

Abstract
Shal (Kv4) α-subunits are the most conserved among the family of voltage-gated potassium channels. Previous work has shown that the Shal potassium channel subfamily underlies the predominant fast transient outward current in Drosophila neurons (Tsunoda, S., and Salkoff, L. (1995) J. Neurosci. 15, 1741–1754) and the fast transient outward current in mouse heart muscle (Guo, W., Jung, W. E., Marionneau, C., Aimond, F., Xu, H., Yamada, K. A., Schwarz, T. L., Demolombe, S., and Nerbonne, J. M. (2005) Circ. Res. 97, 1342–1350). We show that Shal channels also play a role as the predominant transient outward current in Caenorhabditis elegans muscle. Green fluorescent protein promoter experiments also revealed SHL-1 expression in a subset of neurons as well as in C. elegans body wall muscle and in male-specific diagonal muscles. The shl-1 (ok1168) null mutant removed all fast transient outward current from muscle cells. SHL-1 currents strongly resembled Shal currents in other species except that they were active in a more depolarized voltage range. We also determined that the remaining delayed-rectifier current in cultured myocytes was carried by the Shaker ortholog SHK-1. In shl-1 (ok1168) mutants there was a significant compensatory increase in the SHK-1 current. Male shl-1 (ok1168) animals exhibited reduced mating efficiency resulting from an apparent difficulty in locating the hermaphrodite vulva. SHL-1 channels are apparently important in fine-tuning complex behaviors, such as mating, that play a crucial role in the survival and propagation of the species.
Voltage-dependent potassium (K+) (Kv)2 channels are key regulators of membrane excitability. Based on their inactivation kinetics, Kv currents can loosely be divided into two categories as follows: noninactivating or slowly inactivating K+ currents, and rapidly inactivating transient currents also known as “A-type” currents. A-type currents were first described in molluscan neurons by Hagiwara et al. (3) and later by Connor and Stevens (4), Neher (5), and Thompson (6). Classical A-type currents (IA) are voltage-dependent, Ca2+-independent K+ currents that undergo rapid activation and inactivation. The classical A-type currents activate at subthreshold voltages and recover from inactivation quickly compared with other Kv currents. They are also characterized by strong steady-state voltage-dependent inactivation. Repolarization to potentials negative to −50 mV is typically required to restore channel availability. A-type currents have a widespread distribution and are abundantly expressed in neurons and cardiac and smooth muscle cells, where they are thought to play many important physiological roles (2, 7-11).
Connor and Stevens (12) proposed that the IA may determine the interspike interval in repetitively firing neurons. Studies in cerebellar granule cells demonstrated a role for IA in determining the latency to first spike (13). IA has also been demonstrated to limit the back propagation of action potentials into the dendritic arbor (14), to impact long term potentiation in CA1 hippocampal neurons (15), to affect neuronal excitability in the visual cortex (16), and to modulate compartmentalization of membrane excitability in distal dendritic spines (17). Fast transient K+ currents in cardiac muscles contribute both to myocyte excitability (18) and to the fast repolarization phase of the cardiac action potential (2, 8, 19, 20). Finally, the pharmacological block of channels that carry fast transient currents in gastrointestinal smooth muscle in mice supports the conclusion that the window currents carried by these channels contribute to the resting membrane potential and cellular excitability of smooth muscle (21, 22).
The molecular identity of the A-type currents in different tissues has been the subject of extensive investigation. Experiments with transgenic animals carrying dominant negative Shal α-subunit constructs indicate that Shal family channels express the cardiac fast transient outward K+ currents in cardiac myocytes (23). However, the molecular composition of IA currents in many neurons remains uncertain. In Drosophila, a synthetic deletion allowed the physiological analysis of embryonic neurons lacking the shal gene, which showed that the vast majority of neurons express Shal currents (1). It is noteworthy that other Kv channels, like the Kv1 family, are capable of forming A-type currents, which add more complexity in trying to establish the specific contribution of a particular type of KV channel in a particular cell type.
Here we show that the Caenorhabditis elegans shal gene (shl-1) encodes a unique transient current in muscle cells. We also show that the fast transient current carried by Shal channels contrasts with Shaker channels (shk-1) from C. elegans, which express currents that are only slowly inactivating. Both Shal and Shaker channels are expressed in body wall muscle cells and in a variety of neurons. Shal currents recorded both from body wall muscle cells in culture and heterologously expressed in Xenopus oocytes have properties similar to Shal channels from other species with regard to their rapid activation and inactivation, but they differ in that they are active in a more positive voltage range. The phenotype of the shl-1 deletion mutation was manifested by an increase in muscle excitability reflected in abnormal aldicarb sensitivity, abnormal thrashing behavior, and mating deficiencies.
MATERIALS AND METHODS
Molecular Biology
EST yk327e11 (Kohara) encodes a full-length shl-1 cDNA and was obtained already cloned into a pBSc II KS+ plasmid. Three shk-1 isoform (a, c, and d2) expression constructs were created using EST yk442g4, which encodes the tetramerization domain and core of the protein, and includes the entire 3′ end of the gene. yk442g4 was inserted into our Oocyte Express pOX vector. The alternative splice isoform expression constructs were created by inserting an EST encoding the variable 5′ end of the gene into the yk442g4∷pOX using BbvC1. The ESTs used were yk813e07, yk753e1, and yk1164g03, and yk1168a01 and yk736e9 for shk-1 isoforms a, c, and d2. All ESTs were obtained from the laboratory of Yuji Kohara and were sequenced. Intron/exon boundaries were determined by comparing verified EST and genomic sequence. The W363F dominant negative SHL-1 (GenBank™ accession mumber NM_068574) construct was created by site-directed mutagenesis of the full-length shl-1 cDNA, changing the phenylalanine at position 363 to a tryptophan.
Xenopus Oocytes and cRNA Injection
Female Xenopus laevis were obtained from Nasco (Fort Atkinson, WI). Oocytes were collected (according to the guidelines of the Institutional Animal Care and Use Committee of Thomas Jefferson University) under anesthesia (immersion in 0.2% 3-aminobenzoic acid ethyl ester (Sigma) for about 30 min) from frogs that were humanely killed after the final oocyte collection. Before injection, oocytes were defolliculated by digestion with collagenase (2 mg ml−1; Sigma) in calcium-free external solution (see below). The shl-1 cRNA was injected (total RNA injected ~2–100 ng per oocyte) into defolliculated oocytes using a Nanoject microinjector (Drummond, Broomall, PA). Currents were recorded 1–5 days post-injection.
Electrophysiology; Two-electrode Voltage Clamp
Macroscopic whole-oocyte currents were recorded in ND96 plus 1 mm DIDS using the two-electrode voltage clamp technique as in Yuan et al. (24). Conductance-voltage relationship data were obtained using a standard voltage-step protocol stepping from a holding potential of −100 mV to voltages from −80 to +50 mV in steps of 10 mV for a period of either 450 ms (for shl-1 currents, n =7) or 5 s (for shk-1 currents, each isoform had n = 5–6). Steady-state inactivation data for SHL-1 currents (n = 7) were obtained using a protocol stepping from a holding potential of −100 mV to a range from −120 to +10 mV in 10-mV steps for 9 s with a depolarizing step to +40 mV for 250 ms. Steady-state inactivation data for SHK-1 currents (n = 8) was obtained using a protocol stepping from a holding potential at −100 mV to a range from −120 to +10 mV in 10-mV steps for 38 s with a depolarizing step to +40 mV for 1 s. The dominant negative SHL-1 construct cRNA was examined in a 1:1 ratio co-injection with the SHL-1 cRNA using the same protocols described for SHL-1 cRNA oocyte expression. All protocols utilized a 5-s intersweep interval at −100 mV to allow for inactivated channel recovery. Current records were filtered at 1 kHz, acquired digitally using Clampex 9.0 (Axon Laboratories), and analyzed using Clampfit 9.2 (Axon Laboratories). G-V curves were obtained by converting the peak current values from the I-V relationships to conductances by using the equation: G = I (V − EK), where G is the conductance; I is the peak current; V is the command pulse potential; and EK is the K+ reversal potential. Conductance values were normalized and fitted with a Boltzmann equation: G/Gmax = {1 + exp(−(V − V0.5a)/ka)}−1, where G is the peak conductance; Gmax is the maximal peak conductance; V and V0.5a are the command potential and the midpoint of activation, respectively; and ka is the activation slope factor. Steady-state inactivation analysis was performed using the normalized current during the test pulse plotted as a function of the prepulse potential. The data were fitted with the Boltzmann equation: I/Imax = {1 + exp(V − V0.5i/ki)}−1, where I is the peak current; Imax is the peak current when the prepulse potential was −80 mV; V and V0.5i are the prepulse potential and half-inactivation potential, respectively; and ki is the inactivation slope factor.
Electrophysiology; Patch Recordings from Xenopus Oocytes
Macropatch and unitary recordings were conducted as described previously (25) using an Axopatch 200A or 200B amplifier (Axon Instruments, Foster City, CA). Patch pipettes were constructed from Corning Glass 7052 or 7056 (Warner Instrument Corp., Hamden, CT). For the recording of fast currents (e.g. tail current relaxations) and single channel currents, the pipettes (~0.5–1 and 5–10 megohms in the bath solution, respectively) were coated with Sylgard elastomer (Dow Corning Co., Midland, MI). Passive leak current and the capacitive transients were subtracted online using a P/4 procedure. The recordings were filtered at 0.5– 8 kHz (−3 dB, 8-pole Bessel filter; Frequency Devices, Haverhill, MA) and digitized at 2–40 kHz. All experiments were recorded at room temperature (23 ± 1 °C).
Data analysis was conducted using Clampfit 8–9 (Axon Instruments), SigmaPlot 8–9 (Systat Software Inc., Point Richmond, CA), and Origin 7.0 (OriginLab Inc., Northhampton, MA). The voltage dependence of the peak chord conductance (Gp-V relationship), steady-state inactivation, and time-dependent current relaxations were analyzed as described elsewhere (25, 26). The unitary conductance was estimated by applying voltage ramp protocols to evoke the single channel currents (−100 to +100 mV; 0.9 ms/mV). In this case, passive leak current and the capacitive transients were subtracted by using blank sweeps (no unitary currents). Results throughout are expressed as means ± S.E.
Constructs and Transgenic Animals
The expression construct for shl-1 was created using two overlapping PCR products encompassing the entire 10-kb upstream region of the gene. We designed an ~500-bp overlap between the PCR pieces (5′-PCR was 6.4 kb, and 3′-PCR was 7.3 kb), as well as similar overlap with the “core” protein coding genomic DNA fragment (genomic DNA positions 22316–24990 in YAC Y73B6BL) fused in-frame to GFP in the pPD95 GFP expression plasmid. The construct was originally created in a pPD95 plasmid containing a nuclear localization sequence (NLS), and several lines were also created where the NLS had been removed from the same construct. The expression construct for shk-1 was created in part using one long 11-kb PCR fragment encompassing all of the upstream noncoding sequence except for ~800 bp until the next open reading frame. The remainder of the shk-1∷GFP construct was the complete genomic shk-1 gene open reading frame fused in-frame to GFP in a pPD95 GFP expression plasmid. No NLS was present in the construct.
Animals were injected as described in Salkoff et al. (27). We utilized ~90 ng/μl of the XhoI-linearized plasmid DNA of the shl-1 or 100 ng/μl of the SphI-linearized shk-1 transgene core constructs. For the shl-1 expression construct, 75 ng/μl of the Shal 5′-PCR and 91 ng/μl of the Shal 3′-PCR were added. For the shk-1 expression construct, 100 ng/μl of the Shaker PCR was added to the linearized plasmid and the marker plasmid. At least three lines each were examined for shl-1 and shk-1 expression analysis.
Construction of the Dominant Negative Transgenic Lines
The pshl-1∷W363F∷GFP dominant negative transgenic construct was created by using site-directed mutagenesis to alter the tryptophan at position 363 into a phenylalanine in the expression construct pshl-1∷shl-1∷GFP. The dominant negative transgenic plasmid pshl-1∷W363F∷GFP (40 ng/μl) was then combined with the two PCR products encompassing the shl-1 promoter (40 ng/μl each) and the lin-15+ phenotypic marker plasmid (150 ng/μl) and injected into adult hermaphrodite N2 animals. Each of two lines was integrated by exposing ~20 P0 L4 animals of each line to γ-ray irradiation. Two integrated lines, designated 320 and 625, were obtained where >50 L2 animals exhibited 100% GFP expression and non-Muv. Integration was verified by crossing the potential integrated line to the TY1657 marker strain (dpy-5; rol-6; lon-1; bli-6; unc-23). Line 320 integrated onto chromosome X and line 625 integrated onto chromosome IV.
A nonintegrated pmyo-3∷W363F∷GFP transgenic line was also constructed. We subcloned GFP into the pmyo-3 containing plasmid pPD95.52. We then subcloned the W363F dominant negative shl-1 cDNA into the pPD95.52 plasmid, creating a pmyo-3∷W363F plasmid, co-injected into N2 animals with pRF4 (rol-6).
RNAi
Double-stranded RNA (dsRNA) was synthesized by using standard methods described by Fire et al. (28) and Christensen et al. (29). The size and integrity of dsRNA were assayed on Tris boric acid/EDTA-agarose gels. Cells were plated in L-15 control medium or L-15 medium containing 15 μg/ml dsRNA final volume. One hour after plating, the dsRNA was diluted to a final concentration of 5 μg/ml. Media containing the dsRNA were replaced each day. Electrophysiological experiments were performed 2–4 days after plating the cells.
C. elegans Cell Culture
Embryonic cells were isolated and cultured as described (29) with the following modifications. Nematode eggs were not separated from adult carcasses in a sucrose gradient. Cellular debris and carcasses were removed upon filtration. Muscle cells were identified based on their distinctive morphology in cell culture. An integrated myo-3∷GFP-transformed strain, which labeled the body wall muscle cells with GFP, verified the method of identification (29). Recordings were performed 4–6 days after plating.
C. elegans Myocyte Electrophysiology
Whole-cell recordings were obtained by using the patch clamp technique (30). Whole-cell currents in external solution contained the following (in mm): 140 NaCl, 5 KCl, 5 CaCl2, 5, MgCl2, 11 dextrose, 5 Hepes, pH 7.2, with NaOH. The 10 nm Ca2+ and 4 mm Cl− internal solution contained 120 K+ gluconate, 20 KOH, 2 MgCl2, 4 magnesium gluconate, 5 Tris, 0.25 CaCl2, 36 sucrose, 5 EGTA, and 4 Na2ATP. The program EGTA (Ed McClesky, Oregon Health Sciences University, Vollum Institute, Portland) was used to calculate free Ca2+. Whole-cell current traces were obtained by applying voltage steps from −70 to +60 mV in 10-mV increments from a holding potential of −70 mV. Prepulse inactivation curves for shk-1 and shl-1 were obtained by eliciting K+ currents with a test potential to +50 mV applied after a prepulse from −70 to +25 mV, in 5-mV steps. The normalized current during the test pulse was plotted as a function of the prepulse potential. Analysis was performed as above.
Strains
We utilized the slo-2 (nf100) strain isolated in the Salkoff laboratory and the DA2056 shl-1 (ok1168) strain provided by the International C. elegans Gene Knockout Consortium at the Oklahoma Medical Research Foundation and the laboratory of Dr. Leon Avery. The following alleles were utilized in the studies as control strains: unc-51 (e369), unc-10 (md1117), ttx-3 (k55), tax-6 (p675), unc-64 (e246), and unc-64 (OxIs34).
Characterization of the shl-1 (ok1168) Breakpoints
The RB1144 shl-1 (ok1168) mutant was obtained from the C. elegans Knockout Consortium and the laboratory of Dr. Robert Barstead. The Knockout Consortium utilized a PCR-based screen to identify this mutant, with the region screened encompassing ~3 kb. We utilized a series of primers just outside of this region and within the region (primer sequences available upon request) to sequence into the screened 3-kb region to identify the deletion breakpoints. The 10× outcrossed DA2056 shl-1 (ok1168) strain was obtained from Dr. Leon Avery.
Aldicarb Assay
25 age-matched hermaphrodite worms from several strains (N2, unc-64 (OxIs34), integrated pshl-1∷W363F∷GFP line 320, shl-1 (ok1168), integrated pshl-1∷W363F∷GFP line 625, and pmyo-3∷W363F∷GFP line 22) were placed onto NGM-0.25 mm aldicarb plates with a small (1 cm diameter) spot of OP50 bacteria (31, 32). Animals were examined for paralysis every 30 min for 5 h, either by tap response or by lightly prodding the worm at the tail with a platinum wire pick. We assayed three plates per strain in each assay. All assays were performed blind at least three times.
Thrashing Assay
Age-matched hermaphrodite worms from N2, shl-1 (ok1168), or unc-10 (md1117) grown at 15 °C were examined individually in a glass staining dish in 60 μl of M9 saline (33) for~1 min. Assays were recorded using a Nikon SMZ 1500 dissecting scope and a PixelLink A6XX camera, magnification approximately ×20. The Streampix program was used to capture all video data. Body bends, defined as a change in the direction of bending in the middle of the body (33), were counted and data analyzed using Sigmaplot (Jandel, San Rafael, CA). At least 20 animals per strain per assay for two blind assays were examined.
Mating Efficiency and Method
Mating efficiency was scored as described by Hodgkin (34) with the following modifications. Two N2 or DA2056 shl-1 (ok1168) males and two N2 hermaphrodites were placed on a 3.5-cm NGM plate seeded with a small amount (20 μl of actively growing culture) of OP50. Efficiency was scored by counting the ratio of male progeny to the total progeny, with a successful mating plate possessing at least 10% male progeny. 42 mating plates per strain were analyzed.
Turning behavior and vulva location were analyzed using the procedure defined in Loer and Kenyon (35) and Crowder et al. (36). 10 males of either N2 or DA2056 shl-1 (ok1168) were mated to either N2 hermaphrodites or unc-51 (e369) hermaphrodites and were analyzed for turning behavior, followed by location of the vulva. 9 of 10 N2 males were capable of exhibiting good turns (g) followed immediately by a slow search for the vulva using spicules (s). The other N2 male passed the vulva by more than 10% of the length of the hermaphrodite and proceeded to quickly swim back to search again for the vulva, or alternatively, they would turn around the hermaphrodite again for a new approach. None of the DA2056 shl-1 (ok1168) males exhibited any change in turning behavior compared with N2 males. DA2056 shl-1 (ok1168) males demonstrated variability in their ability to locate the vulva. These animals frequently exhibited a process of vulval location exhibited by the following pattern: g/f/g/f/g/p/g/o, where “g” is a good turn; “f” is a fast pass across the vulva without slowing; “p” is a slow pass over the vulva and a temporary pause but relatively quick (<10 s) continuation around the hermaphrodite again; and “o” indicates falling off the hermaphrodite without mating. Animals that mated exhibited a pattern similar to g/f/g/f/g/s/si, where “s” is a slowed approach to the vulva; “si” indicates spicule insertion. We examined 60 males per strain of both N2 and DA2056 shl-1 (ok1168).
RESULTS
Tissue Expression Patterns
Shal channels are the most highly conserved of all α-subunits of voltage-gated potassium channels. From the initiator methionine to amino acid residue 430, which encompasses all membrane spanning domains, Shal α-subunits have 75% identity with Drosophila and 69% identity with human orthologs (37). To examine native Shal (SHL-1) and Shaker (SHK-1) currents, we first determined the tissue expression patterns of these genes. We constructed a full-length cDNA translational fusion of shl-1 with ~5 kb of the upstream promoter region tagged to GFP, pshl-1∷shl-1∷GFP. Two versions of this expression plasmid were constructed, one with and one without a nuclear localization signal. Expression of SHL-1 was observed in posterior intestine, body wall muscle, vulval muscle, male-specific diagonal muscles, and a variety of motor neurons, interneurons, and sensory neurons (Fig. 1, A and C; Table 1). Several neurons and muscle cells expressing SHL-1∷GFP are known to be associated with a variety of behaviors, including thermosensation, chemosensation, dauer formation, egg-laying, male mating behavior, and locomotion. Expression in posterior intestine suggests a potential role for shl-1 in defecation. We also examined tissue expression for the Shaker gene (shk-1) using a translational GFP transgene (see “Materials and Methods”). The SHK-1∷GFP fusion protein was expressed in a variety of interneurons and sensory neurons, as well as body wall muscle (Fig. 1B).
FIGURE 1. Cell-type expression patterns of shl-1 and shk-1.
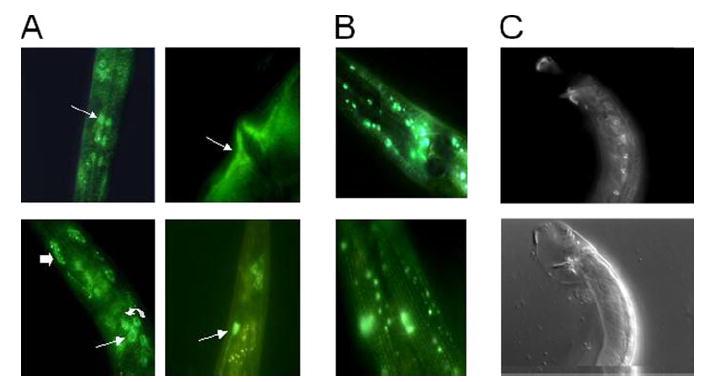
A, GFP expression of the transgene pshl-1∷shl-1∷GFP in N2 animals. Expression was observed in a variety of neurons, including pharyngeal neurons (top left, white arrow), interneurons (bottom left, thin arrow), sensory neurons (bottom left, curved arrow), and phasmid neurons (bottom right, white arrow). In addition, we observed expression in two muscle types, body wall muscle (bottom left, thick arrow) and vulval muscle (top right, white arrow). B, GFP expression of the transgene pshk-1∷shk-1∷GFP in N2 animals. Expression was observed in a variety of neurons in the ganglia of the head (top). Expression throughout body wall muscle cells was visible (bottom) in a striated pattern. C, top, GFP expression of the transgene pshl-1∷shl-1∷GFP in male N2 animals. Expression was observed in the eight male-specific diagonal muscle cells. C, bottom, Nomarski picture of GFP view above.
TABLE 1.
Cell identification for SHL-1 expression pattern
| Non-neuronal cells | |
| Muscle (body wall) | |
| Muscle (vulval) | |
| Intestine | |
| Diagonal muscles (male) | |
|
| |
| Neurons | |
| Sensory neurons | AWC, ASE, ASJ, ADL, ASK, ADF, AFD strongly, ASI, URX, AWB weaker or more variable, PHA, PHB, ADA, ADE |
| Interneurons | RIC, RIM, probably AVB perhaps AIA, AIZ, RIG, PVQ, PVC, PVT |
| Motor neurons | SMD, SMB, RMD L/R but never RMD DL/VL/DR/DL |
| Pharynx | NSM, M3, MC, M4, I2, probably I3, MI, I1 |
Electrophysiological Properties of SHL-1 Expressed in Xenopus Ooctyes
Prior experiments in mammals, Drosophila, lobster, and jellyfish showed that Shal potassium channels carry a fast, transient outward current (37). Our results show that, in general, the properties of Shal channels in C. elegans are similar to those in other systems(supplemental Fig. 1 and Fig. 2, A and C). However, the current-voltage and steady-state inactivation relationships for the C. elegans Shal currents were more depolarized than heterologously expressed Shal currents from other species (38-41). Although prepulse inactivation kinetics of most Shal-Kv4 channels are well described as an exponential time course, some results have hinted at the presence of biexponential prepulse inactivation kinetics (25). In C. elegans SHL-1 prepulse inactivation was clearly biexponential with both time constants decreasing with membrane depolarization (supplemental Fig. 1). SHL-1 channels also exhibit fast deactivation with modest voltage dependence (supplemental Fig. 2).
FIGURE 2. Comparison of conductance voltage and steady-state inactivation properties of SHK-1 and SHL-1 in C. elegans slo-2 (nf100) myocytes or heterologously expressed in Xenopus oocytes.
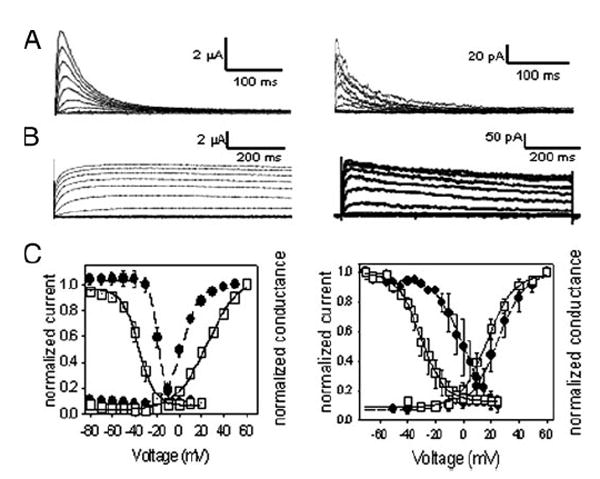
A, left, currents of SHL-1 heterologously expressed in Xenopus oocytes were obtained under voltage clamp at a holding potential of −100 mV with 10-mV steps from −80 to +50 mV for 450 ms with 5-s interpulse intervals. A, right, whole-cell currents from shk-1 RNAi treated slo-2(nf100) myocytes were obtained under voltage clamp at a holding potential of −70 mV with 10-mV steps from −70 to +60 mV with 5-s interpulse intervals. B, left, currents from SHK-1 isoform a heterologously expressed in Xenopus oocytes obtained under voltage clamp at a holding potential of −100 mV with 10-mV steps from −80 to +60 mV. B, right, currents from shl-1(ok1168) myocytes were obtained under voltage clamp at a holding potential of −70 mV with 10-mV steps from −70 mV to +60 mV with 5-s interpulse intervals. C, left, steady-state inactivation and activation data plotted for SHL-1 and SHK-1 heterologously expressed in Xenopus oocytes. □, SHL-1; ■, SHK-1. Best fits for Boltzmann equation are shown as lines through data points. V0.5a = 25.0±1.6 mV (n = 7), ka = 14. 0±1.2 mV;V0.5i = −36.7 ± 0.7 mV (n = 7), ki = 7. 3 ± 0.6 mV. V0.5a = 2. 4 ± 0.8 mV (n = 8), ka = 7. 4 ± 0.7 mV; V0.5i = −19. 5 ± 2.8 mV (n = 8), ki = 3. 7 ± 0.3 mV. Calculated conductance used the equation G = I/(E − EK), where G is the conductance; I is the measured current; E is the test voltage, and EK is estimated at −96 mV. Steady-state inactivation data were obtained using a voltage step protocol stepping from −120 to +20 mV in 10-mV steps, with a holding potential of −100 mV. Normalization was obtained by the following ratio: I/Imax, where Imax is the maximum observed current, and I is the current at any given voltage. C, right, steady-state inactivation and activation data were plotted for SHL-1 and SHK-1 currents observed in cultured C. elegans myocytes. The activation parameter values were V0. 5a = 11.2 ± 1.5 mV, ka = 14.1 ± 1.04 (n = 5); V0.5a = 20. 4 ± 2, ka = 7. 7 ± 1.1 for SHL-1 (□)(n = 3) and SHK-1 (■) (n = 7) currents, respectively. SHL-1 currents inactivated with values of V0.5i = −33.1 mV ± 1.2 and ki = 8.3 ± 0.7 (n = 2), whereas SHK-1 inactivation parameter values were V0. 5i = −6.95±1.7, ki = 5.8±0.5 (n =3). Internal concentrations of Cl− and Ca2+ were 120 mm and 10 nm, respectively. Previously published in Santi et al. (7).
In contrast to shl-1 which appears to produce only one protein product, we cloned and expressed three splice variants of the shk-1 locus termed a, c, and d2 (GenBank™ accession numbers, respectively, CAA88477.2, CAD57716.1, and CAD57717.1). Of the three, only isoforms a and d2 expressed currents when cRNA from those forms was injected into Xenopus oocytes. Ion currents and basic channel properties for SHK-1 isoform a are shown in Fig. 2, B, left, and C, left. The SHK-1 isoform a expresses currents in a more depolarized voltage range than isoform d2. The V0.5a for isoform a is 2.4 ± 0.8 mV with a ka of 7.4 ± 0.7 mV, and the V0.5i is −19.5 ± 2.8 mV with a ki = 3.7 ± 0.3 mV. For isoform d2 the V0.5a is −31.9 ± 1.2 mV; kaa is 7.4 ± 1.1 mV; V0.5i is −62.5 ± 5.9 mV, and ki is 3.4 ± 0.5 mV. Currents of both splice variants inactivate very slowly and can be characterized as delayed rectifiers (Fig. 2B, left).
Electrophysiology and Molecular Dissection of Native SHL-1 and SHK-1 in Cultured Myocytes
Because GFP promoter experiments indicated that SHL-1 and SHK-1 were expressed in body wall muscle cells, we recorded from these cells in culture and analyzed the voltage-dependent current components. Unlike adult body wall muscle cells, Ca2+-dependent inward currents have not been seen in C. elegans myocytes in culture (29). Cultured myocytes are easy to identify because of their characteristic teardrop shape and also because they are significantly larger (three times) than cultured neurons. In addition they can be unequivocally identified by expressing GFP under control of the body wall muscle-specific myosin heavy chain promoter pmyo-3 (42). We sought to examine the native properties of SHL-1 and SHK-1 by isolating each current with three different approaches.
By using cultured myocytes treated with RNAi, we were able to selectively suppress either shl-1 or shk-1 mRNA translation (28) and to record the currents remaining after such treatment (7). Previously, it was shown that SLO-2 K+ channels are abundantly expressed in cultured myocytes but that they required intracellular Ca2+ and Cl− at relatively high levels to be active (43). We thus took advantage of these special requirements of the SLO-2 channels to eliminate the large SLO-2 current component, and we performed all recordings with pipette recording solutions containing low physiological concentrations of Cl− and Ca2+ (see “Materials and Methods”). Patch clamp recordings from wild type (N2) myocytes and from slo-2 (nf100) myocytes demonstrated that two voltage-dependent current components were present in both genotypes, a fast transient component and a slowly inactivating component (7). Those experiments showed that ion currents in N2 cells with low intracellular Cl− and Ca2+ are indistinguishable from slo-2 (nf100) myocytes. The properties we observed of SHL-1 and SHK-1 currents expressed in Xenopus oocytes (Fig. 2, A-C, left) combined with the GFP tissue expression data showing that both shl-1 and shk-1 genes are expressed in body wall muscle (Fig. 1) led us to hypothesize that the macroscopic voltage-dependent outward K+ currents expressed in C. elegans cultured myocytes were SHL-1 and SHK-1 currents. To investigate this hypothesis, we treated the slo-2 (nf100) myocytes with shk-1 RNAi. This resulted in the removal of the slowly inactivating current component, leaving a fast transient current (Fig. 2A, right). Notably, the native fast transient current strongly resembles the SHL-1 current observed when shl-1 cRNA is heterologously expressed in Xenopus oocytes. Conversely, treatment of myocytes with shl-1 RNAi removed the fast transient current component leaving only a slowly inactivating current (7). This slowly inactivating current is virtually identical to that seen in the shl-1 (ok1169) deletion mutant (Fig. 2B, right). This slowly inactivating current resembled the heterologously expressed SHK-1 current. Our data support the conclusion that there are only two normally expressed macroscopic voltage-dependent K+ currents in C. elegans myocytes in culture and that SHL-1 and SHK-1 channels carry these currents.
We characterized the conductance/voltage and steady-state inactivation relationships for the SHL-1 and SHK-1 currents recorded in vivo from myocytes (Fig. 2C, right). Typical of Shal currents in other systems, the V0.5 of activation for the fast transient SHL-1 current was more hyperpolarized than that observed for the slowly inactivating SHK-1 current (11.2 and 20.4 mV, respectively). The SHL-1 current was also less voltage-sensitive, with a slope factor k of 14.1 mV versus 7.7 mV for SHK-1. As expected, the V0.5 of steady-state inactivation was also more hyperpolarized for SHL-1 currents than for SHK-1 currents (−33 and −6.9 mV, respectively). Therefore, the native SHL-1 current is significantly more hyperpolarized in both activation and steady-state inactivation than the SHK-1 current, which is in agreement with the literature for the relative voltage ranges of currents for these channels (37, 44). The conductance/voltage and steady-state inactivation relationships for the SHL-1 and SHK-1 currents recorded in vivo from myocytes (Fig. 2C, right) were similar to those of the two currents expressed heterologously in Xenopus oocytes (Fig. 2C, left) (7).
A second way of separating and analyzing the currents present in myocytes in cell culture was by creating a transgenic animal carrying a dominant negative form of shl-1, which removed or greatly diminished the SHL-1 current. We constructed two transgenes translationally tagged with GFP fused to the shl-1 dominant negative construct that had a site-directed alteration in the pore of the channel (W363F, see “Materials and Methods”). One transgene was under control of the native shl-1 promoter (pshl-1), and the other was under control of the body wall muscle-specific promoter pmyo-3. The pshl-1∷W363F∷GFP transgenic line was then integrated to obtain a stable expression line. The effect of each of these transgenes on the native SHL-1 current was analyzed using the patch clamp technique in cultured myocytes. Cells carrying the dominant negative transgene were identified by their GFP fluorescence. In these cells we observed that the current consisted primarily of slowly inactivating currents. We compared the ratio of the normalized peak current to the sustained current to gauge the effectiveness of removing the SHL-1 component (Fig. 3). This analysis showed a significant reduction in the peak transient current in the animals carrying the dominant negative construct and also in RNAi-treated animals. In these cells a small amount of transient current remained, which may represent a residual component of SHL-1 current as well as a larger component of the slower inactivating SHK-1 current. In 1 of 9 and 1 of 7 cells, for pshl-1∷W363F∷GFP and pmyo-3∷W363F∷GFP, respectively, we observed a more hyperpolarized slowly inactivating current, which may represent expression of variable SHK-1 isoforms (data not shown). Control slo-2 (nf100) cells not carrying the transgene clearly have both the slowly inactivating SHK-1 and fast transient SHL-1 currents. However, cells carrying either of the dominant negative transgenes showed a significant reduction in the amount of the fast transient outward current (Fig. 3).
FIGURE 3. Removal of SHL-1 current in cultured C. elegans myocytes.
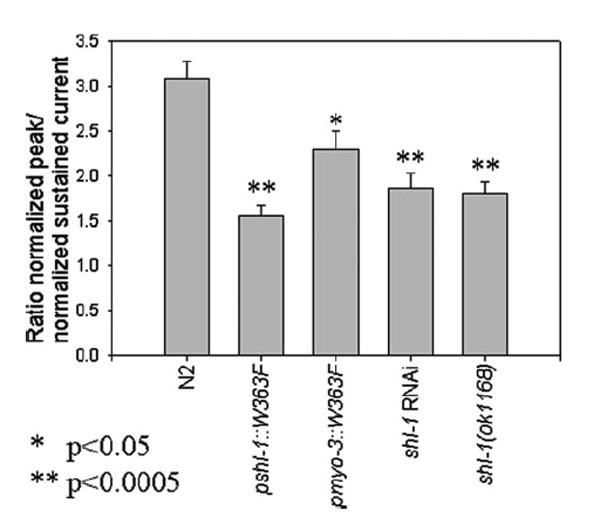
The ratio of the peak current (pA/pF) to the minimum sustained current (pA/pF) is plotted for the wild type N2 strain (3.0831 ± 0.1973) (n = 7), the dominant negative strains pshl-1∷W363F (1. 5623±0. 1101) (n=5) and pmyo-3∷W363F (2.2910±0.0020) (n=5), shl-1RNAi-treated myocytes (1.5723±0.1502) (n=3), and the deletion mutant shl-1(ok1168) (1.8088±0.1324) (n=6). * signifies p < 0.05. ** signifies p < 0.0005.
A deletion mutant for the Y73B6BL.19 shl-1 locus (International C. elegans Knockout Consortium) was the third method by which we characterized native physiological K+ currents in cultured myocytes. DNA sequence analysis showed that both the 5′ and 3′ breakpoints were within intronic sequence. The deletion removed two exons containing the terminal three amino acids of the fourth transmembrane domain and the entire fifth transmembrane domain (Fig. 4A). Furthermore, the deletion resulted in a frameshift adding 16 ectopic amino acid residues and an early stop codon. This early termination resulted in the elimination of membrane-spanning domains 5 and 6, together with the residues encompassing the pore of the channel, and all downstream sequence encompassing the carboxyl cytoplasmic portions of the channel (Fig. 4B). Clearly, the removal of protein domains required for ion conduction in the channel resulted in a loss-of-function. We cultured C. elegans myocytes from the shl-1 (ok1168) deletion mutant strain and examined the currents remaining in these cells using single-electrode whole-cell patch clamp. Our results showed that the fast transient current was completely absent from these cells (Fig. 2B, right, and Fig. 4C, bottom). The currents remaining in these shl-1 deletion cells appeared highly similar to those observed in the majority of our wild-type and slo-2 (nf100) myocytes after treatment with shl-1 RNAi (Fig. 4C, middle). This result is consistent with our molecular analysis of the shl-1 (ok1168) deletion, showing that it cannot form a functional channel and functions as an effective null when homozygous.
FIGURE 4. Characterization of the shl-1 (ok1168) deletion mutant breakpoints.
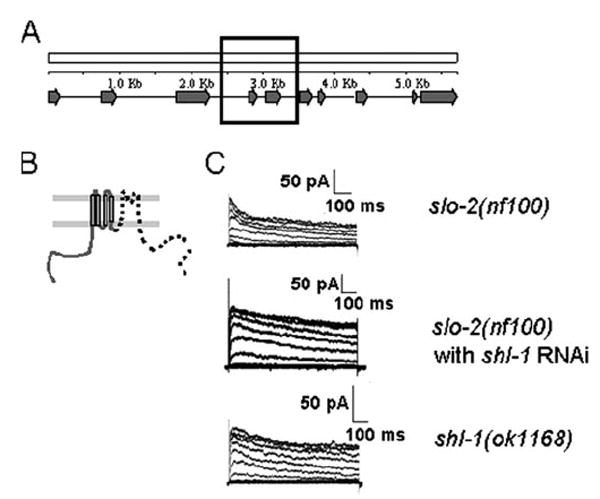
A, schematic of shl-1 genomic locus in C. elegans showing all 10 exons of the gene. The black box represents the genomic area that is deleted in the shl-1(ok1168) deletion mutant. B, pictorial representation of the truncated protein that might be produced by the shl-1(ok1168) mutant. The deletion removes the terminal 3 amino acids of the fourth transmembrane domain, and a frameshift resulting from the splicing of exons 3 and 6 results in missense coding of 16 amino acids ending in a stop codon. The portion of the channel that has been removed is designated by a dashed line. C, slo-2(nf100) myocytes display two voltage-sensitive macroscopic outward currents because of the presence of SHL-1 and SHK-1 channels (n = 7) (top). RNAi removal of SHL-1 current results in retention of a slowly inactivating voltage-sensitive current, SHK-1 (n = 3) (middle). The shl-1(ok1168) deletion mutant (n= 7) results in removal of fast transient current (bottom). Cells were held at −70 mV and stepped from −70 to +60 mV in 10-mV increments.
Definitive proof that SHL-1 and SHK-1 constitute the only voltage-dependent components in C. elegans myocytes results from the demonstration that selective elimination of both SHL-1 and SHK-1 currents produced cells having no macroscopic voltage-dependent outward current (43). To remove the SHL-1 component, we utilized myocytes from the transgenic dominant negative line (Fig. 5B) or the shl-1 deletion mutant (Fig. 5C). To remove the SHK-1 current, we treated both myocyte genotypes with shk-1 RNAi (Fig. 5, B and C). In such cells we observed a complete removal of all macroscopic outward current. Further examination of such cells at high gain revealed only single channel openings (Fig. 5, B and C, right traces). These single channel openings exhibited no detectable voltage dependence because their open probability was not noticeably different at different holding potentials (data not shown). We hypothesize that these single channel currents observed after the removal of all macroscopic currents could be due to any of several other genes encoding nonvoltage-dependent potassium channels that are expressed in C. elegans muscle (27). We therefore conclude that only three K+ channels produce the macroscopic outward K+ currents in cultured C. elegans myocytes, SHL-1, SHK-1, and the previously studied SLO-2 current (24).
FIGURE 5. Removal of all macroscopic outward currents in C. elegans myocytes.
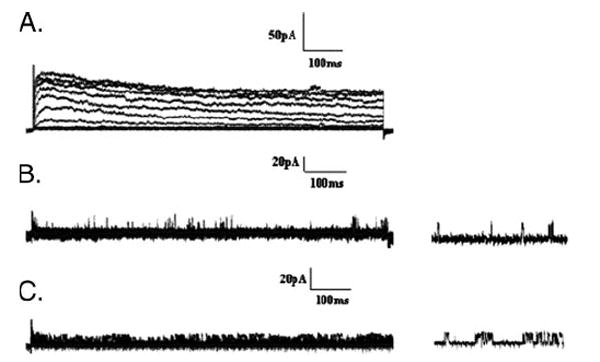
A, currents from slo-2(nf100) cells are shown as a control where both SHL-1 and SHK-1 currents are present. Whole-cell recordings were obtained from a holding potential of −70 mV, and stepped from −70 to +60 mV in 10-mV intervals. B, dominant negative pshl-1∷shl-1∷W363F cells (n = 3) treated with shk-1 RNAi to remove both SHL-1 and SHK-1 macroscopic currents. SLO-2 currents were inhibited by limiting the intracellular Ca2+ and Cl−. There is a complete removal of all macroscopic SHL-1 and SHK-1 currents in these myocytes. Unidentified single channel openings can be seen in these records (right traces). C, shl-1(ok1168) deletion mutant cells (n = 3) treated with shk-1 RNAi as in B.
Prior results have suggested that certain instances of altered expression of Shal channels are accompanied by altered expression of other ion channel types (43). Our results in C. elegans seem to bear this out (Fig. 6). In our studies using the shl-1 deletion mutant, we found that the remaining sustained current, most likely due to SHK-1, may be up-regulated as a result of the complete removal of SHL-1. To test this hypothesis, we compared the sustained current amplitudes in the slo-2 (nf100) myocytes (12.5 pA/pF ± 1.9), N2 myocytes (12.6 pA/pF ± 1.5), and the shl-1 (ok1168) myocytes (25.5 pA/pF ± 8.5). All currents were normalized to account for variable cell size difference (45). There was no significant difference (p=0.66 and p=0.34 respectively) in the amplitudes of normalized sustained currents between N2 myocytes (12.6 pA/pF ± 1.5) and either the shl-1 RNAi-treated myocytes (14.9 pA/pF ± 6.8) or the pshl-1∷W363F∷GFP (16.2 pA/pF ± 7.4) transgenic myocytes. However, there was a significant (p < 0.05) increase in sustained current amplitude in shl-1 (ok1168) myocytes, which is most likely due to up-regulation of the SHK-1 current. Fig. 6 summarizes our findings and shows the relatively larger sustained current component in shl-1 mutant myocytes and control animals (Fig. 6A) and a statistical analysis of these data (Fig. 6B). These results parallel studies in lobster that have demonstrated that changes in Shal (Kv4) channel expression levels can result in changes in whole-cell currents because of the up-regulation of other channel types (46). Notably, in both studies up-regulation of compensatory currents depends on the complete absence of a channel protein product, even if the ion conduction pathway of the protein product is blocked. Therefore the Shal protein may have a functional role in regulation, which is distinct from ion conduction.
FIGURE 6. Up-regulation of SHK-1 current in shl-1 (ok1168) mutant myocytes.
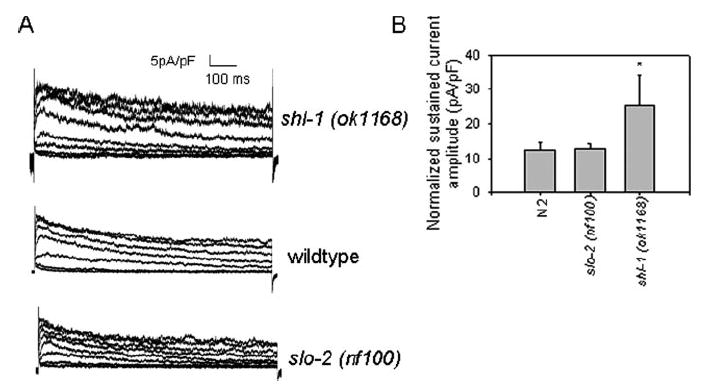
A, current traces from shl-1 (ok1168) myocytes (top), N2 wild-type myocytes (middle), and slo-2 (nf100) myocytes (bottom) shown on the same scale normalized for cell capacitance. B, mean normalized sustained current amplitudes with standard errors for the slo-2 (nf100) myocytes (12.5 pA/pF±1.9), N2 myocytes (12.6 pA/pF±1.5), and the shl-1 (ok1168) myocytes (25.5 pA/pF ± 8.5). * indicates p < 0.05 compared with shl-1 (ok1168) myocytes.
Physiological and Behavioral Analysis of SHL-1
A distinctive feature of C. elegans body wall muscle is that the resting potentials of cells are known to be significantly more positive than the resting potentials of muscle cells in mammalian species (47). This raises the possibility that SHL-1 channels in C. elegans, even though functioning in a more depolarized voltage range than their mammalian (or Drosophila) orthologs, may be active at or near the muscle cell resting potential. A significant Shal “window current” might then contribute substantially to cell resting conductance and thus basal muscle excitability and muscle responsiveness to neurotransmitter. If this were the case, eliminating the Shal current either by the deletion mutant or the dominant negative construct would increase muscle responsiveness to neurotransmitter and might manifest as an increase in sensitivity to aldicarb. Aldicarb is an acetylcholinesterase inhibitor that results in delayed clearance of acetylcholine (ACh) from the synaptic cleft of the neuromuscular junction. In C. elegans, the accumulated ACh results in rigid paralysis of animals with the severity of phenotype depending on the amount of ACh released into the cleft or the responsiveness of the postsynaptic muscle membrane to ACh (48). Thus, to explore the possibility of a heightened responsiveness of the postsynaptic membrane to ACh when the SHL-1 current was removed, we exposed both the shl-1 (ok1168) deletion mutant and the dominant negative transgenic line pshl-1∷W363F∷GFP to aldicarb (0.25 mm) (Fig. 7A). (Note that the expression of the dominant negative transgene was under control of the native shl-1 promoter.) We observed rigid paralysis in the animals with paralysis midpoints around 125 min for the dominant negative transgenic animals and about 132 min for the shl-1 (ok1168) animals (p < 0.0028 and p < 0.0025, respectively). These values were significantly different from those observed for wild-type N2 animals which was ~155 min. We also observed significant (p < 0.05) aldicarb hypersensitivity in both of our dominant negative transgenic lines under the native shl-1 promoter (Fig. 7B). We conclude that the SHL-1 current may well contribute to the postsynaptic muscle response to released ACh.
FIGURE 7. Removal of SHL-1 current in body wall muscle is sufficient to confer aldicarb hypersensitivity with 0.25 mm aldicarb.
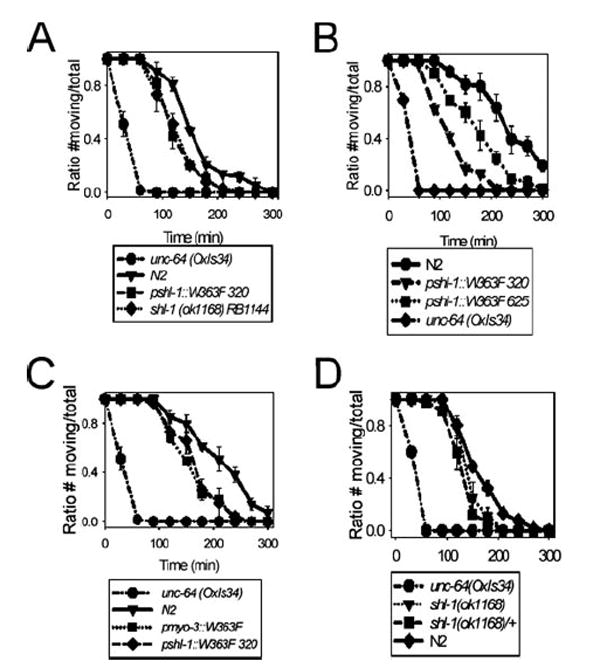
A, both the shl-1(ok1168) deletion mutant and the pshl-1∷W363F∷GFP line 320 carrying the dominant negative construct displayed aldicarb hypersensitivity compared with N2 animals. B, we tested two lines carrying dominant negative constructs integrated on different chromosomes to ensure that the observed aldicarb hypersensitivity phenotype was not because of position effects in these transgenic animals. Both lines carrying integrated constructs (320 on chromosome X and 625 on chromosome IV) displayed aldicarb hypersensitivity. The aldicarb hypersensitivity of the dominant negative transgenic line was not influenced by position effects. C, expression of the dominant negative transgene under control of pmyo-3 (a muscle-specific promoter) resulted in aldicarb hypersensitivity similar to that observed for the dominant negative under the native shl-1 promoter. D, the heterozygous shl-1(ok1168)/+ animals had an intermediate phenotype which fell between N2 and shl-1(ok1168) homozygotes. The positive control for hypersensitivity to aldicarb for all assays was the strain unc-64(OxIs34), a gain-of-function integrated transgenic syntaxin line.
An alternative hypothesis to explain these results is that hypersensitivity is because of a presynaptic effect and that the SHL-1 current controls the release of ACh-containing vesicles in the presynaptic terminal. To differentiate between pre- or postsynaptic causes of aldicarb hypersensitivity, we expressed the dominant negative transgene under control of the body wall muscle-specific myo-3 promoter (49) to limit expression of the dominant negative construct to the muscle. The demonstration that the pmyo-3∷W363F∷GFP transgenic line was also aldicarb-hypersensitive would prove that the absence of the SHL-1 current in muscle alone was sufficient to confer aldicarb hypersensitivity. Indeed, the pmyo-3∷W363F∷GFP transgenic line exhibited almost identical aldicarb hypersensitivity to that exhibited by the transgenic line pshl-1∷W363F∷GFP line 320 (Fig. 7C). Heterozygous shl-1(ok1168)/+ animals exhibited an intermediate level of aldicarb sensitivity (Fig. 7D). This result appears to validate the hypothesis that the increased sensitivity to accumulated ACh in the neuromuscular junction is a postsynaptic effect.
We examined a variety of behaviors in the shl-1 (ok1168) mutant animals in an effort to understand the shl-1 deletion mutant phenotype. shl-1 (ok1168) animals exhibited a defect in aldicarb sensitivity and a slight but significant alteration in thrashing behavior (data not shown). However, observed mating behavior presents a more rigorous behavioral assay that can reveal subtle defects in sensory processing; mating requires complex coordination of sensory information and intricate motor functions. Mating efficiency was determined by observing the number of mating plates possessing male progeny. Hermaphrodite C. elegans produce male progeny at very low penetrance unless mated by males (50). Two N2 or shl-1 (ok1168) males and two N2 hermaphrodites were placed on a small spot (~1 cm diameter) on mating plates. The number of successful mating events in these experiments, as indicated by the number of mating plates with male progeny, was significantly lower on plates with shl-1 (ok1168) male animals than with N2 males (48.8 and 85.4%, respectively) (Table 2). One possible complication in interpreting these results is the phenomenon of feminization whereby worms possessing an XO sex chromosome genotype exhibit female gonads and sexual behavior, instead of that of the male. Conceivably, the SHL-1 protein could be involved in sex determination turning males into phenotypic hermaphrodites (50, 51). To eliminate this possibility, we examined the proportion of males out of total progeny on individual mating plates. The proportion of male progeny on successfully mated N2 hermaphrodite × N2 male plates was not statistically different from the proportion of male progeny on successfully mated N2 hermaphrodite × shl-1 (ok1168) male plates. This eliminated the possibility of feminization producing the observed difference between wild type and mutant mating efficiency. We conclude then that the shl-1 (ok1168) strain exhibits decreased mating efficiency compared with N2 animals.
TABLE 2.
Analysis of mating behavior Vulva location behavior is divided into two categories, shown as “approximate” and “precise.” Approximate refers to the gross ability to locate the vulva. Precise refers to the process by which the vulva is located. Mating efficiency is as described under “Materials and Methods.” n = 10 for both the N2 and the DA2056 shl-1 (ok1168)-strains.
| Strain | Vulva location behavior
|
Mating efficiency | |
|---|---|---|---|
| Approximate | Precise | ||
| DA2056 shl-1 (ok1168) | Circles hermaphrodite and then stops at vulva | Occasional slow search; loses vulva easily | Poor (48.7805%) |
| N2 | Stops at vulva | Slow search using spicules | High (85.3660%) |
As a follow-up to these studies we sought to determine the defective steps in shl-1 mutant mating behavior by carefully observing the detailed behavior of the shl-1 (ok1168) males and comparing it with that of wild-type N2 males (35, 36). These observations showed that mutant males were less effective in locating the vulva of the hermaphrodite. On average, mutant males missed the vulva location by passing it 58.1% of the time compared with wild-type animals, which were unable to locate the vulva 42.1% of the time (p < 0.05). Furthermore, mutant animals unable to locate the vulva on the first pass moved past the vulva by more than 25% of the length of the hermaphrodite. N2 males typically do not move more than ~10% of the length of the hermaphrodite when passing over the vulva. We conclude that shl-1 (ok1168) males are less capable of fine motor control in mating behavior than are N2 animals. Some measure of this deficit in fine motor control in mating may involve the fact that SHL-1 is expressed in male-specific diagonal muscles in the tail region.
DISCUSSION
SHL-1 and SHK-1 C. elegans voltage-gated potassium channels exhibit fast transient and delayed rectifier currents, respectively. In other species, Shal channels are typically described as operating in an hyperpolarized voltage range relative to other voltage-gated K+ channels (37, 52). However, in C. elegans, SHL-1 activates in a more depolarized voltage range than is typically observed for Shal channels in other species (37). Recent work has demonstrated that pyramidal neurons in the rat visual cortex also activate at more depolarized voltages than are typically observed in mammals (16). Nevertheless, SHK-1 currents operate in an even more depolarized voltage range and thus seem to maintain their relative functional relationship to SHL-1 channels, consistent with that found in other species (25, 38-41, 53, 54). Furthermore, there is evidence that C. elegans body wall muscle rests at a rather positive value relative to muscle cells of other species (approximately −20 to −30 mV (47, 55)) which suggests that SHL-1 channels have evolved to serve a specialized purpose in this species. A second gene encoding a transient K+ channel has been reported in C. elegans, kvs-1 (56). KVS-1 channels exhibit an even more depolarized activation and inactivation range than that observed for SHL-1 channels; it also exhibits significantly slower inactivation kinetics. kvs-1 is closer to the Shab (Kv2) family of voltage-gated K+ channels than to the Shal family (57), and this gene is not expressed in body wall muscle.
Native SHL-1 channels have properties similar to the heterologously expressed cloned channels in that they carry fast transient currents with rapid activation and inactivation. However, the native channels activate in a somewhat more hyperpolarized voltage range than the cloned channels (approximately −14 mV). Such differences could conceivably be due to accessory proteins or post-translational modification occurring in native cells. In mammals, modulatory subunits or post-translational modification affects Shal channel voltage sensitivity, and similar mechanisms are known to affect Shaker currents as well. In C. elegans at least one protein similar to a mammalian modulatory subunit has been identified (58).
We showed that the major component of voltage-dependent current remaining in the shl-1 (ok1168) deletion mutant background is encoded by the Shaker gene, shk-1, and has the properties of a delayed rectifier current. We characterized two isoforms produced by the shk-1 locus, isoform a and isoform d2. Both express slowly inactivating currents, but the d2 isoform of SHK-1 functions in a more hyperpolarized voltage range than SHK-1 isoform a and may inactivate slightly faster. When the Shal component was removed either by RNAi in cell culture or in cells of the strain carrying the dominant negative form of Shal, the remaining current seemed to have the properties of SHK-1 isoform d2. However, the current remaining in the shl-1 deletion strain had properties more similar to the SHK-1 isoform a. In this latter case, the SHK-1 currents appeared to be larger. Precedents for this exist in the literature, which show that a second current component may be up-regulated to compensate for the removal of a first component. The compensating component may differ radically in its properties from the original component, which was removed (59, 60). For example, changes in HCN ion channel expression in response to increased Shal subunit expression have been observed previously in lobster stomatogastric ganglion cells (46, 61). In our case, the up-regulation of the SHK-1 current was not observed in either the shl-1 RNAi-treated myocytes nor the dominant negative transgenic line pshl-1∷W363F. Because only the deletion mutant exhibited the increased SHK-1 current, the compensatory expression of this current may be responsive to the lack of all SHL-1 protein, whether functional or not.
Although a complete knockout of SHL-1 and SHK-1 in the slo-2 (nf100) background eliminated all macroscopic outward currents, some unidentified unitary currents remained. We hypothesize that these single-channel currents are likely due to the many two-pore twk channels expressed in body wall muscle (27). However, the macroscopic outward currents in C. elegans cultured myocytes seem to be entirely carried by SLO-2, SHK-1, and SHL-1 channels.
Detailed analysis of shl-1-GFP cell-type expression patterns showed channel expression in cells involved with a variety of behaviors. However, mutant phenotypes were not observed for chemosensation, thermosensation, defecation, or pharyngeal pumping. On the other hand, we observed a significant shl-1 mutant phenotype with regard to the response to aldicarb. The total elimination of the SHL-1 current in all tissues by the deletion mutation, as well as the specific inhibition of SHL-1 current in body wall muscle by selective muscle-specific expression of the dominant negative construct, resulted in aldicarb hypersensitivity. This latter result strongly suggests that the muscle membrane itself is the site that confers aldicarb hypersensitivity. An attractive hypothesis that takes into account the unique properties of the SHL-1 current is that the mutant membrane is more susceptible to depolarization by acetylcholine than the wild-type membrane, because an SHL-dependent component of membrane resting conductance has been removed. This could come about via the SHL-1 window current, which may contribute to the resting membrane K+ conductance controlling basal resting membrane excitability. The fact that the SHL-1 current contributes to the K+ conductance at rest does not necessarily mean that it significantly changes the resting membrane potential. Indeed, if the membrane resting potential is close to EK, the driving force on K+ is so low that a small but significant change in K+ conductance would not have a measurable effect on the resting potential. However, current components that are active at rest need not be very large to have a major impact on the overall excitability of the cell, even if they do not significantly change the resting membrane potential. This is because total cell resting conductance is usually small and represents only a tiny fraction of the overall cell membrane conductance during peak electrical activity. A crucial factor is their contribution to the critical balance of inward and outward currents at the threshold of active responses. Many models show that a change of only ~10% of resting cell conductance can have a major effect on the excitable properties of a cell.
We demonstrated a significant reduction in mating efficiency in shl-1 (ok1168) deletion mutant males, which at least partially involves a reduced ability to locate the vulva of the hermaphrodite. Male copulatory behavior includes input from neurons common to both hermaphrodite and male, as well as male-specific neurons (note that we did not specifically examine male-specific neurons for SHL-1 channel expression). The difficulty of shl-1 (ok1168) animals to effectively mate appears to be due to a deficit in fine motor control or spicule insertion (62, 63), which may result from a mutant alteration of membrane excitability in muscle or neurons or both. The ether-ago-go-related-like potassium channel, UNC-103, is known to impact male-specific muscle contraction leading to failure of spicule insertion into the vulva of the hermaphrodite that reduces mating efficiency (62). Conceivably, SHL-1 functions in a similar manner, and removal of SHL-1 channels results in hypersensitivity of the muscle to ACh, lowering the efficiency of spicule insertion or impeding sensory perception of the vulva.
Loss of the shl-1 gene product produces neither a cell nor an organismal lethal. Nevertheless, its role in fine-tuning complicated behavior such as mating may offer essential survival benefits and may be one factor contributing to the extraordinarily high conservation of the Shal potassium channel.
Supplementary Material
Acknowledgments
We thank Dr. Cornelia Bargmann for the detailed cell expression analysis of the SHL-1 protein. We thank Dr. Michael Nonet for cell expression studies, helpful suggestions, sharing equipment, and the mutant strains for behavioral assays. We thank Dr. Crowder for help with the mating, chemosensation, and thermosensation behavioral assays. We thank Dr. Leon Avery for providing the backcrossed shl-1 (ok1168) strain. We thank Drs. Gonzalo Ferreira, Michael Crowder, Aaron DiAntonio, Jeanne Nerbonne, and Michael Nonet for comments and discussion throughout the course of this work. We thank Dr. Tim Schedl for suggesting the mating phenotype as a direction of further inquiry. We also thank Dr. Aguan Wei and Dr. Alex Yuan for critical comments and suggestions.
Footnotes
This work was supported by National Institutes of Health Grants R24 RR017342-01, R01 GM067154-01A1 (to L. S.), R01 NS032337 (to M. C.), and T32 AA007463 (to T. H.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: Kv, mammalian voltage-gated K+ channel family; unc, uncoordinated mutant; exp, expulsion-defective in defecation mutant; GFP, green fluorescent protein; RNAi, RNA interference; dsRNA, double-stranded RNA; DIDS, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid; NLS, nuclear localization sequence; ACh, acetylcholine; EST, expressed sequence tag.
References
- 1.Tsunoda S, Salkoff L. J Neurosci. 1995;15:1741–1754. doi: 10.1523/JNEUROSCI.15-03-01741.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM. Circ Res. 2005;97:1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- 3.Hagiwara S, Kusano K, Saito N. J Physiol (Lond) 1961;155:471–489. doi: 10.1113/jphysiol.1961.sp006640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connor JA, Stevens CF. J Physiol (Lond) 1971;213:21–30. doi: 10.1113/jphysiol.1971.sp009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neher E. J Gen Physiol. 1971;58:36–53. doi: 10.1085/jgp.58.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson SH. J Physiol (Lond) 1977;265:465–488. doi: 10.1113/jphysiol.1977.sp011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santi CM, Yuan A, Fawcett G, Wang Z-W, Butler A, Nonet ML, Wei A, Rojas P, Salkoff L. Proc Natl Acad Sci USA. 2003;100:14391–14396. doi: 10.1073/pnas.1935976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu H, Li H, Nerbonne JM. J Physiol (Lond) 1999;519:11–21. doi: 10.1111/j.1469-7793.1999.0011o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahn J, Tse TE, Levitan ES. J Neurosci. 2003;23:10859–10866. doi: 10.1523/JNEUROSCI.23-34-10859.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohya S, Tanaka M, Oku T, Asai Y, Watanabe M, Giles WR, Imaizumi Y. FEBS Lett. 1997;420:47–53. doi: 10.1016/s0014-5793(97)01483-x. [DOI] [PubMed] [Google Scholar]
- 11.Birnbaum SG, Varga AW, Yuan L-L, Anderson AE, Sweatt JD, Schrader LA. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- 12.Connor JA, Stevens CF. J Physiol (Lond) 1971;213:31–53. doi: 10.1113/jphysiol.1971.sp009366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shibata R, Nakahira K, Shibasaki K, Wakazono Y, Imoto K, Ikenaka K. J Neurosci. 2000;20:4145–4155. doi: 10.1523/JNEUROSCI.20-11-04145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan L-L, Adams JP, Swank M, Sweatt JD, Johnston D. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramakers GMJ, Storm JF. Proc Natl Acad Sci USA. 2002;99:10144–10149. doi: 10.1073/pnas.152620399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan W, Burkhalter A, Nerbonne JM. J Neurosci. 2005;25:9185–9194. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai X, Lian CW, Muralidharan S, Kao JPY, Tang C-M, Thompson SM. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 18.Sah T, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, Backx PH. J Physiol (Lond) 2003;546:5–18. doi: 10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo W, Xu H, London B, Nerbonne JM. J Physiol (Lond) 1999;521:587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nerbonne JM. J Physiol (Lond) 2000;525:285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amberg GC, Koh SD, Hatton WJ, Murray KJ, Monaghan K, Horowitz B, Sanders KM. J Physiol (Lond) 2002;544:403–415. doi: 10.1113/jphysiol.2002.025163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amberg GC, Koh SD, Imaizumi Y, Ohya S, Sanders KM. Am J Physiol. 2003;284:C583–C595. doi: 10.1152/ajpcell.00301.2002. [DOI] [PubMed] [Google Scholar]
- 23.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Circ Res. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- 24.Yuan A, Dourado M, Butler A, Walton N, Wei A, Salkoff L. Nat Neurosci. 2000;3:771–779. doi: 10.1038/77670. [DOI] [PubMed] [Google Scholar]
- 25.Beck EJ, Covarrubias M. Biophys J. 2001;81:867–883. doi: 10.1016/S0006-3495(01)75747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jerng HH, Covarrubias M. Biophys J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salkoff L, Butler A, Fawcett G, Kunkel M, McArdle C, Paz-Y-Mino G, Nonet M, Walton N, Wang Z-W, Yuan A, Wei A. Neuroscience. 2001;103:853–859. doi: 10.1016/s0306-4522(01)00079-3. [DOI] [PubMed] [Google Scholar]
- 28.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 29.Christensen M, Estevez A, Yin X, Fox R, Morrison R, McDonnell M, Gleason C, Miller DM, III, Strange K. Neuron. 2002;33:503–514. doi: 10.1016/s0896-6273(02)00591-3. [DOI] [PubMed] [Google Scholar]
- 30.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Pfluegers Arch Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 31.Jorgensen EM, Hartwieg E, Schuske K, Nonet ML, Jin Y, Horvitz HR. Nature. 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- 32.Saifee O, Wei L, Nonet ML. Mol Biol Cell. 1998;9:1235–1252. doi: 10.1091/mbc.9.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sulston J, Hodgkin J. In: The Nematode Caenorhabditis elegans. Wood WB, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1988. pp. 587–607. [Google Scholar]
- 34.Hodgkin J. Nature. 1983;304:267–268. doi: 10.1038/304267a0. [DOI] [PubMed] [Google Scholar]
- 35.Loer CM, Kenyon CJ. J Neurosci. 1993;13:5407–5417. doi: 10.1523/JNEUROSCI.13-12-05407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crowder CM, Shebester LD, Schedl T. Anesthesiology. 1996;85:901–912. doi: 10.1097/00000542-199610000-00027. [DOI] [PubMed] [Google Scholar]
- 37.Jerng HH, Pfaffinger PJ, Covarrubias M. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Pak MD, Baker K, Covarrubias M, Butler A, Ratcliffe A, Salkoff L. Proc Natl Acad Sci USA. 1991;88:4386–4390. doi: 10.1073/pnas.88.10.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baro DJ, Coniglio LM, Cole CL, Rodrigues HE, Lubell JK, Kim MT, Harris-Warrick RM. J Neurosci. 1996;16:1689–1701. doi: 10.1523/JNEUROSCI.16-05-01689.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holmqvist MH, Cao J, Hernandez-Pineda R, Jacobson MD, Carroll KI, Sung MA, Betty M, Ge P, Gilbride KJ, Brown ME, Jurman ME, Lawson D, Silos-Santiago I, Xie Y, Covarrubias M, Rhodes KJ, Distefano PS, An WF. Proc Natl Acad Sci USA. 2002;99:1035–1040. doi: 10.1073/pnas.022509299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bähring R, Boland LM, Varghese A, Gebauer M, Pongs O. J Physiol (Lond) 2001;535:65–81. doi: 10.1111/j.1469-7793.2001.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan A, Santi CM, Wei A, Wang Z-W, Pollak K, Nonet M, Kaczmarek L, Crowder CM, Salkoff L. Neuron. 2003;37:765–773. doi: 10.1016/s0896-6273(03)00096-5. [DOI] [PubMed] [Google Scholar]
- 44.Covarrubias M, Wei A, Salkoff L. Neuron. 1991;7:763–773. doi: 10.1016/0896-6273(91)90279-9. [DOI] [PubMed] [Google Scholar]
- 45.Kovac M, Davis WJ, Matera E, Gillette R. J Neurophysiol. 1982;47:909–927. doi: 10.1152/jn.1982.47.5.909. [DOI] [PubMed] [Google Scholar]
- 46.MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM. Neuron. 2003;37:109–120. doi: 10.1016/s0896-6273(02)01104-2. [DOI] [PubMed] [Google Scholar]
- 47.Jospin M, Mariol M-C, Segalat L, Allard B. J Physiol (Lond) 2002;544:373–384. doi: 10.1113/jphysiol.2002.022293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nonet ML, Grundahl K, Meyer BJ, Rand JB. Cell. 1993;73:1291–1305. doi: 10.1016/0092-8674(93)90357-v. [DOI] [PubMed] [Google Scholar]
- 49.Waterston RH. In: The Nematode Caenorhabditis elegans. Wood WB, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1988. pp. 281–336. [Google Scholar]
- 50.Hodgkin J. J Embryol Exp Morphol. 1984;83:103–117. [PubMed] [Google Scholar]
- 51.Hodgkin J. Development (Camb) 1987;101:5–16. doi: 10.1242/dev.101.Supplement.5. [DOI] [PubMed] [Google Scholar]
- 52.Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- 53.Wei A, Covarrubias M, Butler A, Baker K, Pak M, Salkoff L. Science. 1990;248:599–603. doi: 10.1126/science.2333511. [DOI] [PubMed] [Google Scholar]
- 54.Dilks D, Ling HP, Cockett M, Sokol P, Numann R. J Neurophysiol. 1999;81:1974–1977. doi: 10.1152/jn.1999.81.4.1974. [DOI] [PubMed] [Google Scholar]
- 55.Hille B. Ionic Channels of Excitable Membranes. Sinauer Associates Inc.; Sunderland, MA: 1992. pp. 76–78.pp. 115–139.pp. 507 [Google Scholar]
- 56.Bianchi L, Kwok SM, Driscoll M, Sesti F. J Biol Chem. 2003;278:12415–12424. doi: 10.1074/jbc.M212788200. [DOI] [PubMed] [Google Scholar]
- 57.Wei A, Jegla T, Salkoff L. Neuropharmacology. 1996;35:805–829. doi: 10.1016/0028-3908(96)00126-8. [DOI] [PubMed] [Google Scholar]
- 58.Price M, Thompson RJ, Eshcol JO, Wemmie JA, Benson CJ. J Biol Chem. 2004;279:53886–53891. doi: 10.1074/jbc.M407708200. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi E, Ino M, Miyamoto N, Nagasu T. Neurosci Lett. 2004;359:37–40. doi: 10.1016/j.neulet.2004.01.066. [DOI] [PubMed] [Google Scholar]
- 60.Guo W, Li H, London B, Nerbonne JM. Circ Res. 2000;87:73–79. doi: 10.1161/01.res.87.1.73. [DOI] [PubMed] [Google Scholar]
- 61.Baro DJ, Ayali A, French L, Scholz NL, Labenia J, Lanning CC, Graubard K, Harris-Warrick RM. J Neurosci. 2000;20:6619–6630. doi: 10.1523/JNEUROSCI.20-17-06619.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garcia LR, Sternberg PW. J Neurosci. 2003;23:2696–2705. doi: 10.1523/JNEUROSCI.23-07-02696.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia LR, Mehta P, Sternberg PW. Cell. 2001;107:777–788. doi: 10.1016/s0092-8674(01)00600-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.