. The nonparametric linkage statistics examining all individuals simultaneously (NPL-all), is reported.
A parametric analysis was performed as well. An autosomal dominant mode of inheritance with reduced penetrance, as described above (power calculations), was used. Marker allele frequencies were calculated using all spouses (unrelated individuals) coming from the same geographic area. Due to the genetic heterogeneity of the disease, the genome scan data was analyzed, maximizing the heterogeneity LOD score (HLOD) with respect to the proportion of linked families (α). Nonparametric (NPL) and parametric (LOD) scores were calculated for each of the families, and then total NPL and HLOD scores were obtained.
Results
Patients
Fifty-one pedigrees with multiple patients with VUR, originating from the same region of southern Italy (Campania), were available for the study. All persons in the study were ascertained through an index case documented by VCUG/RNC. A total of 143 patients were detected. According to the phenotype, three groups of families were identified: primary VUR; VUR associated with additional abnormalities of kidney/urinary tract such as duplicated collecting system, renal agenesis, or hypospadia; and VUR occurring in syndromes, such as renal-coloboma, branchio-oto-renal syndromes, and reflux associated with congenital ichthyosis (Fig. 1).
A total of 78 primary VUR patients belonging to 25 pedigrees were identified (33 based on a positive VCUG/RNC, 18 based on detection of RN, 21 diagnosed as having both reflux and RN, and six with ESRF). During the course of the study, a new patient affected with RN was detected in family 1 and included in the second stage. Twenty-eight patients were males and 51 were females (ratio M/F = 0.57). The median age at diagnosis was 3.5 years (range 1 month–68 years). Among the 79 patients, 19 were treated surgically, and four out of six who developed ESRF underwent renal transplantation.
The most informative pedigrees are shown in Fig. 2a,b. The pedigrees showed patients in several generations, and male-to-male transmission was observed in some families, supporting an autosomal dominant mode of inheritance. As reported before for primary VUR, we observed several obligate carriers in our families, i.e., families 4, 6, 12, and 13. Family 6 is especially interesting: when assessing the parents (asymptomatic) of the four symptomatic children, both individuals were found to have RN. No consanguinity was reported, but history of ESRF in both branches was described. We are currently recruiting clinical information of those patients (family 6, second generation, Fig. 2). Due to this bilinear transmission, this family was excluded from the linkage analysis.
Fig. 2.
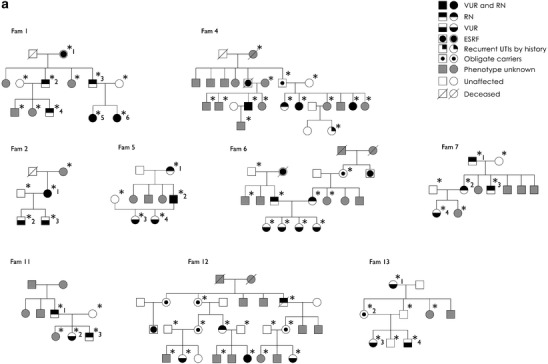
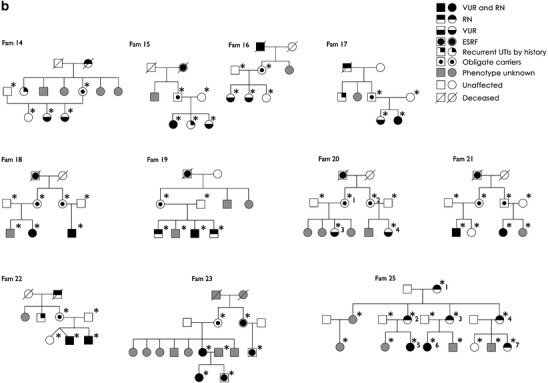
A selection of pedigrees enrolled in the study is shown. Squares indicate males and circles indicate females. Family members with unknown phenotype are in grey, whereas those unaffected (normal voiding cystourethrography or direct radionuclide cystography before age of 5 years and unrelated spouses) are in white. An asterisk highlights the individuals genotyped in the study. A number following the asterisk indicates the patients/obligate carriers reported in Fig. 3. Individuals with urinary tract infections by history and “obligate carriers” are considered as unknown in all analyses. a Nine multiplex pedigrees included in the genome-wide scan are shown. An additional five parent–child trios are not displayed. Family 6 was excluded from the linkage analysis due to the bilineal inheritance. b All 11 multiplex pedigrees included in the fine-typing stage are shown. VUR vesicoureteral reflux, RN reflux nephropathy, ESRF end-stage renal failure, UTIs urinary tract infections
Genome search
We performed a systematic genome-wide scan using 382 microsatellite markers. Results from both parametric and nonparametric linkage analysis excluded most of the genome (data not shown). Furthermore, negative scores were found on both VUR loci, 1p13 [18] (NPL = −0.86, p = 0.86) and 13q33–34 [28] (NPL = −0.21, p = 0.58) previously reported.
We used a nominal p value of < 0.05 to decide whether a region was promising for further study. Table 1 summarizes the genome-wide scan results, showing all regions that yielded a total NPL corresponding to p < 0.05. These regions were located on chromosome 1 (D1S468-D1S255, D1S213-D1S2785), chromosome 3 (D3S3681-D3S1569), and chromosome 4 (D4S402-D4S1597). Furthermore, on chromosome 22q11.22–12.3, patients from a single family showed excess allele sharing (D22S539-D22S280). The best evidence of linkage was observed on chromosome 3p12.3–3q24, which yielded the highest NPL score (2.75, p = 0.008, HLOD = 1.52, α = 0.76) and on chromosome 1p36.32–1p34.3 (NPL = 2.22, p = 0.02, HLOD = 1.13, α = 0.62). Recently, mutations in ROBO2 were described in two VUR/CAKUT patients [29]. ROBO2, which is an ideal functional candidate gene for VUR/CAKUT, maps on chromosome 3p12.3 at the border of our region. Thus, we retrospectively sequenced all gene exons in our index patients, and no mutations were found.
Table 1.
Summary of the genome-wide scan results. Total nonparametric linkage (NPL) scores with p values <0.05
| Pedigrees | D1S468-D1S255 | D1S213-D1S2785 | D3S3681-D3S1569 | D4S402-D4S1597 |
|---|---|---|---|---|
| 1p36.32–1p34.3 | 1q41–1q43 | 3p12.3–3q24 | 4q26–4q32.3 | |
| 4–65 cM | 242–266 cM | 109–158 cM | 117–169 cM | |
| 1 | −0.80 (−1.09) | 0.59 (0.19) | 2.48 (0.87) | −0.37 (0.07) |
| 2 | 1.34 (0.27) | −0.42 (−0.76) | 1.34 (0.28) | 0.44 (0.27) |
| 4 | 1.14 (0.72) | 0.26 (−0.36) | 1.04 (−0.31) | 3.05 (1.3) |
| 5 | 1.9 (0.55) | 1.78 (0.55) | 1.89 (0.58) | 1.69 (0.57) |
| 7 | 1.59 (0.54) | 1.07 (0.42) | 1.75 (0.58) | −0.33 (−1.37) |
| 11 | 1.34 (0.27) | 1.34 (0.27) | 1.34 (0.28) | 0.44 (0.28) |
| 12 | 1.47 (0.19) | 1.58 (0.35) | 0.65 (0.10) | −0.04 (−1.66) |
| 13 | 1.25 (0.54) | 1.97 (0.54) | 1.25 (0.57) | 1.25 (0.57) |
| Total NPL (p) | 2.22 (p = 0.02) | 2.35 (p = 0.02) | 2.75 (p = 0.008) | 2.15 (p = 0.02) |
| Total HLOD (α) | 1.13 (0.62) | 0.61 (0.57) | 1.52 (0.76) | 1.31 (0.51) |
NPL and (LOD) scores per family are shown at each locus. Total NPL (p value) and HLOD (α) obtained from the analysis of all eight pedigrees are displayed. HLOD was obtained maximizing the “heterogeneity” LOD score with respect to the proportion of linked families (α). Negative NPL and LOD scores indicate no linkage or inconclusive result
NPL nonparametric LOD score, HLOD heterogeneity LOD score, α proportion of “linked” families
The NPL score of 2.15 (p = 0.02) on chromosome 4q26–32.3 was mainly due to family 4 (NPL = 3.05, p = 0.062, LOD = 1.3). Yet, allele sharing among the patients of this family was observed also on chromosome 2p23.2–2p15 (D2S165-D2S337, NPL = 3.05, p = 0.062, LOD = 1.3). The only region in which patients from family 12 (Fig. 2) showed excess allele sharing was on chromosome 22q11.22–12.3 (NPL = 3.65, p = 0.015, LOD = 1.49). We then closely inspected the haplotypes from the positive regions in all genotyped families. Families 1, 2, 5, 7, 11, and 13 displayed haplotype sharing among each family’s patients on chromosome 3p12.3–3q24, supporting the linkage to this area. Thus, in six out of eight informative families, we observed allele/haplotype sharing in an overlapping area on chromosome 3q. We noted also on chromosome 1p haplotype sharing in four out of eight multiplex families (families 2, 5, 7, and 11) extending from D1S2667 until D1S255. Therefore, we decided to explore these genomic areas.
Refinement of the chromosome 3 locus
The chromosome 3q region between D3S3681 and D3S1569 was saturated with 26 additional microsatellites markers (average spaced approximately 2 cM). A maximum HLOD = 2.69 (α = 0.95) was reached with an NPL = 2.96, p = 0.001. In a second stage, the fine mapping was extended with 16 additional families with 41 patients. The expansion of pedigrees introduced additional unlinked families, which lowered the maximum HLOD to 1.24 (α = 0.36), giving a total NPL = 2.01 (p = 0.01) (Table 2). Pedigree 25 was the main contributor to the total score, with an NPL = 4.63, p = 0.03, (LOD = 1.45). We then performed a detailed haplotype analysis in all 24 families. Patients from families 2, 5, 7, 13, 20, and 25 shared a common region delimited by markers D3S3641 and D3S1764, containing 36 genes according with the NCBI map, built 36.2. Instead, families 1 and 11 were sharing a more centromeric area not overlapping with the other families (Fig. 3). We did not observe a common “ancestral” haplotype shared across the families.
Table 2.
Summary of fine-typing results in chromosome 1 and 3 (total NPL scores with p values <0.05)
| Pedigrees | D1S468-D1S255 | D3S3681-D3S1569 |
|---|---|---|
| 1p36.32–1p34.3 | 3p12.3–3q24 | |
| 4–65 cM | 109–158 cM | |
| 1 | −0.79 (−2.19) | 1.43 (0.74) |
| 2 | 1.34 (0.28) | 1.34 (0.28) |
| 4 | 1.59 (1.17) | 0.46 (−0.43) |
| 5 | 1.89 (0.56) | 1.79 (0.58) |
| 7 | 1.6 (0.57) | 1.75 (0.57) |
| 11 | 1.34 (0.28) | 1.34 (0.28) |
| 12 | 1.51 (0.05) | 0.81(−0.73) |
| 13 | 1.26 (0.57) | 1.98 (0.57) |
| 14 | 1.41 (0.27) | 0.004 (0.001) |
| 15 | −0.24 (−0.07) | 1.41 (0.28) |
| 16 | −0.07 (−0.02) | 0.0004 (−0.008) |
| 17 | 1.41 (0.28) | 1.41 (0.28) |
| 18 | −0.37 (−0.32) | −0.57 (−0.48) |
| 19 | 0.9 (0.28) | −0.81 (−0.9) |
| 20 | −0.37 (−0.29) | 1.73 (0.48) |
| 21 | −0.48 (−0.45) | −0.58(−0.48) |
| 22 | 1.37 (0.27) | −0.57 (−0.48) |
| 23 | 2.43 (0.86) | −1.41 (−0.96) |
| 25 | 0.22 (0.28) | 4.63 (1.45) |
| Total NPL (p) | 1.70 (p = 0.03) | 2.01 (p = 0.01) |
| Total HLOD (α) | 1.65 (0.55) | 1.24 (0.36) |
NPL and (LOD) scores per family are shown at each locus. Total NPL (p value) and HLOD (α) obtained from the analysis of all eight pedigrees are displayed. HLOD was obtained maximizing the “heterogeneity” LOD score with respect to the proportion of linked families (α). Negative NPL and LOD scores indicate no linkage or inconclusive result
NPL nonparametric LOD score, HLOD heterogeneity LOD score, α proportion of “linked” families
Fig. 3.
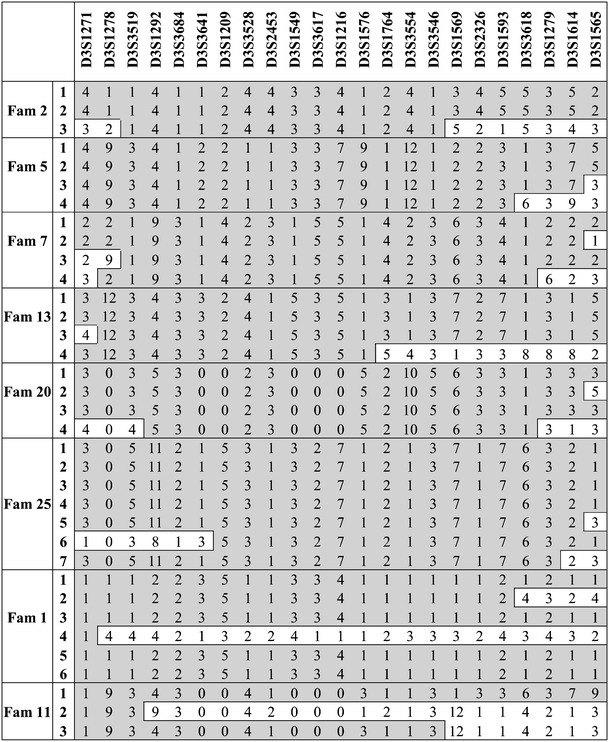
Haplotype analysis in families supporting the chromosome 3 region after fine typing. A selection of microsatellite markers is shown. The shared genomic region is shown in grey; recombinants are shown in white blocks
Refinement of chromosome 1 locus
The 1p36.22-p34.3 region between D1S468 (4.2 cM) and D1S255 (65.5 cM) was saturated with seven extra microsatellite markers (total 12, spaced 4–5 cM) in the complete group of 24 families (Table 2). We observed a maximum HLOD of 1.65 (α = 0.55) and NPL of 1.7 (p = 0.03). The haplotype analysis showed allele sharing in families 2, 5, 7, 11, 19, and 23 between D1S228 (29.8 cM) and D1S255 (65.5 cM). Patients from families 13, 14, and 22 displayed haplotype sharing, but it was limited to the upper or lower part of the region, thus showing no overlapping with the rest of the families.
Candidate genes
There were several interesting genes map on the chromosome 3q region. The PPP2R3A gene encodes the protein phosphatase 2A, one of four major protein phosphatases identified in eukaryotic cells, implicated in the regulation of most major metabolic pathways. It has been shown to be expressed ubiquitously in 15-day-old kidneys, regulating the mitogenic activity in the early embryonic kidney [37]. The FNDC6 gene encodes a type of fibronectin, expressed mainly in fetal kidney [38], implicated in the development of renal basement membrane [39]. RBP1 and RBP2 genes (Retinol-binding-proteins type 1 and 2) are involved in the metabolism of vitamin A, which in its active form of retinoic acid plays a critical role during kidney development, even connecting ureters to bladder [40]. The AGTR1 gene encodes the angiotensin receptor II type 1 (AT1), shown to be related to gross abnormalities in renal morphogenesis in mutant mice (hydronephrosis) as well as a poor proliferation of ureteral smooth cells [41].
Discussion
This study reports the second genome-wide search for primary VUR. Our results suggest the existence of several loci mapping to chromosomes 1, 3, 4, and 22, further supporting the hypothesis that primary VUR is genetically heterogeneous.
We, as others, encountered several pitfalls when studying VUR. We observed clinical variability among and within families and the presence of obligate carriers (individuals who carry and transmit the disease allele but do not manifest any disease sign or symptom). Although the appropriate clinical investigations were performed in several apparently healthy individuals (carriers), no evidence of disease was found, indicating a reduced or age-dependent penetrance. As most of these individuals were recruited during their adulthood, we could not exclude an earlier disease condition that evolved to a spontaneous resolution. To overcome these problems, we first performed a careful clinical evaluation of patients and available relatives. All individuals older than 5 years of age with insufficient or no evidence of VUR were classified as diagnosis unknown, despite the consequent loss of power for the statistical analysis.
We performed the first genome search in VUR that includes families with the same ethnic origin. In order to strengthen the genetic homogeneity of the patients, all families included in this study originated from the same geographic area in the southern part of Italy. Yet, our results strongly support that primary VUR is a (highly) genetic heterogeneous condition. Whereas a large number of our patients and families supported the chromosome 3q locus, one relatively large pedigree (family 12) showed evidence of linkage on chromosome 22. Six of our families supported a locus on chromosome 3q22.2–23 (149.8–153.2 cM). This region is fully overlapping with one of the loci reported by Feather et al. [18]. In their study, the interval on chromosome 3q (from GATA128C02 to D3S1763, 112–176 cM) showed a high α (0.98) with an NPL = 3, p = 0.008. This region was supported by one of their largest pedigrees (with seven patients) that was clearly not linked to the chromosome 1p13 locus. It is interesting that two independent studies have found the same genomic region in distinct groups of patients.
Besides the number of patients included in our genome scan, the results of the statistical analysis are quite modest and do not reach the criteria suggested by Lander and Kruglyak [42] to declare significant linkage. Therefore, some of the identified loci may represent false positive hits and should be interpreted with caution.
The complex disease etiology of primary VUR has shown to be difficult to disentangle. Genetic heterogeneity and lack of knowledge of the true genetic model for VUR are probably the main difficulties in the identification of the genetic etiology of VUR. Although genetic studies in VUR are still in an early phase, we can presume that primary VUR is likely a complex disorder, with a number of not fully penetrant genes causing most of the familiar cases. Finally, primary VUR could be caused by simultaneous gene-environment interactions.
In conclusion, our results show further evidence for the genetic heterogeneity in primary VUR. We will next focus on the refinement of the identified genomic regions and the sequence analysis of the candidate genes according to their tissutal expression and biological function. Replication of the results in additional families will be essential, first to confirm and eventually to evaluate the contribution of these loci to the pathogenesis of primary, nonsyndromic VUR.
Acknowledgments
This work was in part supported by the Nierstichting Nederland (grant number C05.2146) and Fondo per gli Investimenti della Ricerca di Base (FIRB). We gratefully acknowledge patients and family members for donating samples and undergoing several tests for the study. We thank D. Di Pinto and C. Migliaccio for their technical assistance in collecting and preparing DNA samples and F. Sica for assessing and providing clinical data (family 6). We thank T. de Vries Lentsch and P. Marati for the artwork. We also acknowledge Dr. V. Bonifati for his helpful comments and Dr. J. Nauta and Dr. K. Wolffenbuttel for useful discussions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Financial support: This work was in part supported by the Nierstichting Nederland (grant number C05.2146) and Fondo per gli Investimenti della Ricerca di Base (FIRB).
References
- 1.Scott JE, Swallow V, Coulthard MG, Lambert HJ, Lee RE. Screening of newborn babies for familial ureteric reflux. Lancet. 1997;350:396–400. doi: 10.1016/s0140-6736(97)01515-8. [DOI] [PubMed] [Google Scholar]
- 2.Hoberman A, Chao HP, Keller DM, Hickey R, Davis HW, Ellis D. Prevalence of urinary tract infection in febrile infants. J Pediatr. 1993;123:17–23. doi: 10.1016/s0022-3476(05)81531-8. [DOI] [PubMed] [Google Scholar]
- 3.Hodson CJ, Twohill SA. The time factor in the development of sterile renal scarring following high-pressure vesicoureteral reflux. Contrib Nephrol. 1984;39:358–369. doi: 10.1159/000409263. [DOI] [PubMed] [Google Scholar]
- 4.Ardissino G, Dacco V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E, Marra G, Edefonti A, Sereni F. Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics. 2003;111:e382–e387. doi: 10.1542/peds.111.4.e382. [DOI] [PubMed] [Google Scholar]
- 5.Craig JC, Irwing LM, Knight JF, Roy LP. Does treatment of vesicoureteric reflux in childhood prevent end-stage renal disease attributable to reflux nephropathy? Pediatrics. 2000;105:1236–1241. doi: 10.1542/peds.105.6.1236. [DOI] [PubMed] [Google Scholar]
- 6.Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995;9:358–364. doi: 10.1038/ng0495-358. [DOI] [PubMed] [Google Scholar]
- 7.Cheong HI, Cho HY, Kim JH, Yu YS, Ha IS, Choi Y. A clinico-genetic study of renal coloboma syndrome in children. Pediatr Nephrol. 2007;22:1283–1289. doi: 10.1007/s00467-007-0525-z. [DOI] [PubMed] [Google Scholar]
- 8.Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157–1649. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- 9.Murer L, Benetti E, Artifoni L. Embriology and genetics of primary vesico-ureteral reflux. Pediatr Nephrol. 2007;22:788–797. doi: 10.1007/s00467-006-0390-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaefer M, Curran M, Treves ST, Bauer S, Hendren WH, Peters CA, Atala A, Diamond D, Retik A. Sibling vesicoureteral reflux in multiple gestation births. Pediatrics. 2000;105:800–804. doi: 10.1542/peds.105.4.800. [DOI] [PubMed] [Google Scholar]
- 11.Mebust WK, Foret JD. Vesicoureteral reflux in identical twins. J Urol. 1972;108:635–636. doi: 10.1016/s0022-5347(17)60824-3. [DOI] [PubMed] [Google Scholar]
- 12.Tobenkin MI. Hereditary vesicoureteral reflux. South Med J. 1964;57:139–147. doi: 10.1097/00007611-196402000-00004. [DOI] [PubMed] [Google Scholar]
- 13.King LR. Vesicoureteral reflux: a radiographic sign common to multiple diseases. JAMA. 1972;220:854. [PubMed] [Google Scholar]
- 14.Kenda RB, Fettich JJ. Vesicoureteric reflux and renal scars in asymptomatic siblings of children with reflux. Arch Dis Child. 1992;67:506–508. doi: 10.1136/adc.67.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parekh DJ, Pope JC, 4th, Adams MC, Brock JW., 3rd Outcome of sibling vesicoureteral reflux. J Urol. 2002;167:283–284. [PubMed] [Google Scholar]
- 16.Van den Abbeele AD, Treves ST, Lebowitz RL, Bauer S, Davis RT, Retik A, Colodny A. Vesicoureteral reflux in asymptomatic siblings of patients with known reflux: radionuclide cystography. Pediatrics. 1987;79:147–153. [PubMed] [Google Scholar]
- 17.Chapman CJ, Bailey RR, Janus ED, Abbott GD, Lynn KL. Vesicoureteric reflux: segregation analysis. Am J Med Genet. 1985;20:577–584. doi: 10.1002/ajmg.1320200403. [DOI] [PubMed] [Google Scholar]
- 18.Feather SA, Malcolm S, Woolf AS, Wright V, Blaydon D, Reid CJ, Flinter FA, Proesmans W, Devriendt K, Carter J, Warwicker P, Goodship TH, Goodship JA. Primary, nonsyndromic vesicoureteric reflux and its nephropathy is genetically heterogeneous, with a locus on chromosome 1. Am J Hum Genet. 2000;66:1420–1425. doi: 10.1086/302864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanna-Cherchi S, Reese A, Hensle T, Caridi G, Izzi C, Kim YY, Konka A, Murer L, Scolari F, Ravazzolo R, Ghiggeri GM, Gharavi AG. Familial vesicoureteral reflux: testing replication of linkage in seven new multigenerational kindreds. J Am Soc Nephrol. 2005;16:1781–1787. doi: 10.1681/ASN.2004121034. [DOI] [PubMed] [Google Scholar]
- 20.van Eerde AM, Koeleman BP, van de Kamp JM, de Jong TP, Wijmenga C, Giltay JC. Linkage study of 14 candidate genes and loci in four large Dutch families with vesico-ureteral reflux. Pediatr Nephrol. 2007;22:1129–1133. doi: 10.1007/s00467-007-0492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasch A, Hoefele J, Grimminger H, Hacker HW, Hildebrandt F. Multiple urinary tract malformations with likely recessive inheritance in a large Somalian kindred. Nephrol Dial Transplant. 2004;19:3172–3175. doi: 10.1093/ndt/gfh514. [DOI] [PubMed] [Google Scholar]
- 22.de Vargas A, Evans K, Ransley P, Rosenberg AR, Rothwell D, Sherwood T, Williams DI, Barratt TM, Carter CO. A family study of vesicoureteric reflux. J Med Genet. 1978;15:85–96. doi: 10.1136/jmg.15.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Middleton GW, Howards SS, Gillenwater JY. Sex-linked familial reflux. J Urol. 1975;114:36–39. doi: 10.1016/s0022-5347(17)66937-4. [DOI] [PubMed] [Google Scholar]
- 24.Groenen PM, Vanderlinden G, Devriendt K, Fryns JP, Van de Ven WJ. Rearrangement of the human CDC5L gene by a t(6;19)(p21;q13.1) in a patient with multicystic renal dysplasia. Genomics. 1998;49:218–229. doi: 10.1006/geno.1998.5254. [DOI] [PubMed] [Google Scholar]
- 25.Izquierdo L, Porteous M, Paramo PG, Connor JM. Evidence for genetic heterogeneity in hereditary hydronephrosis caused by pelvi-ureteric junction obstruction, with one locus assigned to chromosome 6p. Hum Genet. 1992;89:557–560. doi: 10.1007/BF00219184. [DOI] [PubMed] [Google Scholar]
- 26.Ogata T, Muroya K, Sasagawa I, Kosho T, Wakui K, Sakazume S, Ito K, Matsuo N, Ohashi H, Nagai T. Genetic evidence for a novel gene(s) involved in urogenital development on 10q26. Kidney Int. 2000;58:2281–2290. doi: 10.1046/j.1523-1755.2000.00412.x. [DOI] [PubMed] [Google Scholar]
- 27.Groenen PM, Garcia E, Debeer P, Devriendt K, Fryns JP, Van de Ven WJ. Structure, sequence, and chromosome 19 localization of human USF2 and its rearrangement in a patient with multicystic renal dysplasia. Genomics. 1996;38:141–148. doi: 10.1006/geno.1996.0609. [DOI] [PubMed] [Google Scholar]
- 28.Vats KR, Ishwad C, Singla I, Vats A, Ferrell R, Ellis D, Moritz M, Surti U, Jayakar P, Frederick DR, Vats AN. A locus for renal malformations including vesico-ureteric reflux on chromosome 13q33–34. J Am Soc Nephrol. 2006;17:1158–1167. doi: 10.1681/ASN.2005040404. [DOI] [PubMed] [Google Scholar]
- 29.Lu W, van Eerde AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, Kim HG, Fan Y, Xi Q, Li QG, Sanlaville D, Andrews W, Sundaresan V, Bi W, Yan J, Giltay JC, Wijmenga C, de Jong TP, Feather SA, Woolf AS, Rao Y, Lupski JR, Eccles MR, Quade BJ, Gusella JF, Morton CC, Maas RL. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet. 2007;80:616–632. doi: 10.1086/512735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polito C, La Manna A, Rambaldi PF, Nappi B, Mansi L, Di Toro R. High incidence of a generally small kidney and primary vesicoureteral reflux. J Urol. 2000;164:479–482. [PubMed] [Google Scholar]
- 31.Duckett JW, Bellinger MF. A plea for standardized grading of vesicoureteral reflux. Eur Urol. 1982;8:74–77. doi: 10.1159/000473484. [DOI] [PubMed] [Google Scholar]
- 32.Mattoo TK. Medical management of vesicoureteral reflux. Pediatr Nephrol. 2007;22:1113–1120. doi: 10.1007/s00467-007-0485-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chertin B, Puri P. Familial vesicoureteral reflux. J Urol. 2003;169:1804–1808. doi: 10.1097/01.ju.0000058428.00284.d5. [DOI] [PubMed] [Google Scholar]
- 34.Ott J. Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA. 1989;86:4175–4178. doi: 10.1073/pnas.86.11.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukhopadhyay N, Almasy L, Schroeder M, Mulvihill WP, Weeks DE. Mega 2: data handling for facilitating genetic linkage and association analysis. Bioinformatics. 2005;21:2556–2557. doi: 10.1093/bioinformatics/bti364. [DOI] [PubMed] [Google Scholar]
- 36.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 37.Svennilson J, Durbeej M, Celsi G, Laestadius A, da Cruz e Silva AF, Ekblom P, Aperia A. Evidence for a role of protein phosphatases 1 and 2A during early nephrogenesis. Kidney Int. 1995;48:103–110. doi: 10.1038/ki.1995.273. [DOI] [PubMed] [Google Scholar]
- 38.Laitinen L, Vartio T, Virtanen I. Cellular fibronections are differentially expressed in human fetal and adult kidney. Lab Invest. 1991;64:492–498. [PubMed] [Google Scholar]
- 39.Ekblom P. Formation of basement membranes in the embryonic kidney: an immunohistological study. J Cell Biol. 1981;91:1–10. doi: 10.1083/jcb.91.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batourina E, Choi C, Paragas N, Bello N, Hensle T, Costantini FD, Schuchardt A, Bacallao RL, Mendelsohn CL. Distal ureter morphogenesis depends on epithelial cell remodeling mediated by vitamin A and Ret. Nat Genet. 2002;32:109–115. doi: 10.1038/ng952. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki Y, Ichikawa I. Role of the angiotensin receptor in the development of the mammalian kidney and urinary tract. Comp Biochem Physiol A Mol Integr Physiol. 2001;128:89–97. doi: 10.1016/s1095-6433(00)00299-3. [DOI] [PubMed] [Google Scholar]
- 42.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:541–547. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]