

Summary
Positive transcription elongation factor b (P-TEFb) phosphorylates the C-terminal domain of RNA polymerase II (RNA pol II), facilitating transcriptional elongation. Beside its participation in general transcription, P-TEFb is recruited to specific promoters by some transcription factors such as c-Myc or MyoD. The P-TEFb complex is composed of a cyclin dependent kinase (cdk9) subunit and a regulatory partner (cyclin T1, cyclin T2, or cyclin K). Since cdk9 has been shown to participate in differentiation processes, such as muscle cell differentiation, we studied a possible role of cdk9 in adipogenesis. In this study we show that the expression of the cdk9 p55 isoform is highly regulated during 3T3-L1 adipocyte differentiation at RNA and protein levels. Furthermore, cdk9, as well as cyclin T1 and cyclin T2, show differences in nuclear localization at distinct stages of adipogenesis. Overexpression of cdk9 increases the adipogenic potential of 3T3-L1 cells, whereas inhibition of cdk9 by specific cdk inhibitors, and dominant negative cdk9 mutant impairs adipogenesis. We show that the positive effects of cdk9 on the differentiation of 3T3-L1 cells are mediated by a direct interaction with and phosphorylation of PPARγ which is the master regulator of this process, on the promoter of PPARγ target genes. PPARγ-cdk9 interaction results in increased transcriptional activity of PPARγ and therefore increased adipogenesis.
Keywords: 3T3 Cells, Adipogenesis, drug effects, physiology, Animals, CHO Cells, Cell Differentiation, Cell Division, drug effects, Cricetinae, Cyclin-Dependent Kinase 9, antagonists & inhibitors, metabolism, physiology, Dichlororibofuranosylbenzimidazole, pharmacology, Gene Expression Regulation, Mice, PPAR gamma, metabolism, Phosphorylation, Positive Transcriptional Elongation Factor B, metabolism, Protein Binding
Keywords: Adipogenesis, PPARγ, Nuclear receptors, P-TEFb, RNA pol II, Transcription
Introduction
Positive transcription elongation factor b (P-TEFb) facilitates transcription elongation through phosphorylation of RNA pol II carboxyl-terminal domain (CTD). The active core of P-TEFb comprises cyclin-dependent kinase 9 (cdk9) and a C-type regulatory cyclin (cyclin T1, T2, K) (reviewed in (1)). Cdk9 exist in mammalian cells in two isoforms, p42 cdk9 and p55 cdk9 with a tissue-specific expression pattern, responding to different signals (2). Both isoforms phosphorylate Ser-2 of multiple heptapeptide repeats of CTD RNA pol II releasing the repression action of the DRB sensitivity-inducing factor (DSIF) and the negative elongation factor (NELF). P-TEFb is required not only as a basic transcription elongation factor, but it is also recruited by some transcription factors to activate transcriptional elongation from specific promoters. For example, the requirement of cyclin T1/cdk9 complex by the HIV-1 Tat protein to efficiently elongate the viral RNA has been extensively studied (3, 4). Other transcription factors targeting P-TEFb to the promoters of its target genes include MyoD (5), c-Myc (6), STAT3 (7), NFκB (8), androgen receptor (AR) (9), and the aryl hydrocarbon receptor (10), implicating cyclin T/cdk9 complex in the regulation of cellular processes such as heat-shock response, antigen presentation and processing, apoptosis, muscle cell differentiation, cell growth, or cell proliferation. Moreover, interaction of c-Myc with cyclin T1 and cdk9 results in efficient transcription of c-Myc target genes, and inhibition of cdk9 with DRB, at a concentration that is not inhibiting the general transcription process, results in the block of c-Myc induced proliferation and apoptosis (6). Of particular interest is the participation of P-TEFb in differentiation processes. Interaction of cdk9 with MyoD results in increased differentiation of muscle cells. Interestingly, MyoD interacted with, and was phosphorylated by cyclin T2/cdk9 complex, whereas c-Myc and NFκB were shown to recruit the cdk9/cyclin T1 complex (5). Hence, the specificity of the interaction might be dictated by the cyclin T component of the P-TEFb complex (1). Cdk9 is also required for the monocyte differentiation program. In these cells, cyclin T1 is highly induced upon stimulation with PMA (11). High levels of expression of cyclin T1 and T2, as well as cdk9 in differentiated tissues suggest the participation of P-TEFb in the activation or maintenance of specific differentiation processes (12, 13). Interestingly, white adipose tissue showed very high level of expression of cyclin T1 (12). All these studies prompted us to investigate the role of P-TEFb in the adipocyte differentiation process.
Adipogenesis is a particular system, which involves two major events: preadipocyte proliferation and adipocyte differentiation. Upon reaching confluence, proliferative preadipocytes become growth arrested by contact inhibition. Those cells re-enter cell cycle after hormonal induction, arrest proliferation again and undergo terminal adipocyte differentiation. This last stage is characterized by the coordinated expression of specific genes that will finally determine the specific adipocyte phenotype of the cells. The major regulator of terminal adipocyte differentiation is the peroxisome proliferator-activated receptor gamma (PPARγ reviewed in (14, 15)), which is induced during the initial phases of these terminal stages of adipogenesis by C/EBPβ and δ, as well as by E2F1 (16) PPARγ upon activation by either fatty acid derivatives or antidiabetic thiazolidinediones, drives the expression of several adipocyte-specific genes, such as the fatty acid binding protein (aP2) thus transforming the cell into the characteristic lipid rich adipocyte (17). Subsequent studies have demonstrated that ectopic expression of PPARγ further induces adipocyte differentiation (18). This pivotal role of PPARγ in adipocyte differentiation is also highlighted by the phenotype observed in humans with mutations in the PPARγ gene and by PPARγ deficient mice which are essentially devoid of white adipose tissue (19).
In this study we show that cyclin T1, T2, and cdk9 are expressed in adipocytes. Furthermore, cdk9 activity is increased during adipocyte differentiation. We show that inhibition of cdk9 activity impairs adipogenesis, whereas ectopically expressed cdk9 is highly adipogenic in 3T3-L1 cells. The adipogenic effects of cdk9 are mediated by the induction of PPARγ transcriptional activity through direct phosphorylation. Finally, we show that PPARγ is complexed with P-TEFb on promoters of PPARγ target genes.
Results
P-TEFb is expressed and active in differentiated adipocytes
While evaluating the participation of the P-TEFb complex in adipocyte differentiation we found that cdk9 was expressed at all stages of differentiation of 3T3-L1 preadipocytes, both at the RNA and protein level (Fig. 1A–B). Interestingly, western blot analysis as well as RNA quantification showed that the expression of p55 cdk9 protein isoform was strongly increased during adipocyte differentiation, whereas p42 cdk9 expression was unchanged (Fig. 1A–B). No differences in the expression of cyclin T1 or cyclin T2 were observed (data not shown). aP2 mRNA expression was quantified as a marker of differentiation (Fig. 1C). To further study the expression of P-TEFb during adipogenesis, immunofluorescence analyses were performed. During early stages of differentiation of 3T3-L1 cells cdk9 was evenly distributed between the cytoplasm and the nucleus (Fig. 1D; days 0 and 1). Starting at day 3, cdk9 expression became more prominent in the nuclear compartment (Fig. 1D). Strikingly, at this stage of differentiation coexpression of cdk9 with PPARγ in the nucleus of differentiating cells could be observed (Fig. 1D; days 3,8). Similar results were observed when the expression of cyclins T1 and T2 were analyzed (supplemental Fig. 1). Furthermore, immunoprecipitated cdk9 from differentiated adipocytes was able to phosphorylate purified recombinant RNA pol II CTD, showing a gradual increase in cdk9 activity as, as measured by the level of RNA pol II CTD phosphorylation, indicating that cdk9 was indeed active in adipocytes (Fig. 1E). This was consistent with the increase in nuclear localization of cdk9 at the same stages of adipocyte differentiation (Fig. 1D).
Figure 1. Expression of cdk9 during 3T3-L1 differentiation.
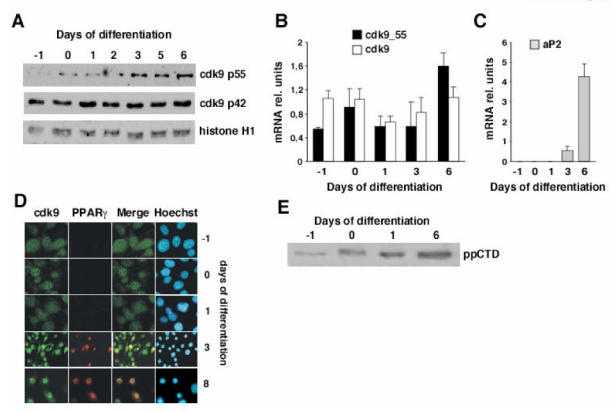
(A) Western blot analysis of nuclear extracts prepared at different days of adipocyte differentiation of 3T3-L1 cells. The proteins detected with specific antibodies are indicated. Histone H1 is used as a load control marker
(B–C) Quantification of mRNA expression levels by real time PCR of the long form (cdk9p55) and total cdk9 (B), and of the adipogenic marker aP2 (C) at the indicated times of differentiation. Results were normalized by the expression level of 18s RNA
(D) Comparative analysis of PPARγ and cdk9 expression by immunofluorescence in 3T3-L1 cells induced to differentiate. Days of differentiation indicate proliferating (day -1), confluent (day 0), re-entry into cell cycle (day 1), early differentiation (day 3), and terminally differentiated (day 8) cells. PPARγ expressing cells are labeled in red whereas cdk9 expressing cells are in green. Nuclei were visualized with Hoechst staining
(E) In vitro kinase assay using immunoprecipitated cdk9 from 3T3-L1 cells at different stages of differentiation. Purified human RNA polymerase II carboxy-terminal domain (CTD) was used as substrate.
These results suggested a role of P-TEFb in adipose tissue biology and differentiation.
DRB inhibition of cdk9 results in impaired clonal expansion and terminal adipocyte differentiation
To further assess the role of cdk9 in the differentiation of 3T3-L1 cells, cdk9 activity was inhibited using 5,6-dichloro-1-β-ribofuranosyl-benzimidazole (DRB). 3T3-L1 cells treated with either vehicle or DRB were compared for their ability to differentiate into adipocytes. After 8 days in differentiation media, normal lipid accumulation was observed in control cells whereas a dose-dependent decrease in lipid accumulation was observed in cells treated with DRB, as showed by Oil Red O staining (Fig. 2A). Quantitative RT-PCR performed on differentiated 3T3-L1 cells confirmed a DRB dose-dependent decrease in expression of the PPARγ target gene aP2, which is a marker of adipocytes (Fig. 2B). 3T3-L1 preadipocytes re-enter cell cycle after hormonal induction of differentiation (the clonal expansion phase). Since this is a required event of these cells before terminal differentiation into adipocytes, we next explored the hypothesis that cdk9 participates in adipogenesis through the control of cell cycle during the clonal expansion phase. A decrease in cell proliferation during the clonal expansion phase (days 1 and 2) was observed, as measured by BrdU incorporation assays, in cells treated with DRB compared to cells treated with vehicle (Fig. 2C). This indicated that inhibition of cdk9 by DRB suppressed, at least in part the clonal expansion phase of adipogenesis. Furthermore, the decrease in cyclin B1 mRNA expression at day 1 of differentiation in cells treated with DRB suggested that cdk9 could participate in the control of the G2/M transition (Fig. 2D). No differences in the expression of cyclin D1 or DHFR were observed at this stage, whereas a significant reduction in the expression of these genes was observed at day 2 of differentiation (Fig. 2D). To further elucidate whether the role of cdk9 was limited to the regulation of the clonal expansion phase of adipocyte differentiation, cdk9 activity was inhibited starting at day 3 of differentiation. At this particular stage 3T3-L1 cells are quiescent and have already gone through the clonal expansion phase (data not shown). Five days after (day 8) treatment, oil-red-O staining indicated an inhibitory dose-dependent effect of DRB in lipid accumulation (Fig. 2E). A dose-dependent decrease in aP2 mRNA expression in DRB treated cells further demonstrated inhibition of adipogenesis (Fig. 2F). These results suggested that cdk9 has a dual role in adipogenesis, i.e. a first role promoting the clonal expansion phase, and a second role promoting terminal differentiation.
Figure 2. Effects of Cdk9 on adipogenesis.
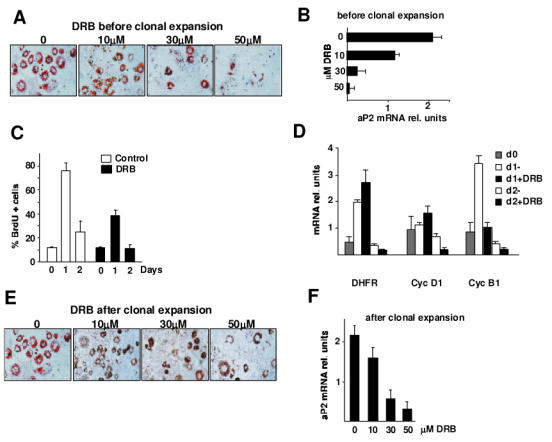
(A–E). Representative micrographs of oil-red-O staining of 3T3-L1 cells differentiated in vitro for 8 days in the presence or absence of the indicated concentrations of the specific cdk9 inhibitor DRB added either at the induction of differentiation (A) or two days after induction (E). mRNA of differentiated cells was analysed for the expression of the adipocyte marker aP2 by quantitative PCR in response to DRB added either before (B) or after (F) the clonal expansion phase. Results were normalized by the expression of the β̃actin RNA. Cell cycle status of the cells was analysed by quantification of BrdU incorporation either in the absence or presence of 30 μM DRB added before the clonal expansion phase (C). mRNA expression of cyclin B1, DHFR, and cyclin D1 was quantified at different times of adipocyte differentiation (d0, d1, d2) in the absence or in the presence of DRB added before the clonal expansion phase (D).
Impaired expression of cdk9 modulates adipogenesis
The observed effects of DRB in adipogenesis could be the result of the inhibition of kinases other than cdk9. To prove the implication of cdk9, a 3T3-L1 cell line overexpressing a dominant negative, kinase dead cdk9 (DNcdk9) was created. Oil-red-o staining indicated that after 8 days in differentiation medium, these cells lost their capacity to differentiate into adipocytes, compared to cells expressing an empty vector (Fig. 3A). Overexpression of DNcdk9 was assessed by PCR assay (Fig. 3A, bottom panel). Inhibition of adipogenesis was further proved by decreased aP2 mRNA expression in DNcdk9 cells (Fig. 3B).
Figure 3. Cdk9 promotes adipogenesis.
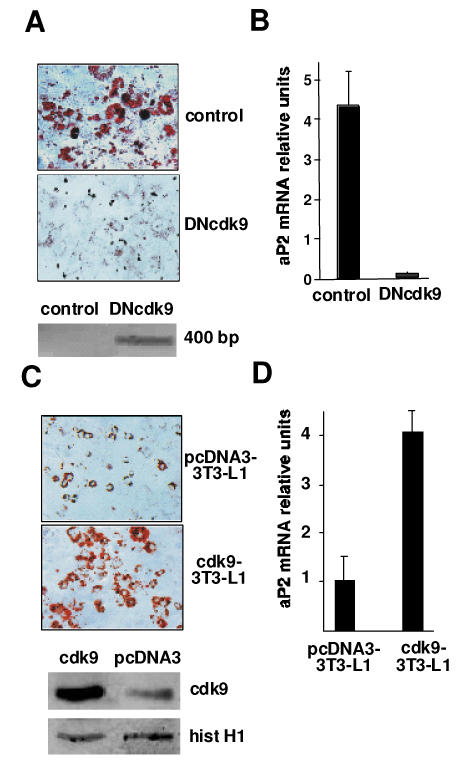
(A) Micrographs of Oil Red O staining of 3T3L1 adipocytes expressing either an empty vector (control) or a vector encoding a dominant negative cdk9 mutant (DNcdk9) 8 days after induction of differentiation. The expression of DNcdk9 is visualized by PCR using T7 and cdk9_42 reverse primers (see Materials and methods) (lower panel)
(B) mRNA expression of aP2, analysed by real-time PCR, of cells used in (A). Results were normalized by the expression of the 18s RNA
(C) Representative micrographs of Oil Red O staining of 3T3L1 adipocytes expressing either an empty vector (pcDNA3-3T3L1) or a vector encoding cdk9 (cdk9-3T3L1) 4 days after induction of differentiation. Overexpression of cdk9 in cdk9-3T3L1 cells is visualized by Western blot (lower panel). Histone H1 was used as a load control marker
(D) mRNA expression of aP2, analysed by real-time PCR, of cells used in (C). Results were normalized by the expression of the 18s RNA.
Since inhibition of cdk9 activity resulted in decreased adipocyte differentiation, we next tested whether overexpressing cdk9 could accelerate this process. To test this hypothesis, we created stable cell lines expressing the pcDNA3-cdk9 and control vector. Overexpression of cdk9 in these cells was demonstrated by western blot analysis (Fig. 3C, bottom panel). After 4 days of differentiation only a small proportion of cells expressing a control vector were differentiated (Fig. 3A–B), whereas a strong increase in lipid accumulation and up-regulation of adipogenic mRNA markers was observed in cells over expressing cdk9 (Fig. 3C–D), further demonstrating that cdk9 is an adipogenic factor.
Cdk9 increases PPARγ activity
Since P-TEFb has an impact on adipogenesis we tested whether these effects could be mediated by PPARγ which is the master regulator of adipocyte differentiation. To analyse the influence of cdk9 on PPARγ activity we performed cotransfection experiments using a PPARγ responsive, luciferase-based, reporter construct (PPRE-TK-Luc) and expression vectors for PPARγ and cdk9. A cdk9 dose-dependent induction of luciferase activity was observed in the presence of PPARγ both in the presence and absence of the PPARγ agonist rosiglitazone (Fig. 4A). To further prove that the effects of cdk9 on the PPARγ responsive promoter were mediated by PPARγ a chimeric Gal4-PPARγ construct containing the Gal4 DNA-binding domain fused to either the PPARγ AB that contains the ligand-independent PPARγ transactivating domain, or the PPARγ LBD that contains the ligand dependent transactivating domain were used in combination with a Gal4 responsive, luciferase-based reporter construct (UAS-TK-Luc). Consistent with the results using a PPRE-TK-Luc, a robust dose-dependent increase in luciferase activity was observed when cdk9 was cotransfected with the Gal4-PPARγ AB construct (Fig. 4B). No effects of cdk9 were observed when the Gal4-PPARγ LBD construct was used either in the presence or absence of rosiglitazone (Fig. 4B). These results demonstrated that cdk9 modulates PPARγ activity. This was further proved by the observation that cdk9 inactivation by cotransfection of a dominant-negative cdk9 expression vector resulted in the attenuation of PPARγ activation by rosiglitazone (Fig. 4C). Moreover, inactivation of cdk9 kinase activity by DRB resulted in a complete inhibition of PPARγ activity (Fig. 4D). Finally to ascertain that cdk9 activates PPARγ-mediated transcription, chromatin immunoprecipitation studies on the aP2 promoter, which is a known PPARγ target gene, were performed in both non-differentiated and differentiated 3T3-L1 adipocytes. A 200 bp fragment of the mouse aP2 promoter, containing the binding site of PPARγ was amplified by PCR when anti-cdk9, anti-PPARγ or anti-acetylated histone H4 antibodies were used to immunoprecipitate chromatin from differentiated 3T3-L1 cells (Fig. 4E, upper panel). No amplification product was observed when immunoprecipitated chromatin from confluent, non-differentiated 3T3-L1 preadipocytes was used as template, or when chromatin was immunoprecipitated using an irrelevant antibody, or when a non-PPRE containing region of the aP2 or GAPDH promoter were used as template (Fig. 4E), demonstrating the specificity of the binding. The results of the ChIP assays demonstrate that the complex cdk9-PPARγ is present on the promoter of a PPARγ target gene. Furthermore, the presence of acetylated histone H4 on the PPARγ binding site of the aP2 promoter suggests that, in the presence of cdk9, this promoter is active. These results suggest that P-TEFb’s stimulatory role during adipogenesis is likely the result of its ability to modulate PPARγ activity.
Figure 4. Cdk9 modulates PPARγ-mediated transactivation in vitro and in vivo.
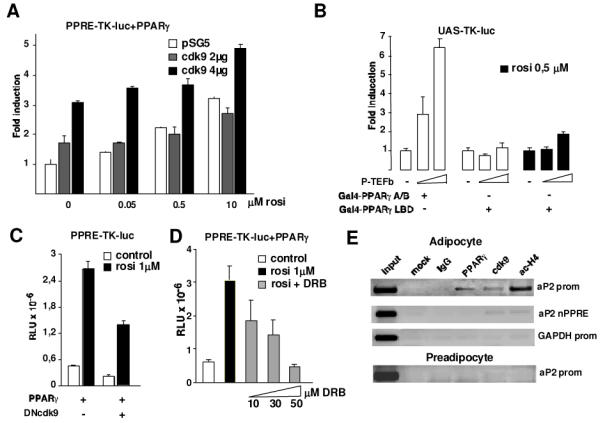
(A) Activity of the PPRE-TK-luc reporter transfected in NIH-3T3 cells in the presence or absence of the PPARγ agonist rosiglitazone. Cells were transfected with an expression vector encoding PPARγ and with the indicated amounts of expression vector coding for cdk9. The luciferase activity was measured and normalized to the expression of a β-gal-encoding plasmid. Activity is presented relative to the values obtained in cells transfected with PPARγ and treated with DMSO
(B) Relative luciferase activity as determined after transfection of NIH-3T3 cells with the Gal4-responsive reporter construct UAS-TK-Luc. Cells were transfected with an expression vector for Gal4-PPARγ A/B fusion protein or Gal4-PPARγLBD fusion protein in the absence or presence of increasing concentrations of a cdk9 and cyclin T1 expression vectors (collectively termed P-TEFb in the figure) and in the absence or presence of rosiglitazone as indicated. Results were normalized for the expression of a β-gal reporter. Values are the mean of 3 independent experiments
(C) NIH-3T3 cells were transfected with PPRE-TK-luc and expression vectors coding for PPARγ and a dominant negative form of cdk9 (DNcdk9) in the presence or absence of PPARγ agonist rosiglitazone
(D) Same transfection as in (A) using PPRE-TK-luc and PPARγ vectors. Cells were treated with increasing concentrations of the cdk9 inhibitor DRB (10–50 μM). Results were normalized to the expression of a β-gal-encoding plasmid
(E) Chromatin immunoprecipitation (ChIP) assay demonstrating binding of cdk9 to the aP2 promoter. Cross-linked chromatin from either confluent preadipocytes (down panel) or 3T3L1 adipocytes differentiated during 7 days (upper panels) was incubated with antibodies against PPARγ cdk9, acetylated histone H4, with purified rabbit IgGs or without any antibody (mock). Immunoprecipitates were analyzed by PCR using primers specific for the aP2 promoter region containing a PPRE (aP2 prom), a region of the aP2 promoter non-containing the PPRE, or the GAPDH promoter. The input, included in the PCR, represents 20% of the total chromatin.
Cdk9 physically interacts with PPARγ
To test whether the induction of PPARγ activity in the presence of P-TEFb is the consequence of a direct interaction between PPARγ and cdk9, nuclear extracts from COS cells transfected with cdk9 and PPARγ expression vectors were immunoprecipitated with cdk9, or irrelevant antibodies. PPARγ protein could be detected in cdk9 immunoprecipitates as assessed by western-blot analysis (Fig. 5A), suggesting that a PPARγ-cdk9 complex is present in these cells. To identify the PPARγ domain responsible for the interaction, GST- PPARγ fusion proteins containing different PPARγ domains were incubated with in-vitro translated 35S-radiolabeled cdk9 (Fig. 5B). Interestingly we found that cdk9 bind to both the A/B and DE domains of PPARγ (Fig. 5C–D). In particular, cdk9 bound to a region from aa 1–72 and 292–399. Same results were observed when cyclin T1 or T2 were used instead of cdk9 (data not shown). These results further demonstrated a direct interaction between cdk9 and PPARγ
Figure 5. PPARγ interacts with cdk9.
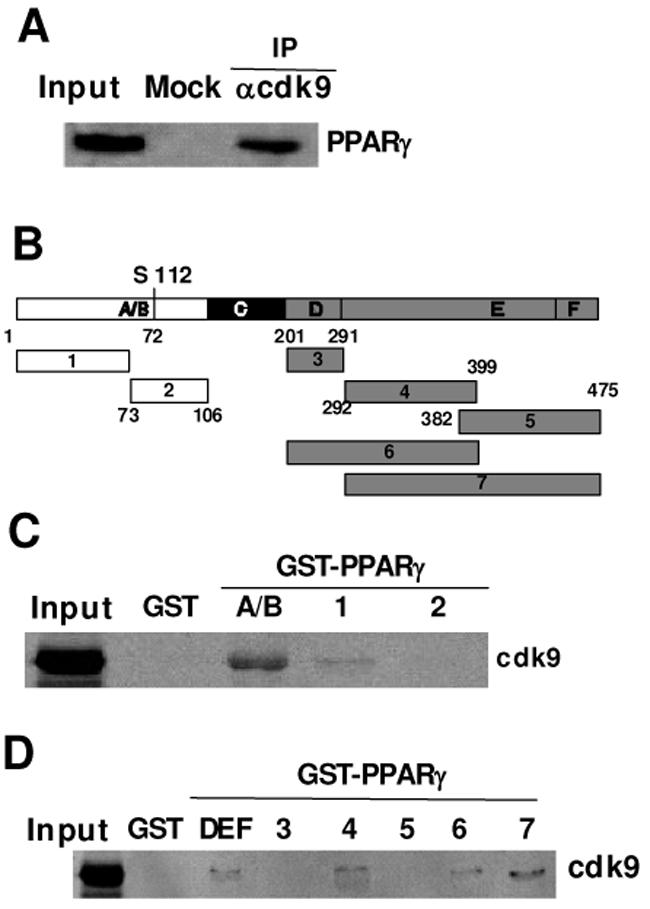
(A) Coimmunoprecipitation of PPARγ and cdk9 from Cos cells transfected with PPARγ and cdk9 expression vectors. Extracts were immunoprecipitated with a cdk9 antibody or purified rabbit IgGs (mock) and revealed by an anti-PPARγ antibody. One twentieth of total extract is shown as a control (input)
(B) Schematical representation of the deletion GST-PPARγ constructs used in the subsequent experiments
(C–D) GST pull-down assay showing the interaction of cdk9 with the A/B (C) or DBF (D) domains of PPARγ In vitro translated 35S-radiolabeled cdk9 protein was incubated with glutathione-sepharose bound GST- PPARγ (deletion constructs) fusion proteins or GST alone. Bound proteins were separated by SDS-PAGE and detected by autoradiography. Input represents total in vitro translated protein.
Cdk9 phosphorylates PPARγ
Cdk9 is a cyclin dependent kinase that modulates the activity of its substrates through phosphorylation. We wanted therefore to test whether modulation of PPARγ activity implicated its phosphorylation by cdk9. Overexpression of cdk9 and cyclin T in Saos cells, resulted in the accumulation of the phosphorylated form of PPARγ as assessed by western blot analysis (Fig. 6A), suggesting that PPARγ could be a target for cdk9. Moreover, immunoprecipitated cdk9 from 293 cells was able to in vitro phosphorylate purified full length GST-PPARγ whereas neither GST alone or GST-PPARγ LBD, which contains the PPARγ ligand binding domain were phosphorylated by cdk9 (Fig. 6B). In contrast, GST-PPARγ A/B, which contains the PPARγ A/B domain was indeed phosphorylated by cdk9 (Fig. 6B). The PPARγ A/B domain contains S112 which has been shown to be phosphorylated by other serine-threonine kinases, such as cdk7. To investigate whether this residue was the target for cdk9, S112 was replaced by an alanine (S112A). GST-PPARγ A/B-S112 was not phosphorylated by cdk9 (Fig. 6B, right panel). These results demonstrate that cdk9 phosphorylates PPARγ at S112. To further elucidate how PPARγ phosphorylation could influence its transcriptional activity, we correlated the expression of the PPARγ target gene aP2 with the phosphorylation status of PPARγ As expected, 3T3-L1 adipocytes treated with rosiglitazone increased the expression of aP2 (Fig. 6C). This effect was blunted by the addition of the PPARγ antagonist GW9662 (Fig. 6B). Strikingly, maximal aP2 induction correlated with the accumulation of the PPARγ phosphorylated forms (Fig. 6D). Similarly, the observed decrease in aP2 expression upon incubation with GW9662 correlated with a switch to non-phosphorylated PPARγ (Fig. 6D). Furthermore, incubation of 3T3-L1 adipocytes with the cdk9 inhibitor DRB resulted in a decrease in aP2 mRNA expression to the levels observed in cells treated with the PPARγ antagonist GW9662 (Fig. 6E). Interestingly, PPARγ phosphorylation was inhibited when cells were incubated with DRB (Fig. 6F). Quantification of the relative intensity of the bands demonstrated that the observed reduction in PPARγ phosphorylation levels was similar to what observed using the PPARγ antagonist GW9662 (Fig. 6D and 6F). Over all these results suggested that phosphorylation by cdk9 increases PPARγ activity. Further supporting this hypothesis was the observation that, in differentiated adipocytes, phosphorylated PPARγ is complexed with the aP2 promoter as demonstrated by ChIP assays (Fig. 6G). Coincident with phospho-PPARγ is the presence of cdk9 and acetylated histone H4 in the aP2 PPRE, indicating that under these conditions the promoter is active (Fig. 6G).
Figure 6. Cdk9 phosphorylates and activates PPARγ.
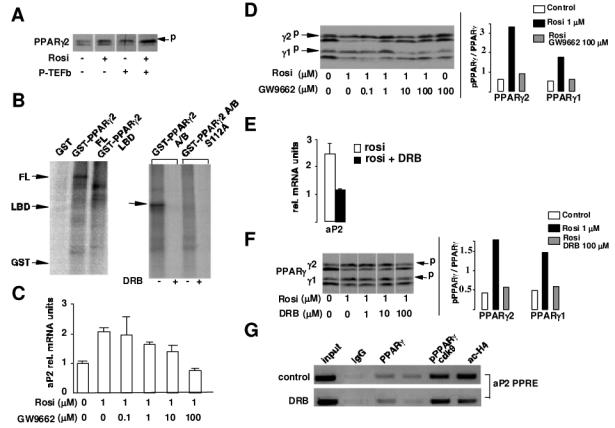
(A) Western blot analysis of PPARγ phosphorylation status in Saos cells transfected with expression vectors for PPARγ cdk9/cyc T1, or both in the absence or presence of rosiglitazone. Two hours before harvesting cells were treated with either rosiglitazone (1μM) or the DMSO vehicle. The phospho PPARγ slow migrating form is indicated by p
(B) In vitro kinase assay using purified GST, GST-PPARγ full-length, GST-PPARγ LBD, GST-PPARγ A/B, or GST-PPARγ A/B S112A fusion proteins as a substrate and immunoprecipitated cdk9 from 293 cells transfected with cdk9. Migration of the different constructs in the SDS gels is indicated by arrows. Specificity of the kinase reaction is verified by the use of the cdk9 kinase inhibitor DRB (250μM)
(C) RT-PCR measuring aP2 mRNA levels in 3T3-L1 adipocytes treated for 6h with rosiglitazone and increasing concentrations of the PPARγ antagonist GW9662. Results were normalized for β-actin expression
(D) Western blot analysis of 3T3-L1 adipocytes, treated for 2h with the indicated concentrations of rosiglitazone and GW9662, showing PPARγ phosphorylation status (left panel). A p indicates migration of phospho-PPARγ The relative intensity of the bands of phosphorylated/non phosphorylated PPARγ is quantified in the right panel
(E) QPCR analysis of aP2 mRNA expression in 3T3-L1 adipocytes treated with either rosiglitazone at 1μM or rosiglitazone and DRB (30 μM). Results were normalized for β-actin expression
(F) Western blot analysis of PPARγ phosphorylation (slow migrating forms) upon incubation of 3T3-L1 cells with rosiglitazone or rosiglitazone and the cdk9 inhibitor DRB for 2h at the indicated concentrations. The relative intensity of the bands of phosphorylated/non phosphorylated PPARγ is quantified in the right panel
(G) Chromatin immunoprecipitation (ChIP) assay demonstrating binding of phosphorylated PPARγ to the aP2 promoter. Cross-linked chromatin from 3T3-L1 adipocytes differentiated during 5 days was incubated with antibodies against PPARγ phospho-PPARγ cdk9, acetylated histone H4, or with purified rabbit IgGs. Immunoprecipitates were analyzed by PCR using primers specific for the aP2 promoter region containing a PPRE (aP2 prom). The input, included in the PCR, represents 20% of the total chromatin.
Cdk9 has a role in adipocyte biology
Besides its participation in adipocyte differentiation PPARγ also plays a major role in adipocyte biology through regulation of genes implicated mainly in lipogenesis. Since cdk9 modulates PPARγ activity we asked whether cdk9 could also participate in these processes. First, we analysed the mRNA expression of lipogenic genes such as aP2, lipoprotein lipase (LPL), stearoyl CoA desaturase (SCD1), and fatty acid synthase (FAS). DRB treatment of 3T3-L1 adipocytes resulted in the inhibition of the expression of aP2, LPL, SCD1, and FAS (Fig. 7A), whereas no changes in the expression of genes implicated in lipolysis were observed (data not shown). These results suggested that similar to PPARγ cdk9 could also be implicated in lipogenesis. Interestingly, expression of cdk9 was strongly increased in adipose tissue of mice fed a high fat diet (Fig. 7B). Furthermore, genetically obese db/db mice expressed high levels of cdk9 mRNA in adipose tissue compared to control non-obese mice (Fig. 7C).
Figure 7. Cdk9 participation in adipose tissue biology.
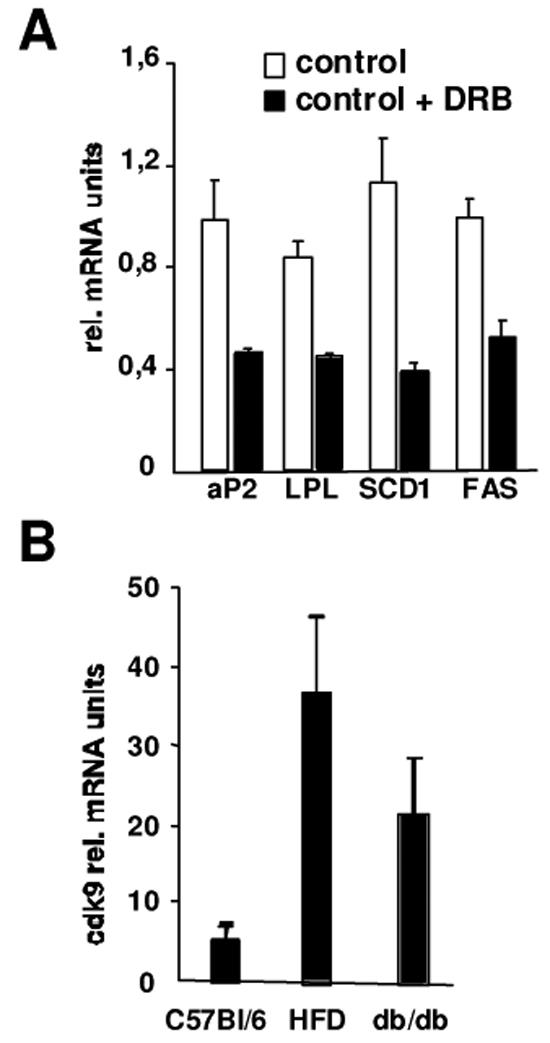
(A) mRNA expression of lipogenic genes in 3T3-L1 adipocytes treated or not with DRB measured by qPCR. DRB was added for 24h in a concentration of 50μM. Results are normalized for the expression of β-actin mRNA
(B) Expression of cdk9 mRNA measured by real-time PCR in the adipose tissue of mice fed a chow diet (C56B1/6), a high fat diet (HFD), or in the adipose tissue of obese db/db mice. Results are normalized for the expression of β-actin mRNA.
Discussion
General transcription is regulated by the activity of RNA pol II. The P-TEFb complex plays a crucial role in the control of transcriptional elongation through phosphorylation of CTD of RNA pol II. Increasing evidence suggest that P-TEFb also participates in the transcriptional regulation of specific genes through recruitment to specific promoters by particular transcription factors. Such is the case for c-Myc (20), NFκB (8), AR (9), and MyoD (5). As a result of the regulation of specific genes, P-TEFb coordinates cellular processes ranging from apoptosis, monocyte differentiation, or myogenesis (reviewed in (21)). In this study, we analysed the participation of cdk9 in adipocyte differentiation and we found that cdk9 was required for adipogenesis. This is supported by several observations. First, expression of cdk9p55 is highly upregulated during adipocyte differentiation (Fig. 1). Second, cdk9 activity is increased during adipogenesis (Fig. 1). Third, overexpression of cdk9 results in increased adipogenesis, whereas inhibition of cdk9 impairs the adipogenic potential of the cells (Fig. 2–3). Finally, cdk9 increases PPARγ activity through direct interaction with PPARγ on the promoters of PPARγ target genes (Fig. 4). The central role of PPARγ in adipose tissue differentiation and biology has been largely demonstrated. Consequently, any factor modulating PPARγ activity, and such is the case for cdk9, will have a major impact on adipocyte differentiation.
A similar scenario has been observed for the transcription factor MyoD. In this case, cdk9 phosphorylates MyoD and increases its transcriptional activity (5). Similarly, PPARγ is also phosphorylated by the P-TEFb complex, which may account for the observed effects of cdk9 on PPARγ activity. Interestingly, PPARγ is phosphorylated at S112, exactly the same residue previously reported to be phosphorylated by MAPK and cdk7 (22–24). The effects of PPARγ phosphorylation on PPARγ activity are so far inconsistent. MAPK phosphorylation of PPARγ has been reported to inhibit its activity (22, 23), whereas phosphorylation by cdk7 has been shown to be required for PPARγ-mediated transactivation (24). Our results are in agreement with this last study, and show that PPARγ phosphorylation by cdk9 results in PPARγ activation. Three different observations support this hypothesis. First, cdk9 mediates PPARγ activation in transient transfection assays (Fig. 4). Second, PPARγ phosphorylation correlates with PPARγ maximal activity in transactivation of target genes (Fig. 6). Finally, phosphorylated PPARγ is complexed to the active promoters of target genes together with cdk9 (Fig. 6). How can phosphorylation of PPARγ at the same residue by different kinases result in either activation or repression of this transcription factor? One possible explanation is cofactor recruitment. The interaction of PPARγ with either kinase could modulate the interaction of PPARγ with different cofactors. In the case of MAPK, the interaction could result in the recruitment of corepressors, whereas for cdk7 and cdk9, the interaction could result in the recruitment of coactivators and the general transcriptional machinery. Coactivators recruitment is likely not the case, since we did not observe any difference in cofactors recruitment to PPARγ in the absence or presence of cdk9 (data not shown). Alternatively, phosphorylation of PPARγ might be required for optimal activation. The effects observed upon MAPK inactivation on PPARγ could be indirect, such as replacement of PPARγ by a more potent transcription factor. Unfortunately no data is available on the presence of PPARγ on the promoters of its target genes when MAPK inhibitors are used. Since phosphorylation of PPARγ coincides with RNA pol II CTD phosphorylation, we believe that PPARγ could tether the P-TEFb complex to the RNA pol II complex, facilitating phosphorylation and therefore elongation of the transcription of its target gene. This is the mechanism that has been suggested to explain the positive effects on the transcription of AR (9), AhR (10), or NFκB (8) mediated by the interaction of cdk9 with these transcription factors. In contrast to this, another mechanism of regulation of pTEFb activity was recently described for the glucocorticoid receptor (GR). In this scenario GR inhibits pTEFb activity by competing for binding to RelA and p50 complex on the promoter of the IL-8 gene, resulting in the repression of this gene (25).
The analysis of the expression of cdk9 during adipocyte differentiation showed that the recently identified cdk9 isoform, p55, which is transcribed from an alternative promoter (2) increases during differentiation. This pattern of expression of p55cdk9 is suggestive of being a PPARγ target gene. Interestingly, we have identified a highly conserved PPARγ responsive element in the promoter of p55cdk9 (data not shown). Regulation of cdk9 expression by PPARγ would result in a positive feed-back loop increasing the availability of cdk9 to activate PPARγ-mediated transcription.
During adipocyte differentiation, which is a highly coordinated process, only a subset of genes are activated resulting in the specific adipocyte phenotype (reviewed in (15)). This selective transcriptional activation supports the idea that the effects of cdk9 are rather gene specific than an effect on general transcription in the context of adipogenesis, and likely in other differentiation processes. Most interesting is the fact that cdk9 could participate in adipose tissue biology in addition to adipocyte differentiation. In differentiated adipocytes cdk9 inactivation results in the inhibition of the expression of genes implicated in lipogenesis, which is consistent with the increased expression of cdk9 that we found in mouse models of obesity. This may have important therapeutical implications, for instance in the context of obesity. Studies on the effects of specific cdk9 inhibitors in adiposity in mice are currently ongoing. In summary, in this study we provide evidence that the nuclear receptor PPARγ uses the P-TEFb complex to stimulate the transcription of its target genes to drive adipogenesis.
Materials and methods
Materials
All chemicals, except if stated otherwise, were purchased from Sigma (St-Louis, MO). Pioglitazone was a kind gift of Takeda Pharmaceuticals (Osaka, Japan) and rosiglitazone was kindly provided by Novo Nordisk A/S (Bagsvaerd, Denmark). Anti-cdk9 C-20 antibody, anti-cyclin T2 S-14 antibody, anti-PPARγ E-8 antibody, anti-PPARγ H-100 antibody, and anti-histone HI FL-219 antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-cdk9 ab6544 antibody, anti-cyclin T1 ab2098 antibody, and the acetylated histone H4 (Lys 9) antibody were from Abeam Ltd (Cambridge, UK). Anti-phosphoPPARγ (Ser112) antibody was purchased from Euromedex (Mundolsheim, France), The RNA polymerase II carboxy-terminal domain (CTD) purified protein was purchased from ProteinOne (Bethesda, MD).
Plasmids and oligonucleotides
PBSK and pcDNA3 vectors were purchased from Stratagene (La Jolla, CA). GST-PPARγ AB, GST-PPARγ DE, PPRE-TK-Luc, and the PPARγ expression vector were previously described (26, 27). GST-PPARγ deletion mutants were generated by PCR and inserted into pGEX-4T-1 vector. A pCMV β-galactosidase vector was used as an internal control for transfection efficiency in mammalian cells. PCMV Sp6-cdk9, pCMV Sp6.1-cyclinT2 and pCMV Sp6-cyclin T1 expression vectors were purchased from MRC geneservice (Cambridge, UK), a distributor for the Mammalian Gene Collection. The pCMV-dominant negative cdk9 HA-tagged (DNcdk9) vector was a kind gift from Monsef Benkirane. Dominant negative cdk9 is containing a point mutation at nucleotide 563, which converts Asp to Asn. Following plasmids were kindly provided from Karsten Kristiansen: upstream activating sequence UAS-TK-luc reporter, pSG5-cdk9, pRc/CMV-cyclin T1, pcDNA3.1-PPARγ2, Gal4-PPARγ A/B, Gal4-PPARγ LBD, pGEX-5X-2 GST-PPARγ2 full-length, GST-PPARγ2 LBD, GST-PPARγ2 A/B, and GST-PPARγ2 A/B S112A. Oligonucleotides used in ChIP assays to amplify the mouse aP2 promoter in PPRE region are 5′-GAGCCATGCGGATTCTTG- 3′ and 5′-CCAGGAGCGGCTTGATT GTTA- 3′; for nonPPRE region of the aP2 promoter: 5′ CAGCCCCACATCCCCACAGC and 3′ GGATGCCCAACAACAGCCACAC; and for mouse GAPDH promoter amplification: 5′ AAGGCTGGTGCTGTGGAGAAACTG and 3′ GTCCCCTTGCAACATACATAACTG. Oligonucleotides used for real time PCR experiments are the following in 5′ to 3′ orientation: cdk9p42 forward, GCCAAGATCGGCCAAGGCAC; cdk9p42 reverse, CAGCCCAGCAAGGTCATGCTC; cdk9p55 forward, CCTCTGCAGCTCCG GCTCCC; cdk9p55 reverse, CACTCCAGGCCCCTCCGCGG; TV promoter GTAATACGACTCACTCACTATAGGG; aP2 forward, AACACCGAGATTTCCTTCAA; aP2 reverse, AGTCACGCCTTTCATAACACA; 18s forward, GTTCCGACCATAAACGATGCC; 18s reverse, TGGTGGTGCCCTTCCGTC AAT; LPL forward, GAGACCAAGAGAAGCAGCAAGATGT; LPL reverse, AGTCGGGCCAGCTGAAGTAGGAGT; SCD1 forward, TGGGTTGGCTGCTTGTG; SCD1 reverse, GCGCTGGGCAGGATGAAG; FAS forward, TGCTCCCAGCTGCAGGC; FAS reverse, GCCCGGTAGCTCTGGGTGTA.
RNA isolation, Reverse transcription and Real Time PCR
RNA was purified using an RNeasy Mini Kit of Qiagen (Hilden, Germany). Reverse transcription of total RNA was performed at 37°C using the M-MLV reverse transcriptase (Invitrogen SARL, France) and random hexanucleotides primers (Promega, Madison, WI), followed by a 15 min inactivation at 70°C. Quantitative PCR was carried out by Real Time PCR using a LightCycler and the DNA double strand specific SYBR Green I dye for detection (Roche, Basel, Switzerland). Results were then normalized to 18s levels.
Cell culture, Cell differentiation and Transfections
NIH-3T3, COS, 293 and 3T3-L1 cells were grown in DMEM supplemented with 10% foetal bovine serum (FBS), 100U/ml penicillin and 100 μ/ml streptomycin in a humidified atmosphere of 5% CO2 at 37°C. 3T3-L1 cells were differentiated with DMEM, 10% serum, 0,5mM 3-Isobutyl-1 methylxanthine (IBMX), 10 μg/ml insulin, 1 μM dexamethasone and 100nM pioglitazone for 2 days. From the day 3 on, cells were incubated with DMEM, 10% serum, 10 μg/ml insulin, and 100nM pioglitazone. Oil Red O staining was performed as described (28). Transfections were performed using the Jet PEI reagent (Qbiogene, Irvine, CA). Rosiglitazone was used at the concentration of 1 μM. The luciferase and β-galactosidase activities were measured as described (29). For stable transfections, Jet PEI was mixed with plasmid DNA, according to the manufacturer’s instructions, and the mixtures were left on the cells in the incubator for 4 hr. Twenty-four hr following transfections, G418 or puromycin (Invitrogen SARL, France) was added at a final concentration of 1mg/ml or 2.5 μg/ml respectively. The medium plus G418 or puromycin was replaced 3 times/week until no surviving cells were observed.
Protein expression assays
For nuclear extracts cells were homogenized in a lysis buffer containing 10mM Hepes pH7.9, 10mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% Nonidet P-40 (MP Biomedicals, Aurora, OH), and 1 mM dithiothreitol. A protease inhibitor cocktail was added (Sigma). Lysates were centrifuged and resuspended in a buffer containing 20 mM Hepes pH 7.9, 1 mM EDTA, 1 mM EGTA, 0.4M NaCl, 1 mM dithiothreitol and protease inhibitor cocktail. Protein concentrations were determined by Bradford method (Bio-Rad, Hercules, CA). SDS-PAGE and electrotransfer was performed as described (30). The membranes were blocked 1h in blocking buffer (PBS, 0,5% Tween-20, 5% skimmed milk). Filters were first incubated overnight at 4°C with the indicated primary antibodies, and then for 1h at room temperature with a peroxidase conjugate secondary antibody. The complex was visualized with enhanced chemiluminescence (ECL) (Interchim, France).
Immunofluorescence in 3T3-L1 cells
For all immunofluorescence experiments cells were grown on coverslips. After fixation and permeabilization with 100% methanol, cells were incubated with antibodies directed against cdk9 (Abeam Ltd, UK), cyclin T1 (Abeam Ltd, UK) and PPAR gamma (Santa Cruz Biotechnology, CA). For cyclin T2 detection cells were fixed with 4% formaldehyde, then permeabilized with 0.1% Triton X-100 (Sigma) and incubated with cyclin T2 antibody (Santa Cruz Biotechnology, CA). Preparations were then incubated with a combination of Texas Red -conjugated anti-mouse IgG and FITC -conjugated anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), or FITC-conjugated anti-goat IgG (Santa Cruz Biotechnology, CA).
Coimmunoprecipitation and chromatin immunoprecipitation (ChIP) assays
For co-immunoprecipitation assays whole cell extracts were precleared with protein A-agarose beads (Roche) during 30 minutes at room temperature, an aliquote of the precleared lysates was saved as input. Extracts were then centrifuged (5 minutes at 3000rpm) and supernatants were immunoprecipitated with the indicated specific antibodies overnight at 4°C, rabbit IgGs (Sigma, St-Louis, MO) were used as negative control (mock). Immunoprecipitates were then washed twice with IP buffer (150mM NaCl, 1% NP40, 50mM Tris/HCl pH8 and protease inhibitor cocktail) and three times with washing buffer (0,25M KCl in PBS) and subjected to SDS-PAGE electrophoresis. Chromatin immunoprecipitation assays were performed as described previously (31). Briefly, proteins were formaldehyde cross-linked to DNA in confluent 3T3-L1 preadipocytes before induction of differentiation or in cells induced with differentiation medium for 7 days. Proteins were then immunoprecipitated using the indicated antibodies, rabbit IgGs or beads were used as mock, DNA was extracted from the immunoprecipitates and PCR amplification was performed using promoter-specific oligonucleotide primers.
Pull-down assays
In vitro translation of pCMV-cdk9 was performed with 35S-methionine (Perkin Elmer, Boston, MA) in a TNT-coupled transcription-translation system, as described by the manufacturer (Promega, Madison, WI). GST fusion or GST alone were expressed in Escherichia Coli, and purified on glutathione-sepharose-4B beads (Amersham Biosciences, Uppsala, Sweden). For in vitro binding GST, GST-PPARγ AB, GST-PPARγ DE and GST-PPARγ deletion mutants were incubated with the labeled protein in 1 ml binding buffer containing 300mM NaCl, 0.5% Triton-X-100, 50mM Tris pH8, and 2mM EDTA at room temperature for 1 hr. Beads were washed five times with the same buffer. The proteins were visualized by autoradiography after SDS-PAGE electrophoresis.
Kinase assays
Immunoprecipitated cdk9 from 3T3-L1 cells at different stages of differentiation was used as kinase. Immunoprecipitates were washed once with kinase buffer (25mM Tris/HCl pH7.5, 150mM NaCl, 10mM MgC12, 1mM DTT and protease inhibitor cocktail) in the presence of a protease inhibitor cocktail (Sigma, St-Louis, MO) and phosphatase inhibitors (5mM Na4P2O7, 50mM NaF, 1mM vanadate). Kinase assay was performed during 30 minutes at 37°C in the presence of 40 □M ATP and 8μCiγ33P-ATP; purified RNA polymerase CTD or GST, GST-PPARγ2 A/B, GST-PPARγ A/B S112A, GST-PPARγ LBD or GST-PPARγ2 full-length were used as substrates. Reaction was stopped boiling the samples during 5 minutes in the presence of denaturing sample buffer. Samples were then subjected to SDS-PAGE electrophoresis; gels were then dried in a gel dryer for 1h at 80°C and exposed to an X-ray film.
Supplementary Material
Acknowledgments
Helpful discussions with various members of the Fajas and the Kristiansen labs are greatly appreciated. This work was supported by grants from INSERM, CHU de Montpellier, Association pour la Recherche contre le Cancer, Association de Langue Française pour l’ Etude du Diabète et des Maladies Métaboliques, and Fondation pour la Recherche Médicale (LF) and the Carlsberg Foundation, the Novo Nordisk foundation, and the Danish Natural Science Research Council (KK). I.I. is supported by a Ph. D. fellowship from the Ligue Nationale Contre le Cancer.
References
- 1.Garriga J, Grana X. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene. 2004;337:15–23. doi: 10.1016/j.gene.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Shore SM, Byers SA, Maury W, Price DH. Identification of a novel isoform of Cdk9. Gene. 2003;307:175–82. doi: 10.1016/s0378-1119(03)00466-9. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y, Pe’ery T, Peng J, et al. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11:2622–32. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mancebo HS, Lee G, Flygare J, et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997;11:2633–44. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simone C, Stiegler P, Bagella L, et al. Activation of MyoD-dependent transcription by cdk9/cyclin T2. Oncogene. 2002;21:4137–48. doi: 10.1038/sj.onc.1205493. [DOI] [PubMed] [Google Scholar]
- 6.Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM. c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene. 2003;22:5707–11. doi: 10.1038/sj.onc.1206800. [DOI] [PubMed] [Google Scholar]
- 7.Giraud S, Hurlstone A, Avril S, Coqueret O. Implication of BRG1 and cdk9 in the STAT3-mediated activation of the p21wafl gene. Oncogene. 2004;23:7391–8. doi: 10.1038/sj.onc.1207972. [DOI] [PubMed] [Google Scholar]
- 8.Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8:327–37. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 9.Lee DK, Duan HO, Chang C. Androgen receptor interacts with the positive elongation factor P-TEFb and enhances the efficiency of transcriptional elongation. J Biol Chem. 2001;276:9978–84. doi: 10.1074/jbc.M002285200. [DOI] [PubMed] [Google Scholar]
- 10.Tian Y, Ke S, Chen M, Sheng T. Interactions between the aryl hydrocarbon receptor and P-TEFb. Sequential recruitment of transcription factors and differential phosphorylation of C-terminal domain of RNA polymerase II at cyp1a1 promoter. J Biol Chem. 2003;278:44041–8. doi: 10.1074/jbc.M306443200. [DOI] [PubMed] [Google Scholar]
- 11.Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J Virol. 1998;72:9881–8. doi: 10.1128/jvi.72.12.9881-9888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Luca A, Russo P, Severino A, et al. Pattern of expression of cyclin T1 in human tissues. J Histochem Cytochem. 2001;49:685–92. doi: 10.1177/002215540104900602. [DOI] [PubMed] [Google Scholar]
- 13.De Luca A, Tosolini A, Russo P, et al. Cyclin T2a gene maps on human chromosome 2q21. J Histochem Cytochem. 2001;49:693–8. doi: 10.1177/002215540104900603. [DOI] [PubMed] [Google Scholar]
- 14.Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–389. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 15.Fajas L, Fruchart JC, Auwerx J. Transcriptional control of adipogenesis. Current Opinions in Cell Biology. 1998;10:165–173. doi: 10.1016/s0955-0674(98)80138-5. [DOI] [PubMed] [Google Scholar]
- 16.Fajas L. Adipogenesis: a cross-talk between cell proliferation and cell differentiation. Annals of Medicine. 2003;35:79–85. doi: 10.1080/07853890310009999. [DOI] [PubMed] [Google Scholar]
- 17.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARg2: tissue-specific regulator of an adipocyte enhancer. Genes & Development. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 18.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARg2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 19.Fajas L, Debril MB, Auwerx J. Peroxisome proliferator-activated receptor-gamma: from adipogenesis to carcinogenesis. J Mol Endocrinol. 2001;27:1–9. doi: 10.1677/jme.0.0270001. [DOI] [PubMed] [Google Scholar]
- 20.Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem. 2002;277:40156–62. doi: 10.1074/jbc.M207441200. [DOI] [PubMed] [Google Scholar]
- 21.Napolitano G, Majello B, Lania L. Role of cyclinT/Cdk9 complex in basal and regulated transcription (review) Int J Oncol. 2002;21:171–7. [PubMed] [Google Scholar]
- 22.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP-kinase mediated phosphorylation of PPARg. Science. 1996;274:2100–2103. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 23.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. JBC. 1997;272:5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 24.Compe E, Drane P, Laurent C, et al. Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Mol Cell Biol. 2005;25:6065–76. doi: 10.1128/MCB.25.14.6065-6076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luecke HF, Yamamoto KR. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev. 2005;19:1116–27. doi: 10.1101/gad.1297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelman L, Zhou G, Fajas L, Raspe E, Fruchart JC, Auwerx J. p300 interacts with the N-and C- terminal part of PPARg2 in a ligand-independent and -dependent manner respectively. JBC. 1999;274:7681–7688. doi: 10.1074/jbc.274.12.7681. [DOI] [PubMed] [Google Scholar]
- 27.Fajas L, Egler V, Reiter R, et al. The retinoblastoma-histone deacetylase 3 complex inhibits the peroxisome proliferator-activated receptor gamma and adipocyte differentiation. Developmental Cell. 2002;3:903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- 28.Hansen JB, Petersen RK, Larsen BM, Bartkova J, Alsner J, Kristiansen K. Activation of peroxisome proliferator activated receptor g bypasses the function of the retinoblastoma protein in adipocyte differentiation. JBC. 1999;274:2386–2393. doi: 10.1074/jbc.274.4.2386. [DOI] [PubMed] [Google Scholar]
- 29.Fajas L, Auboeuf D, Raspe E, et al. Organization, promoter analysis and expression of the human PPARg gene. JBC. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- 30.Rocchi S, Picard F, Vamecq J, et al. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Mol Cell. 2001;8:737–47. doi: 10.1016/s1097-2765(01)00353-7. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–16. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.