

Abstract
Neurofibrillary tangles (NFT) constitute one of the cardinal histopathological features of Alzheimer’s disease (AD). To explore in vivo molecular processes involved in the development of NFTs, we compared gene expression profiles of NFT-bearing entorhinal cortex neurons from 19 AD patients, adjacent non-NFT-bearing entorhinal cortex neurons from the same patients, and non-NFT-bearing entorhinal cortex neurons from 14 non-demented, histopathologically normal controls (ND). Of the differentially expressed genes, 225 showed progressively increased expression (AD NFT neurons > AD non-NFT neurons > ND non-NFT neurons) or progressively decreased expression (AD NFT neurons < AD non-NFT neurons < ND non-NFT neurons), raising the possibility that they may be related to the early stages of NFT formation. Immunohistochemical studies confirmed that many of the implicated proteins are dysregulated and preferentially localized to NFTs, including apolipoprotein J, interleukin-1 receptor-associated kinase 1, tissue inhibitor of metalloproteinase 3, and casein kinase 2, beta. Functional validation studies are underway to determine which candidate genes may be causally related to NFT neuropathology, thus providing therapeutic targets for the treatment of AD.
Keywords: Alzheimer’s disease, Neurofibrillary tangles, Microarray, Gene expression, Dementia, Neurodegeneration, NFT, Laser capture microdissection
1. Introduction
Alzheimer’s dementia (AD) is the most common form of age-related cognitive impairment. AD is characterized pathologically by extracellular amyloid plaques and intracellular neurofibrillary tangles (NFT). Recent experiments suggest a synergy, if not a direct causal link, between amyloid pathology and NFTs in Alzheimer’s disease [27]. However, NFTs can occur without concomitant amyloid pathology in a broad class of neurodegenerative diseases, collectively known as “tauopathies” [16]. Furthermore, the extent of NFT formation correlates with the onset and severity of dementia in these disorders [16]. This indicates that NFTs themselves are intricately involved in neurodegenerative processes and in the resulting onset of dementia. Halting, or even slowing, the development of NFTs could have significant therapeutic impact for millions of individuals currently afflicted with AD or with any of the tauopathies. As a result, significant focus has been directed at understanding the molecular basis of NFT formation with the goal of identifying relevant therapeutic targets and treatment strategies.
NFTs are composed primarily of an aggregated, hyperphosphorylated form of the microtubule organizing protein tau. In normal neurons, tau associates with microtubules and promotes microtubule stability by suppressing dynamic instability. Tau protein has been implicated in numerous cellular processes, including neurite outgrowth [4,5,19], microtubule transport [11], and oligodendrocyte maturation [13,18]. When hyperphosphorylated, as in AD, tau protein self-aggregates to form NFTs. Aggregation of tau protein prevents its normal microtubule dependent functions, and is thought to lead to loss of microtubule integrity and subsequent neuronal cell dysfunction, cell death, and eventual clinical dementia. Thus, understanding the causes of tau dysfunction and NFT formation could lead to viable treatment strategies to prevent NFT-induced neuronal cell death and dementia.
Gene expression profiling is a well-documented, established approach used to identify pathogenic correlates of disease processes. One of the key limitations of expression profiling research in neurodegenerative diseases has been overcoming the variability of gene expression resulting from the cellular heterogeneity of brain tissues and from inter-individual genetic variability. We have applied a single-cell expression profiling strategy to identify significantly altered, neuronally expressed genes that could contribute to NFT formation. NFT-containing neurons (AD NFT neurons) and adjacent histopathologically normal neurons (AD non-NFT neurons) from the entorhinal cortices of AD affected individuals were evaluated first using a within-subjects design. We then performed an additional comparison of histopathologically normal neurons from age-matched, non-demented individuals (ND non-NFT neurons) to facilitate the identification of gene expression changes that may occur prior to the onset of overt NFT pathology and, as such, may be early contributors to NFT formation. The entire data set is publicly available for download at http://www.tgen.org/neurogenomics/data. A large number of genes were identified that exhibited significant alterations in expression in relation to NFT pathology. These genes serve as preliminary gene candidates that could function as mediators of NFT formation. Functional validation efforts are now ongoing to clarify which of these candidates might serve as useful targets for subsequent AD treatment strategies.
2. Materials and methods
2.1. Tissue staining, laser capture microdissection, and RNA isolation
Entorhinal cortex samples were obtained from brain banks within the Alzheimer’s Disease Center (ADC) program funded by the National Institute on Aging. The Arizona ADC, the Duke University ADC and the Washington University ADC contributed frozen brain samples, which met stringent inclusion criteria, to this study. The Arizona ADC contributed 15 AD samples and 14 non-demented controls and 2 AD samples were contributed from both the Duke University ADC and the Washington University ADC. Human brain tissue was obtained from donors who were enrolled in ADC tissue donation programs, prospectively followed until death, and autopsied according to previously published protocols [15]. Tissues were then banked and used for this study according to procedures approved by the Institutional Review Boards. Samples were selected with a post-mortem interval (PMI) less than three hours. AD cases consisted of 9 males and 10 females with an average age of 84.7 years (±7.5). Control cases consisted of nine males and six females with an average age of 80.1 years (±7.9). Entorhinal cortex from these 19 mid stage AD cases (Braak stage III–IV) and 14 non-demented controls (Braak stage 0–II) was sectioned at 10 μm thickness and mounted onto standard, uncoated glass slides (Fisher Scientific). Slides were then stained with a combination of Thioflavin-S (Sigma) and neutral red (Fisher Scientific) using the following protocol: slides were immersed sequentially into xylene for 10 s, 95% EtOH for 5 s, 75% EtOH for 5 s, H2O for 5 s, 0.1% Thioflavin-S for 2 min, H2O for 5 s, 1% neutral red for 30 s, H2O for 5 s, 75% EtOH for 5 s, 95% EtOH for 5 s, 100% EtOH for 5 s, and xylene for 5 s. Slides were then air dried for 2 min prior to laser capture microdis-section (LCM). LCM was performed using CapSure Macro LCM caps (Arcturus) and the AutoPix (Arcturus) instrument with a laser setting that varied (depending on individual caps and tissue topography) from 60 mV with a pulse duration of 1500 μs to 100 mV with a pulse duration of 3000 μs. Neurofibrillary tangles were identified in the layer II stellate cell islands by the bright green fluorescence of thioflavin-S staining and normal neurons were identified by their characteristic size and shape (pyramidal neurons), lack of thioflavin-S staining, and location within layer II. From each AD sample, 1000 neurons were collected. The RNA quality of the tissue sample from each patient was assessed by standard agarose gel electrophoresis prior to LCM by extracting total RNA from an unstained section to ensure and quality control the LCM process. Cell extracts were prepared from the LCM samples using the PicoPure RNA isolation kit (Arcturus) following the standard protocol. Extracts were then stored at −80 °C until all samples for a given neuronal population were completed. Total RNA was isolated using the PicoPure RNA isolation kit following the standard protocol. DNase I treatment was performed as described in the manual.
2.2. RNA amplification and array hybridization
Double round amplification and labeling of the cRNA was performed following the standard Affymetrix protocol (www.affymetrix.com). Quality of the labeled cRNA was assessed by gel electrophoresis.
Labeled cRNA (10 μg) was hybridized to either Affymetrix U133A arrays (10 AD cases) or, after they became available, to the Human Genome U133 plus 2.0 arrays (9 AD and 14 ND) following standard protocols. Standard fluidics protocols were used to wash and stain the arrays (Euk genome WS2V5). Arrays were scanned using the GeneChip Scanner 3000 (Affymetrix).
2.3. Data analysis
Using Microarray Suite 5.0 (Affymetrix; MAS 5.0) arrays were globally normalized to a median signal intensity of 150 to enable interarray comparisons. All genes that did not show expression across the data set (called absent or marginal by MAS 5.0) were removed. Data was imported into Gene-Spring 6.1 software and correlation coefficients derived in pair-wise comparisons of individual samples. Ten paired AD NFT and AD non-NFT samples were hybridized to U133A arrays and a replicate set of nine additional paired AD NFT and AD non-NFT samples was hybridized to U133 plus 2.0 arrays. These two sets of paired data samples were analyzed separately and the most significant genes from both analyses were retained. The permutational p-values for the difference between paired samples were calculated using a web-based program written in CGI/PERL and is available for use at http://biostat.mc.vanderbilt.edu/twiki/bin/view/Main/BonnieLaFleur. These paired permutation tests use algorithms developed by Fisher [12], and described in detail in a paper by Manly [24]. Permutation tests for paired samples are similar to their parametric counterpart (the paired t-test) in that the test statistic is based on the difference between the two correlated samples (in this case NFT and non-NFT samples on the same individuals). The actual permutational p-value is obtained by randomizing the signs of the differences and calculating the paired t-test statistic for each permutation. The permutational p-value is the proportion of permutation test statistics that equal or exceed the original test statistic out of 2n total permutations. Genes with permutational p-values < 0.05 were retained for further consideration. Genes that were significant on both array platforms were considered to be of special interest, given that the later array generation contains substantially more information.
The second analysis strategy, designed to identify early contributors to NFT pathology, was to input all of the data from the U133 plus 2.0 arrays (14 ND non-NFT, 9 AD non-NFT, and 9 AD NFT neuronal samples) into GeneSpring 6.1 software and perform a one-way ANOVA, followed by post hoc permutational t-tests, to identify genes that were either consistently induced or repressed across the continuum of pathological states from ND non-NFT to AD non-NFT to AD NFT neurons, which generated a list of 225 consistently altered genes.
2.4. Immunohistochemistry
Frozen sections from independent cases were selected for validation of gene candidates at the protein level. Glycol preservative was removed by washing 3× in PBS for 10 min each. Antigen retrieval was performed by incubating samples at 55 °C for 10 min in 10 mM citrate buffer. Sections were washed in PBS 3× for 10 min each. Blocking was done by incubating in 3% BSA in PBST for 30 min to 1 h at room temperature (RT) and rinsing 1× in PBST. Primary antibody in PBST with 0.25% BSA was applied at the predetermined dilution and incubated overnight at 4 °C. Tissue was washed 3× 10 min in PBST. Secondary antibody in PBST with 0.25% BSA was applied for 2–4 h at RT and then washed 3× 10 min in PBST. The second primary antibody in PBST with 0.25% BSA was then applied and incubated overnight at 4 °C. Sections were washed 3× 10 min in PBST and secondary applied in PBST/0.25% BSA. Sections were washed 3× 10 min in PBST, mounted onto slides, and air-dried 2 h to overnight. To reduce background autoflourescence the slides were treated with a 1–3 min incubation in Sudan Black, followed by two successive 10-s rinses in 70% EtOH and one rinse in H2O. Slides were mounted with 10× PBS with glycerol. For each antibody used, staining was performed in triplicate.
3. Results
3.1. Reduction of inter-individual expression variability and tissue heterogeneity
Laser capture microdissection (LCM) allows the isolation of individual cell types from a heterogeneous tissue sample. We have used LCM to isolate neurofibrillary tangle-containing neurons and histopathologically normal neurons from the layer II stellate cell islands of the entorhinal cortex of 19 AD affected individuals, as well as histopathologically normal neurons from 14 non-demented control individuals, all matched for age and stage of disease progression (three cohorts of cell types; Braak stage III/IV for AD cases and 0–II for controls). Fig. 1A and C show representative dissections of tangle-bearing and normal neurons, respectively. Extremely high quality cRNA was obtained from these isolated cell populations for hybridization to microarrays (Fig. 1B).
Fig. 1.
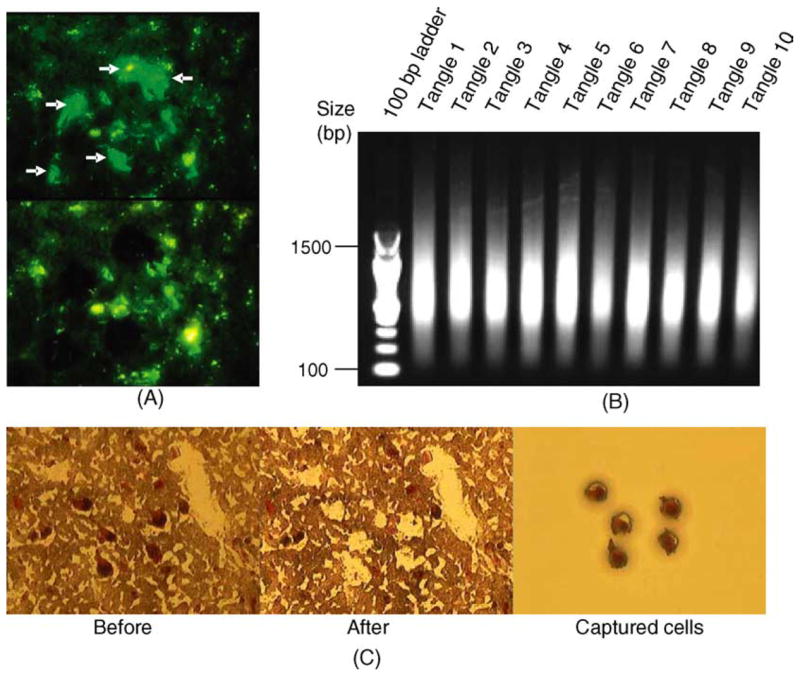
(A) LCM dissected tangle-bearing vs. non-tangle-bearing neurons from the identical AD patient, from the identical brain region. (B) LCM dissected neurons collected from 10 independent AD cases yielded excellent quality RNA following double round amplification and were used to generate expression profiles. Only RNA isolated from tangle-containing neurons is shown. (C) Visualization and isolation of histopathologically normal pyramidal neurons from non-demented control individuals using neutral red staining.
The use of LCM eliminates the inter-individual variability in gene expression profiles that is a significant source of variability in expression profiling experiments of complex human diseases. For our initial comparison of AD NFT neurons to AD non-NFT neurons, we hybridized cRNA samples prepared from LCM dissected neurons of 10 AD cases to Affymetrix U133A arrays (20 arrays total). An analysis of scatter plots comparing expression profiles on a sample-to-sample basis showed that correlation coefficients for the expression profiles from intra-individual comparisons of AD NFT neurons versus AD non-NFT neurons ranged from 0.828 to 0.938, with an average value of 0.913 (Fig. 2). This contrasts with inter-individual comparisons of AD non-NFT neurons or AD NFT neurons, where correlation coefficients varied from 0.552 to 0.926 and 0.561 to 0.920, with average values of 0.849 and 0.873, respectively. We obtained similar results in a replication of these experiments wherein we isolated and expression profiled NFT-containing neurons and non-NFT neurons from an additional set of 9 AD cases on the whole genome Affymetrix U133 plus 2.0 array, containing probes for 38,572 unique genes and an additional 841 potential full-length transcripts. The R2 values averaged 0.853 for intra-individual comparisons (range 0.744–0.960), 0.750 (range 0.552–0.920) for inter-individual comparisons of AD non-NFT neurons, and 0.729 (range 0.561–0.920) for inter-individual comparisons of AD NFT neurons. The genes that are consistently altered across these pairs of NFT-containing and NFT-free samples should be robust correlates of NFTs, as inter-individual “SNP noise” has been eliminated.
Fig. 2.
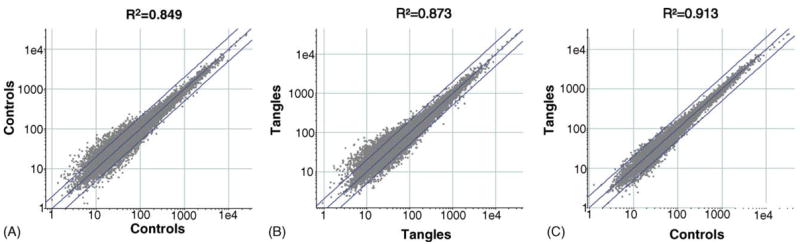
LCM reduces inter-individual gene expression variability. Intra-individual expression variability in histopathologically normal neurons and tangle-bearing neurons is low compared to inter-individual variability between histopathologically normal control neurons and neurofibrillary tangle-containing neurons from different individuals. Shown in panels A and B are scatterplots comparing the expression profiles of inter-individual differences between control neurons and tangle-containing neurons with an R2 value of 0.849 for controls and 0.873 for tangled neurons. To generate the R2 value for control neurons, five of the ten control samples were randomly selected and compared to the remaining five samples. Plotted are the mean signal intensity values for each gene across five control samples relative to the mean of the remaining five samples. The R2 value for tangle-containing neurons was derived similarly. Shown in panel C is the inter-individual comparison of tangle-containing neurons to control neurons (R2 = 0.913). Plotted is the mean signal intensity for each gene across all ten control non-NFT samples relative to all ten NFT-containing neuron samples. Results for a replicate data set of nine control neurons and nine tangle-containing neurons showed a comparable pattern with R2 values of 0.852 for the intra-individual comparison and 0.759 and 0.735 for inter-individual comparisons of control neurons and tangle-containing neurons, respectively.
3.2. Expression profiles of AD NFT neurons differ from AD non-NFT neurons
To identify genes associated with NFT pathology, we performed permutational, paired T statistics treating each AD NFT sample and corresponding AD non-NFT neuronal sample as patient-matched pairs (see Section 2). Shown in Fig. 3 are those genes that showed statistically significant fold changes across replicate analyses performed on Affymetrix U133A and U133 plus 2.0 arrays. These genes all showed consistent expression differences across each of the data sets and had a 99% confidence interval for the observed expression ratio that did not include 1 (unchanged value). For the purposes of validating expression differences at the protein level, we extended the statistical cutoff to the 95% confidence interval at p < 0.05 to include additional genes that had suggested biological relevance to AD or neurodegeneration (see below).
Fig. 3.

Statistically significant genes are consistently dysregulated across the data sets. Shown are heat maps illustrating fold change values for the indicated genes in each patient-matched pair of histopathologically normal neurons (control) and neurofibrillary tangle-containing neurons (tangled). The left heat map represents genes from the initial analysis on Affymetrix U133A arrays and the right heat map shows the results for the replicate data set analyzed on U133 plus 2.0 arrays. Genes that were repressed in tangled neurons relative to controls are colored in green and induced genes are colored in red. Provided are the unique probe IDs for each gene, the gene symbol, average fold change between classes, and permutational P values for each replicate data set. Cluster representation was generated using GeneCluster (http://rana.lbl.gov/EisenSoftware.htm). log2-fold-differences for each sample pair for each gene were imported into GeneCluster and complete linkage clustering performed without clustering either arrays or genes.
3.3. Gene expression analysis of ND non-NFT neurons identifies significant alterations that occur prior to NFT formation
The differentially expressed genes that are identified in comparisons of AD NFT neurons and AD non-NFT neurons were anticipated to fall into two broad classes, those that may cause NFT formation and those that may result from NFT formation. Additionally, thioflavin-S identifies only late stage NFTs. Thus there may be important pre-NFT molecular changes present in our AD non-NFT neuronal population that contribute to NFT formation. To facilitate the identification of these pre-NFT changes, we have compared expression profiles of 14 control non-NFT neuronal (ND non-NFT) samples to nine age-matched mid-stage AD NFT and AD non-NFT neuronal samples using the whole genome Affymetrix U133 plus 2.0 array. Genes that are the most likely early contributors to NFT formation would be anticipated to show dysregulation prior to NFT formation. These genes should have consistently increasing or decreasing expression when comparing ND non-NFT neurons to AD non-NFT neurons and then to AD NFT neurons. Using a one way ANOVA and post hoc permutational t-tests (p < 0.05), we have identified 225 genes that satisfy these criteria of consistently increasing or decreasing alterations across all three data sets (see Supplemental Data for complete list). The top 100 genes from this analysis are shown in Fig. 4. These genes were found to include IRAK1, APOJ, CD44, PAK7, and CAPN7. Clearly genes that are induced or repressed in AD non-NFT neurons compared to ND non-NFT neurons are also of interest and we present for public download the entire data set to enable these types of independent analyses and testing of multiple additional hypotheses (http://www.tgen.org/neurogenomics/data).
Fig. 4.

Significant neuronal gene expression changes occur in Alzheimer’s disease prior to NFT formation. We performed a one-way ANOVA to identify statistically significant genes that show consistently increasing or decreasing expression across the data sets of ND non-NFT neurons, AD non-NFT neurons, and AD NFT neurons. Shown are the top 100 genes from this analysis. Cluster representation was generated by GeneCluster software. Prior to figure generation, data was mean centered for each gene by subtracting the mean gene expression from the expression values so that the mean expression for each gene is 0. Data was subsequently normalized on a gene-by-gene basis by multiplying each value by a scale factor S, such that the sum of the squares of the values for each gene is 1.0. Data shown include the probe ID, gene symbol, ANOVA p-value, the average fold change for the comparison of AD non-NFT neurons vs. control non-NFT neurons, the average fold change for AD NFT neurons vs. AD non-NFT neurons, and the average expression values for each gene across all three data sets.
3.4. Immunohistochemistry confirms the association of numerous candidate proteins with NFTs
We have performed immunohistochemistry on independent tissue samples to validate selected candidate genes from the above expression profiling analyses with the combined purposes of demonstrating neuronal expression and qualitative expression differences consistent with the expression profiling observations. All of the genes selected for validation have either been implicated previously in AD or have resonance with pathologic processes that occur in AD.
Apolipoprotein J (APOJ) mRNA was significantly induced 1.74-fold when comparing AD NFT to AD non-NFT neurons and was upregulated 1.7-fold when comparing AD non-NFT neurons to ND non-NFT neurons. APOJ has been heavily implicated in neurodegenerative disorders, including AD, prion diseases, and Parkinson’s disease [6,17,28]. Consistent with the gene array results, APOJ immunoreactivity co-localized with NFT neuronal perikarya and neuropil threads in AD cases. It was less evident in cells without NFT immunoreactivity in these patients, and was rare in ND samples. Indeed, in ND sections APOJ immunoreactivity was preferentially localized to cells with the morphology of glia. Numerous previous reports have observed glial immunoreactivity for APOJ in rodent brain and in cultures of human glia (Fig. 5A1–A3).
Fig. 5.

Immunoreactivity for the protein products of selected candidate genes in AD and ND entorhinal cortex and their relationship to NFT immunoreactive neurons and neuropil threads. The left panels show immunoreactivity for the candidate proteins (green fluorophor) in AD. The middle panels show NFT immunoreactivity (red fluorophor) in the same field using a PHF1 mouse monoclonal antibody (generous gift of Dr. Peter Davies, Albert Einstein College of Medicine) or pS396 rabbit polyclonal antiserum (Biosource). The right panels show immunoreactivity for the target proteins in ND. All staining was performed in triplicate or more, and the results from multiple AD cases were compared to insure that the micrographs given here were representative. All micrographs were taken at the same magnification (40× objective) and reduced proportionately. Because NFTs occur with much lower frequencies in ND cases, and because of space limitations, PHF1 immunohistochemistry is not shown for ND cases, but was performed. (A1–A3) Clusterin; (B1–B3) casein kinase 2; (C1–C3) TIMP3; (D1–D3) IRAK1.
The mRNA for casein kinase 2, beta polypeptide (CSNK2B) was upregulated 1.34-fold in AD NFT versus AD non-NFT neurons. Our immunostaining results confirm both the upregulation observed in our expression profiling results and prior results showing colocalization of casein kinase 2 to NFTs [3,25]. We observed a robust association of CSNK2B with >90% of NFTs (Fig. 5B1–B3) and labeled NFT-containing neurons in ND cases as well (arrow in Fig. 5B3). Neurons without NFTs were less intensely stained, but exhibited clear immunoreactivity (arrowheads in Fig. 5B1). These findings are similar to the pattern observed in the array data.
Tissue inhibitor of metalloproteinase 3 (TIMP3) showed consistent upregulation across the data sets. TIMP3 was induced 1.52-fold in AD NFT neurons relative to AD non-NFT neurons and showed a 3.3-fold upregulation in AD non-NFT neurons relative to ND non-NFT neurons. Results showed that TIMP3 was expressed most strongly in AD NFT neurons and neuropil threads, with clear but less intense immunoreactivity in AD non-NFT neurons. TIMP3 immunostaining of ND non-NFT cells could also be observed, although there generally appeared to be fewer such cells in the ND cases (Fig. 5C1–C3). These findings are consistent with the differential expression results and suggest that upregulation of TIMP3, which appears to occur prior to overt pathology, may be an important factor in the progressive development of NFTs.
Interleukin-1 receptor-associated kinase 1 (IRAK1) was upregulated 1.7-fold in AD NFT neurons relative to AD non-NFT neurons and induced 1.97-fold in AD non-NFT neurons relative to ND non-NFT neurons. IRAK1 showed strong co-localization with virtually all NFT neurons in AD samples (Fig. 5D1–D2) as well as in the occasional NFT neuron in ND samples (arrow in Fig. 5D3). There was also pronounced co-localization with aggregates of NFT-positive neuropil threads that are likely to be contained in the corona of classical Aβ plaques (arrowheads in Fig. 5D1).
The CD44 antigen is a transmembrane hyaluronic acid binding protein implicated in neuronal regeneration and neurite outgrowth. CD44 mRNA was upregulated 1.86-fold in AD NFT neurons compared to AD non-NFT neurons and 3.5-fold in AD non-NFT neurons compared to ND non-NFT neurons. Immunostaining results showed that expression of CD44 appeared to be most consistent in AD NFT neurons and, especially, neuropil threads. Neurons with NFTs in ND cases could also be observed (arrow in Fig. 6E3), along with many other non-NFT neurons.
Fig. 6.
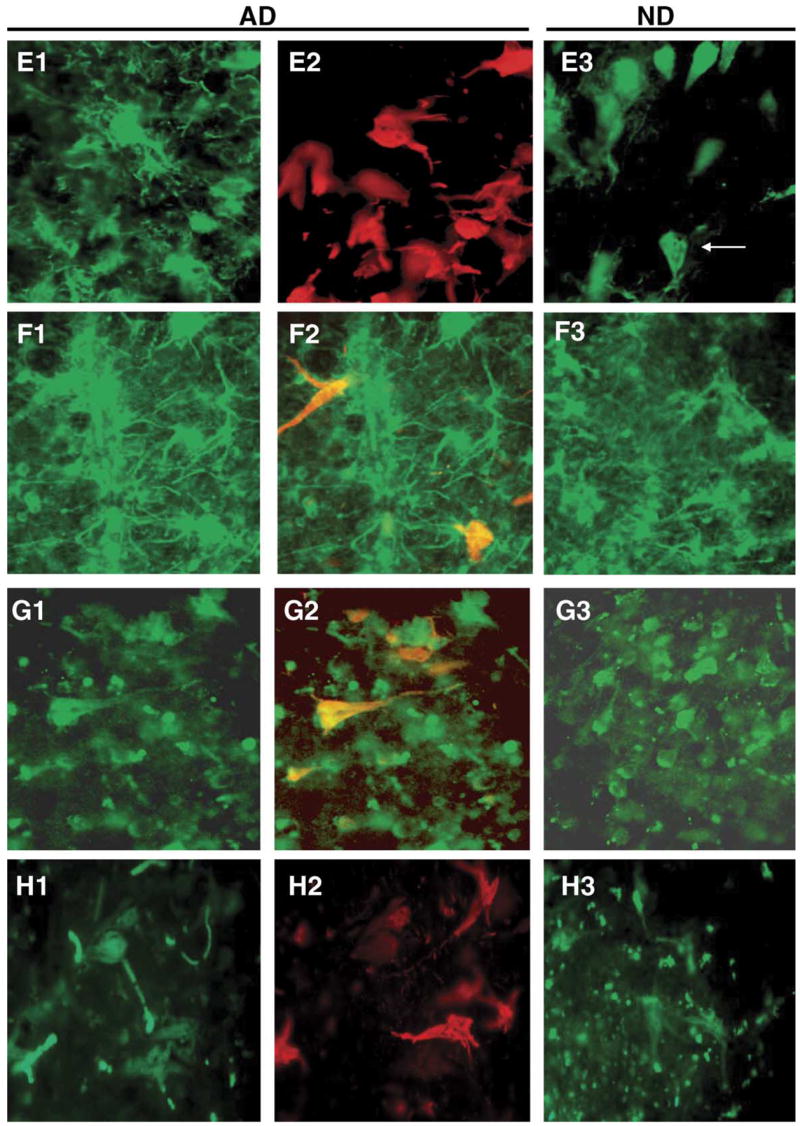
Immunoreactivity for the protein products of selected candidate genes in AD and ND entorhinal cortex and their relationship to NFT immunoreactive neurons and neuropil threads. The left panels show immunoreactivity for the candidate proteins (green fluorophor) in AD. For candidate genes that were downregulated in AD (CAPN7; panels F1–F3 and PAK7; panels G1–G3), the middle panels are merged images of immunoreactivity for the target gene (green fluorophor) and NFTs (red fluorophor) to better illustrate the decreased co-localization with NFTs. The right panels show immunoreactivity for the target proteins in ND. All staining and imaging was performed as in Fig. 4. (E1–E3) CD44; (F1–F3) CAPN 7; (G1–G3) PAK7; (H1–H3) RAP2A.
Calpain 7 (CAPN7) was repressed −1.54-fold in AD NFT neurons compared to AD non-NFT neurons and was similarly significantly repressed −1.28-fold in AD non-NFT neurons relative to ND non-NFT neurons. The calpain family of calcium activated cysteine proteases, which now contains 14 members, is implicated in diverse human diseases, from Alzheimer’s disease to diabetes and cancer [14]. Calpain 7 immunoreactivity could be observed on NFT neurons, but was more intense and prolific in non-NFT cells in both AD and ND cases, especially non-NFT cells with the morphology and smaller size of cortical interneurons. Staining of NFT neurons could also be observed, but was typically somewhat more diffuse (Fig. 6F1–F3). These qualitative differences parallel the AD NFT down-regulation of calpain 7 mRNA observed in the expression analyses.
p21-activated kinase 7 (PAK7) is a serine/threonine kinase that has been shown to bind directly to cdc42 and to promote neurite outgrowth [8]. PAK7 is also microtubule-associated and promotes microtubule stabilization [7]. PAK7 was significantly repressed −1.24 fold in AD NFT neurons compared to AD non-NFT neurons and −2.77-fold in AD non-NFT neurons compared to ND non-NFT neurons. ND control sections immunoreacted for PAK7 revealed many stained cells with the morphology and size of neurons (Fig. 6G1–G3). AD NFT neurons were also immunoreactive, although staining intensities often appeared to be qualitatively weaker. These results are consistent with the repression identified in expression profiling results and suggest potential important roles for this kinase in NFT pathogenesis.
RAP2A is a member of the RAS oncogene superfamily that regulates integrin-mediated cell adhesion, cell spreading, and actin polymerization. Although not implicated in AD, the known function of this protein in the same cytoskeletal regulatory pathways as PAK7 and CAPN7 made this protein a candidate for validation. RAP2A was upregulated 1.25-fold in AD NFT neurons relative to AD non-NFT neurons and 1.31-fold in AD non-NFT neurons compared to ND non-NFT neurons. Immunostaining results demonstrated that, within the sensitivity of this qualitative assay, RAP2A is expressed in both tangled and non-tangled neurons approximately equally (Fig. 6H1–H3).
While apparently very diverse processes appear to be implicated by these genes, the results of our protein validation (in the absence of an integrated theory of pathogenesis of NFTs at this time) show that the expression profiling strategy we have implemented has identified true gene dysregulation events, which are correlated to the histopathology of interest.
4. Discussion
Relative to the application of genomics to the study of neurodegenerative diseases, variability in gene expression profiles arises from four primary sources: inter-individual genetic differences, tissue heterogeneity, technical variability, and heterogeneity of the disease process. A critical benefit of the LCM approach to genomics in neurodegenerative diseases is that it allows the identification of gene expression correlates that are specifically related to the cell type and pathology of interest. By isolating the pathologically affected cell type and adjacent, histopathologically unaffected cells from the same individual, one can generate gene expression profiles that contain no inter-individual variability. Thus, lists of differentially expressed genes are more likely to represent differences that are specifically associated with pathology. In this study, we have shown that LCM enables the identification of specific expression correlates of neurofibrillary tangle pathology, which serve as preliminary candidate mediators of NFT pathogenesis. However, this technology can be applied to the study of any disease which shows a detectable pathology.
Expression profiling of three cohorts of LCM dissected neurons (ND non-NFT neurons, AD non-NFT neurons, and AD NFT neurons) and subsequent validation by immunohistochemistry indicates that numerous cellular genes and proteins are dysregulated in NFTs, with many proteins showing preferential localization to NFT-containing neurons. These findings suggest that multiple cellular processes are dysfunctional during NFT formation. One or more of these processes are the likely early contributors to the formation of NFTs and subsequent neuronal cell death and thus may be excellent potential therapeutic targets for common, sporadic AD.
In mouse models of AD, apolipoprotein J has been shown to promote the formation of extracellular amyloid plaques [10]. Truncated, intracellular forms of APOJ protein have been shown to alter mitochondrial function and, when localized to the nucleus, to induce apoptosis [9,29]. APOJ has been suggested to be both a pro- and anti-apoptotic factor. The distinction appears to be that extracellular forms of the protein promote cell survival and intracellular forms promote apoptosis. The antibody that we used does not allow us to distinguish the specific forms of clusterin protein that co-localize to NFTs. However, because NFTs are intracellular deposits, we speculate that the intracellular form of APOJ colocalizes to NFTs, consistent with the pro-apoptotic role of this form of APOJ protein. Nonetheless, the observation that APOJ is elevated in AD and associated with NFT containing neurons provides insight into the findings that APOJ promotes both amyloid aggregation and cell death in response to β-amyloid. Our findings provide additional in vivo evidence implicating APOJ as an important factor in NFT formation and neuronal cell death in AD.
A second candidate gene, tissue inhibitor of metalloproteinase 3 (TIMP3), has been implicated in neuronal cell death following ischemic injury [32]. TIMP3 is a metalloproteinase inhibitor that functions as a pro-apoptotic factor through its ability stabilize death receptors and their ligands on the cell surface by inhibiting the metalloproteinases that inactivate them [1,30]. TIMP3 has not yet been directly implicated in AD. In contrast, TIMP1 and TIMP2 proteins are elevated in the cerebrospinal fluid of patients with diverse neurodegenerative diseases [22] and in the brains of patients with progressive supranuclear palsy [21]. Further, matrix metalloproteinase 9, which is a target of TIMP inhibition, has been reported to be upregulated in plasma of AD patients [23] and has been implicated in the degradation of toxic β-amyloid species [2]. Loss of TIMP3 function has recently been shown to increase inflammation in the liver of TIMP3 knockout mice [26]. The observation that TIMP3 is upregulated in AD brain and associated with NFTs may reflect a compensatory mechanism to curb the inflammation known to accompany neurodegeneration in AD. Our findings showing co-localization of TIMP3 to NFTs provide additional evidence that TIMP3 may be important in NFT formation in AD.
The third validated candidate interleukin-1 receptor-associated kinase 1 (IRAK1), is an important mediator of interleukin 1 signaling. Inflammatory mechanisms, including complement activation and the release of numerous cytokines (IL1, IL6, and TNFα), are increased significantly in AD brain. Interestingly, IL-1 released from microglia promotes the phosphorylation of neuronal tau protein, suggesting a pathogenic role for microglia and IL-1 signaling in NFT formation and AD progression [20,31]. The upregulation of IRAK1 in AD NFT neurons provides a candidate pathway whereby IL-1 signaling leads to increased tau phosphorylation and, potentially, to NFT formation and neuronal cell death in AD. This makes IRAK1 and interesting candidate target to decrease tau phosphorylation and, possibly, NFT formation in AD.
Ongoing functional validation studies using siRNA and overexpression approaches will clarify a potential role for these proteins in NFT formation and AD pathogenesis and allow us to begin targeting early inducers of NFTs with therapeutics.
5. Summary
We have described results of a single-cell expression profiling approach to identifying genes whose dysregulation is associated with NFT pathology. This approach has both confirmed a few known associations and has identified several novel genes specifically associated with NFTs. Immunohistochemical approaches have confirmed the preferential localization of many of these genes at the protein level and have implicated diverse cellular processes in the formation of NFTs. These results lay the ground-work for more extensive validation efforts to delineate the precise functions of these candidate genes in NFT formation and neuronal cell death in AD. This approach can be utilized in a wide range of neurodegenerative diseases to isolate and characterize specific pathologically affected cell types, thereby garnering valuable new information about the genetic dysregulation that occurs during disease pathogenesis.
Acknowledgments
We would like to thank the National Institute on Aging’s Alzheimer’s Disease Centers program and the National Alzheimer’s Coordinating Committee for their invaluable assistance in accessing banked samples. We also thank Zhouwen Liu for assistance with statistical analyses and Phanindra Tangirala and Steven Day for their efforts to make this data publicly available. This study was supported in part by grants from the National Institute on Aging (#1-RO1-AG023193 to DAS; P30 AG19610 to EMR; P50 AG05681 to JCM; P01 AG03991 to JCM; AG05128 for the Duke University ADC), the National Alzheimer’s Coordinating Center (U01AG016976), and the Arizona Alzheimer’s Research Center (to EMR) under a collaborative agreement from the National Institute on Aging.
References
- 1.Ahonen M, Poukkula M, Baker AH, Kashiwagi M, Nagase H, Eriksson JE, et al. Tissue inhibitor of metalloproteinases-3 induces apoptosis in melanoma cells by stabilization of death receptors. Oncogene. 2003;22:2121–34. doi: 10.1038/sj.onc.1206292. [DOI] [PubMed] [Google Scholar]
- 2.Backstrom JR, Lim GP, Cullen MJ, Tokes ZA. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-beta peptide (1–40) J Neurosci. 1996;16:7910–9. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baum L, Masliah E, Iimoto DS, Hansen LA, Halliday WC, Saitoh T. Casein kinase II is associated with neurofibrillary tangles but is not an intrinsic component of paired helical filaments. Brain Res. 1992;573:126–32. doi: 10.1016/0006-8993(92)90121-o. [DOI] [PubMed] [Google Scholar]
- 4.Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–3. doi: 10.1038/343461a0. [DOI] [PubMed] [Google Scholar]
- 5.Caceres A, Potrebic S, Kosik KS. The effect of tau antisense oligonucleotides on neurite formation of cultured cerebellar macroneurons. J Neurosci. 1991;11:1515–23. doi: 10.1523/JNEUROSCI.11-06-01515.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer’s disease. Microsc Res Tech. 2000;50:305–15. doi: 10.1002/1097-0029(20000815)50:4<305::AID-JEMT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.Cau J, Faure S, Comps M, Delsert C, Morin N. A novel p21-activated kinase binds the actin and microtubule networks and induces microtubule stabilization. J Cell Biol. 2001;155:1029–42. doi: 10.1083/jcb.200104123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dan C, Nath N, Liberto M, Minden A. PAK5, a new brain-specific kinase, promotes neurite outgrowth in N1E-115 cells. Mol Cell Biol. 2002;22:567–77. doi: 10.1128/MCB.22.2.567-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Debure L, Vayssiere JL, Rincheval V, Loison F, Le Drean Y, Michel D. Intracellular clusterin causes juxtanuclear aggregate formation and mitochondrial alteration. J Cell Sci. 2003;116:3109–21. doi: 10.1242/jcs.00619. [DOI] [PubMed] [Google Scholar]
- 10.DeMattos RB, O’Dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, et al. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2002;99:10843–8. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143:777–94. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher RA. The design of experiments. 9. New York: Hafner Press; 1974. [Google Scholar]
- 13.Gorath M, Stahnke T, Mronga T, Goldbaum O, Richter-Landsberg C. Developmental changes of tau protein and mRNA in cultured rat brain oligodendrocytes. Glia. 2001;36:89–101. doi: 10.1002/glia.1098. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Wang KK. The calpain family and human disease. Trends Mol Med. 2001;7:355–62. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 15.Hulette CM, Welsh-Bohmer KA, Crain B, Szymanski MH, Sinclaire NO, Roses AD. Rapid brain autopsy. The Joseph and Kathleen Bryan Alzheimer’s disease research center experience. Arch Pathol Lab Med. 1997;121:615–8. [PubMed] [Google Scholar]
- 16.Iqbal K, Grundke-Iqbal I. Inhibition of neurofibrillary degeneration: a promising approach to Alzheimer’s disease and other tauopathies. Curr Drug Targets. 2004;5:495–502. doi: 10.2174/1389450043345254. [DOI] [PubMed] [Google Scholar]
- 17.Kempster S, Collins ME, Aronow BJ, Simmons M, Green RB, Edington N. Clusterin shortens the incubation and alters the histopathology of bovine spongiform encephalopathy in mice. Neu-roreport. 2004;15:1735–8. doi: 10.1097/01.wnr.0000134990.97051.22. [DOI] [PubMed] [Google Scholar]
- 18.Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;22:698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knops J, Kosik KS, Lee G, Pardee JD, Cohen-Gould L, McConlogue L. Overexpression of tau in a nonneuronal cell induces long cellular processes. J Cell Biol. 1991;114:725–33. doi: 10.1083/jcb.114.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synapto-physin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–11. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorenzl S, Albers DS, Chirichigno JW, Augood SJ, Beal MF. Elevated levels of matrix metalloproteinases-9 and -1 and of tissue inhibitors of MMPs TIMP-1 and TIMP-2 in postmortem brain tissue of progressive supranuclear palsy. J Neurol Sci. 2004;218:39–45. doi: 10.1016/j.jns.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Lorenzl S, Albers DS, LeWitt PA, Chirichigno JW, Hilgenberg SL, Cudkowicz ME, et al. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J Neurol Sci. 2003;207:71–6. doi: 10.1016/s0022-510x(02)00398-2. [DOI] [PubMed] [Google Scholar]
- 23.Lorenzl S, Albers DS, Relkin N, Ngyuen T, Hilgenberg SL, Chirichigno J, et al. Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer’s disease. Neurochem Int. 2003;43:191–6. doi: 10.1016/s0197-0186(03)00004-4. [DOI] [PubMed] [Google Scholar]
- 24.Manly BF. Randomization and Monte Carlo methods in biology. New York: Chapman and Hall; 1991. [Google Scholar]
- 25.Masliah E, Iimoto DS, Mallory M, Albright T, Hansen L, Saitoh T. Casein kinase II alteration precedes tau accumulation in tangle formation. Am J Pathol. 1992;140:263–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36:969–77. doi: 10.1038/ng1413. [DOI] [PubMed] [Google Scholar]
- 27.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Sasaki K, Doh-ura K, Wakisaka Y, Iwaki T. Clusterin/apolipoprotein J is associated with cortical Lewy bodies: immunohistochemical study in cases with alpha-synucleinopathies. Acta Neuropathol (Berl) 2002;104:225–30. doi: 10.1007/s00401-002-0546-4. [DOI] [PubMed] [Google Scholar]
- 29.Scaltriti M, Santamaria A, Paciucci R, Bettuzzi S. Intracellular clusterin induces G2–M phase arrest and cell death in PC-3 prostate cancer cells1. Cancer Res. 2004;64:6174–82. doi: 10.1158/0008-5472.CAN-04-0920. [DOI] [PubMed] [Google Scholar]
- 30.Smith MR, Kung H, Durum SK, Colburn NH, Sun Y. TIMP-3 induces cell death by stabilizing TNF-alpha receptors on the surface of human colon carcinoma cells. Cytokine. 1997;9:770–80. doi: 10.1006/cyto.1997.0233. [DOI] [PubMed] [Google Scholar]
- 31.Tanji K, Mori F, Imaizumi T, Yoshida H, Satoh K, Wakabayashi K. Interleukin-1 induces tau phosphorylation and morphological changes in cultured human astrocytes. Neuroreport. 2003;14:413–7. doi: 10.1097/00001756-200303030-00023. [DOI] [PubMed] [Google Scholar]
- 32.Wallace JA, Alexander S, Estrada EY, Hines C, Cunningham LA, Rosenberg GA. Tissue inhibitor of metalloproteinase-3 is associated with neuronal death in reperfusion injury. J Cereb Blood Flow Metab. 2002;22:1303–10. doi: 10.1097/01.WCB.0000040943.89393.c1. [DOI] [PubMed] [Google Scholar]