

Abstract
Arenaviridae is a family of enveloped viruses some of which are capable of causing hemorrhagic fever syndromes in humans. In this report, we demonstrate that treatment of host cells with the tyrosine kinase inhibitor genistein inhibits infection of cells with the New World arenavirus Pichindé (PICV). The greatest degree of inhibition was observed in pre-treated target cells, but modest inhibition of infection was also seen when drug was added to cultures up to 48 hours after infection. We show that PICV induced phosphorylation of the activating transcription factor-2 protein (ATF-2) and cyclic adenosine monophosphate response element-binding protein (CREB) is inhibited following genistein treatment. Lastly, genistein treatment also inhibited transduction of cells with pseudotyped retrovirus particles expressing envelope proteins of the Old World arenavirus Lassa virus. These results demonstrate that kinase activity is required for arenavirus infection and that therapeutics designed to inhibit kinase activity should be explored.
Keywords: arenavirus, Lassa virus, Pichindé virus, genistein, CREB, ATF-2
Arenaviridae are enveloped, single-stranded bipartite RNA viruses that include members capable of causing hemorrhagic fever syndromes in humans. Because of the potential for aerosol production, person-to-person spread and the ability to cause lethal or debilitating disease in humans, several of these viruses are currently listed as CDC biothreat agents and NIAID category A priority pathogens. Ribavirin and supportive care are the only therapeutic options for arenavirus hemorrhagic fevers in humans. However, there remains a 14–19% mortality rate among severely ill patients infected with the Old World arenavirus Lassa (LASV) despite oral or intravenous therapy (McCormick et al., 1986). Studying LASV has traditionally been difficult due to the BSL-4 laboratory restrictions and due to the fact that mice are poor models for hemorrhagic fevers (Peters, 1997). Because of this, we have used the Clade A New World arenavirus Pichindé (PICV), which produces a pathology in guinea pigs similar to that seen in human Lassa fever (Jahrling et al., 1981), as a model system.
The Old World arenavirus LASV has been shown to enter and infect cells by binding to the cellular α-dystroglycan receptor (Cao et al., 1998), while no receptor has been identified for PICV. However, a new report demonstrated that the New World pathogenic arenaviruses Junin, Machupo, and Guanarito utilize the cellular Transferrin receptor 1 (TfR 1) as the receptor (Radoshitzky et al., 2007). Once these viruses have bound to the cellular receptor, protein phosphorylation likely plays an important role and downstream cell signaling events may be required for priming the cell to facilitate viral replication. We have previously investigated the global kinase/phosphorylation response to PICV infection (Bowick et al., 2007). By comparing the activity of the macrophage kinome following PICV infection of guinea pigs, we have shown the predicted phosphorylation states of a number of proteins from cell surface receptors to downstream transcription factors. Specifically, we have observed activation of kinases that phosphorylate ATF-2 in PICV-infected guinea pigs (Bowick et al., 2007). The transcription factor CREB, a member of the AP-1 transcription factors, has also been implicated in cell signaling induced by PICV infection (Bowick et al., 2006). We have also shown involvement of the epidermal growth factor receptor (EGFR), which is known to phosphorylate Eps15 (Torrisi et al., 1997), and serum response factor, which lies downstream of the Ras/Raf/meiosis-specific serine/threonine-protein kinase (MEK) pathway, in signaling networks induced by PICV infection (Bowick, 2007). These two pathways may well have central roles in specific entry mechanisms (Torrisi, 1999; Daaka, 1998).
We aimed to determine whether inhibition of tyrosine kinase activity in vitro inhibited viral entry and/or productive infection. Genistein is a tyrosine kinase inhibitor that appears to inhibit infection of some viruses like Simian virus 40 (SV40) (Akiyama et al., 1987; Damm et al., 2005; Pelkmans et al., 2002). In this report, we employed genistein to examine the role of cellular tyrosine kinase activity in arenavirus infection. We hypothesize that entry and infection of the arenaviruses PICV and LASV requires cellular phosphorylation events. Arenavirus-induced tyrosine kinase activity may provide a potential target for therapeutic strategies aimed at inhibiting arenaviral infection.
When cells were pre-treated with genistein for 1 hour prior to addition of virus, we observed a decrease in PICV nucleoprotein (NP) and glycoprotein expression compared to untreated cells, which may be due an inhibition in viral infection or a reduction in viral replication (Fig 1A). The genistein pre-treatment did not alter cellular TfR levels in Vero cells. Plaque assay analyses showed a 90% reduction in viral titers in genistein pre-treated Vero cells infected with PICV when compared to untreated or vehicle-treated Vero cells (Fig 1B). Sindbis virus (SIN) titers were not affected by genistein pre-treatment as previously shown (Lecot et al., 2005) (Fig 1C). Moreover, using trypan blue staining, no cellular toxicity was observed in Vero cells treated with 100 μM of genistein (data not shown). We next studied whether treating Vero cells with genistein post infection (p.i) inhibited viral infection. At a multiplicity of infection of 1.0, we observed a 66.5% reduction in viral titers when cells were exposed to drug beginning at time 0 relative to infection, 56.9% reduction when cells treated with genistein 30 min p.i., and a 33.6% reduction when cells were treated with genistein 8 h p.i. (Fig 1D). When genistein was added to cultures 48 hours after infection, 73% reduction in virus yield was observed (Fig 1E). These data suggest that genistein treatment of cells post viral infection may inhibit events downstream of entry. Altogether, these data demonstrate that a reduction in PICV infection occurs when cells are treated with genistein either pre- or post-infection.
Fig 1.


PICV infection is inhibited in cells pre-treated with genistein. Vero cells were pre-treated with 100 μM of genistein for 1 h prior to incubation with PICV (moi 1.0). For western blot experiments, genistein remained present throughout the experiment. (A) Genistein-treated and untreated Vero cells were infected with PICV for 72 h and western blot analyses were performed using the following primary antibodies: monoclonal antibody 3B3.1 (recognizes the PICV NP), the polyclonal anti-peptide antibody GPC (59–79) (recognizes the glycoprotein precursor (GPC) and GP1) (both donated by Dr. M. Buchmeier), and the TfR antibody (Zymed Laboratories, Inc, San Francisco, CA). (B) Supernatants from PICV infected, genistein-treated and untreated Vero cells were harvested 72 h p.i. and analyzed by plaque assays on Vero cells. (C) Supernatants from SIN-infected, genistein (100 μM) pre-treated or untreated Vero cells were harvested 24 h post-infection and analyzed by plaque assay analyses on BHK-21 cells. (D) Vero cells were treated with genistein at various timepoints compared to PICV infection (moi 1.0) and supernatants containing virus were harvested at 72 h after genistein treatment for plaque assays. (E) Vero cells were treated with genistein 48 h after infection, washed to remove any drug, and incubated for an additional 48 h. Supernatants containing virus were harvested for plaque assays. An asterisk represents a p<0.05 as determined by a Student’s t-test when comparing PICV-infected cells to genistein-treated, PICV-infected cells.
In order to further characterize the effect of genistein treatment on the cellular response to PICV infection, we investigated the effect of genistein on two transcription factors which have been implicated in PICV infection, and on global levels of tyrosine phosphorylation. We assayed whole cell extracts prepared from uninfected Vero cells, or cells which had been infected with PICV with or without genistein treatment using antibodies to phospho-ATF-2 Thr-71, phospho-CREB Ser-133, and phosphotyrosine. Figure 2 shows the inhibition of phosphorylation of the transcription factors ATF-2 and CREB following infection and treatment with genistein, compared to infection alone. Total levels of phospho-tyrosine were also reduced in genistein-treated infected cells compared to untreated infected cells (data not shown). Interestingly, levels of phosphorylated ERK (p42 and p44 MAPK) were not inhibited by genistein treatment; conversely pERK levels appeared to increase in genistein-treated, infected cells (figure 2).
Fig 2.
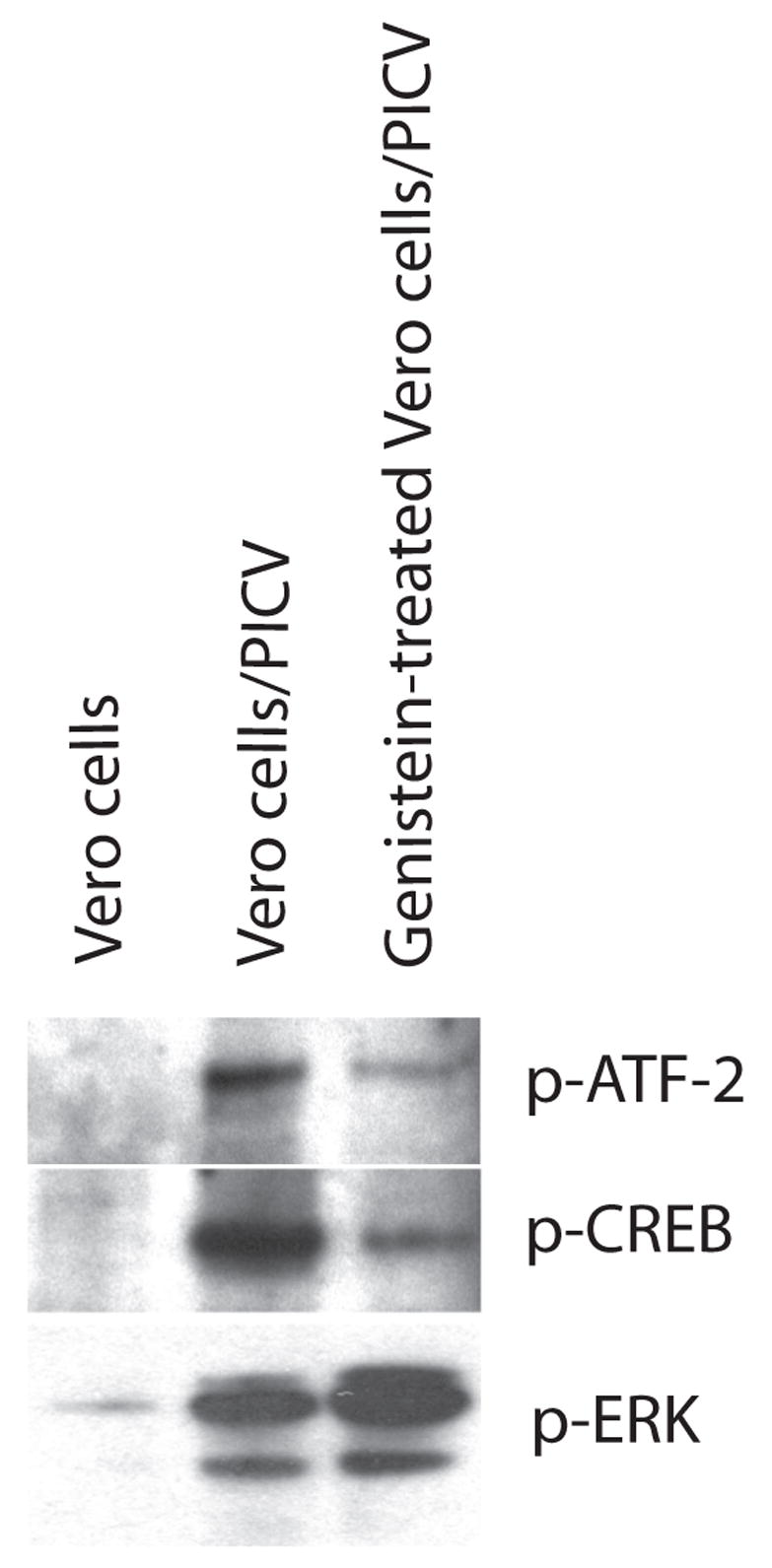
Genistein has different effects on PIC virus-induced tyrosine phosphorylations of ATF-2, CREB, and ERK. Whole cell extracts from uninfected Vero cells, PICV-infected Vero cells (moi 1.0, 4 hours pi), or genistein-treated/PICV-infected Vero cells were immunoblotted using antibodies against the phosphorylated forms of ATF-2 (Thr-71), CREB (Ser-133), or ERK (Thr-202 and Tyr-204) (all purchased from Cell Signaling, Danvers, MA). In experiments involving genistein, cells were pre-treated with 100 μM of genistein for 1 h prior to the addition of PICV and drug remained in the culture throughout the experiment.
To determine whether genistein inhibits the entry of the Old World arenavirus LASV, we utilized a replication-deficient LASV-MLV retroviral pseudotype system. Pseudotypes were generated by co-transfecting 293FT cells using the TranIT-LT-1 transfection method with the following plasmids: the pLGP which encodes the LASV(Josiah) complete glycoprotein (GPC) envelope (donated by Dr. M. Buchmeier, The Scripps Research Institute, CA), the pGAG-POL encoding the MLV gag/pol (Kolokoltsov et al., 2005) (a gift from Dr. R. Davey, UTMB, Galveston, TX), and the pψ β-gal plasmid encoding the β-galactosidase marker gene construct (Strategene, La Jolla, CA). Marker gene expression in pseudotype transduced Vero cells was inhibited by genistein in a dose dependent manner (Fig 3). However, LASV-MLV transduction was not affected by pre-treating cells with 0.1% DMSO (the genistein carrier) indicating that the inhibition observed was due to the drug and not the carrier. Overall, these results suggest that LASV entry requires tyrosine kinase activity.
Fig 3.

Entry of the LASV-MLV pseudotype is inhibited in genistein-treated cells. Genistein-treated and untreated Vero cells were incubated with LASV-MLV pseudotypes for 48 h. After the incubation, the cells were fixed with an X-gal stain containing: 0.05 M potassium ferrocyanide and potassium ferricyanide, 2 mM X-gal, and PBS. β-gal expressing cells were counted and expressed as colony forming units/ml (CFU/ml). (A) Vero cells were treated with various concentrations of genistein for 1 h and infected with the LASV-MLV pseudotype. (B) Vero cells were treated with 100 μM of genistein or 0.1% DMSO and transduced with the LASV-MLV pseudotype. An asterisk represents a p<0.05 as determined by a Student’s t-test.
We have previously shown that PICV infection of guinea pigs resulted in ERK phosphorylation and an activation of kinases that phosphorylate ATF-2 in guinea pig peritoneal cells sequential days after infection (Bowick et al, 2007). Here, we show that PICV infection induces ATF-2 phosphorylation in cultured cells and that this phosphorylation is sensitive to genistein. We have also shown in this study that PICV infection results in phosphorylation of CREB (cAMP response element binding). The transcription factor CREB binds to cAMP response elements in DNA. This protein has been implicated in a cellular signaling network induced by PICV infection (Bowick et al, 2006). The reduced serine phosphorylation observed following genistein treatment could be due to reduced upstream activation or genistein-mediated inhibition of tyrosine phosphorylation of upstream signaling intermediates. The increased phosphorylation of ERK1 and ERK2 seen following genistein treatment and infection could be as a result of other genistein-mediated effects activating the ERK pathway, or as a result of genistein inhibiting an ERK antagonistic pathway. However, the lack of a reduction in ERK phosphorylation demonstrates that there is a degree of specificity to the kinase inhibition mediated by genistein.
Results from our study suggest that arenavirus entry events, as well as later events in the viral life cycle, are inhibited by genistein. The fact that a moderate degree of virus inhibition was observed when cultured cells infected at a high moi and treated at various time points post-infection offers some support for therapeutic potential of this approach. A published report utilized guinea pigs to demonstrate the use of genistein for the treatment of allergic inflammation without causing any detrimental effects on the animals (Duan et al., 2003). Thus, we can potentially utilize the LASV and PICV-guinea pig model to study the effects of genistein in vivo. Future studies will focus on determining which specific tyrosine kinases genistein inhibits during arenaviral infection and the drug efficacy in an animal model. Altogether, this study demonstrates that tyrosine kinase activity is required for cellular infection with the arenaviruses LASV and PICV, and may lead to a design of potential anti-arenavirus therapeutics aimed at disrupting cellular tyrosine kinase activity.
Acknowledgments
This work was supported by the NIAID Training in Emerging and Reemerging Infectious Diseases T32 AI07536 and NIH UO1-AI054827.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. Journal of Biological Chemistry. 1987;262:5592–5595. [PubMed] [Google Scholar]
- Bowick G, Fennewald S, Elsom B, Aronson J, Luxon B, Gorenstein D, Herzog N. Differential signaling networks induced by mild and lethal hemorrhagic fever virus infections. Journal of Virology. 2006;80:10248–10252. doi: 10.1128/JVI.01384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowick G, Fennewald S, Scott E, Zhang L, Elsom B, Aronson J, Spratt H, Luxon B, Gorenstein D, Herzog N. Identification of differentially activated cell-signalling networks associated with Pichinde virus pathogenesis by using systems kinomics. Journal of Virology. 2007;81:1923–1933. doi: 10.1128/JVI.02199-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Henry M, Borrow P, Yamada H, Elder J, Ravkov E, Nichol S, Compans R, Campbell K, Oldstone M. Identification of alpha-dystoglygan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science. 1998;282:2079–2081. doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- Damm E, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A. Clathrin- and caveolin-1-independent endocytosis: entry of simain virus 40 into cells devoid of caveolae. Journal of Cell Biology. 2005;168:477–488. doi: 10.1083/jcb.200407113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Kuo I, Selvarajan S, Chua K, Bay B, Wong W. Antiinflammatory effects of genistein, a tyrosine kinas inhibitor, on a guinea pig model of asthma. American Journal of Respiratory and Critical Care Medicine. 2003;167:185–192. doi: 10.1164/rccm.200205-420OC. [DOI] [PubMed] [Google Scholar]
- Jahrling P, Hesse R, Rhoderick J, Elwell M, Moe J. Pathogenesis of a Pichinde virus strain adapted to produce lethal infections in guinea pigs. Infection and Immunity. 1981;32:872–880. doi: 10.1128/iai.32.2.872-880.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolokoltsov A, Weaver S, Davey R. Efficient functional pseudotyping of oncoretroviral and lentiviral vectors by Venezuelan equine encephalitis virus envelope proteins. Journal of Virology. 2005;79:756–763. doi: 10.1128/JVI.79.2.756-763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecot S, Belouzard S, Dubuisson J, Rouille Y. Bovine viral diarrhea virus entry is dependent on clathrin-mediated endocytosis. Journal of Virology. 2005;79:10826–10829. doi: 10.1128/JVI.79.16.10826-10829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick J, King I, Webb P, Johnson K, O’Sullivan R, Smith E, Tripple S, Tong T. Lassa fever: Effective therapy with Ribavirin. New England Journal of Medicine. 1986;314:20–26. doi: 10.1056/NEJM198601023140104. [DOI] [PubMed] [Google Scholar]
- Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–539. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- Peters C. Viral hemorrhagic fevers. In: Nathanson N, Ahmed R, Gonzalez-Scarano F, Griffin D, Holmes K, Murphy F, Robinson H, editors. Viral Pathogenesis. Lippincott-Raven; Philadelphia: 1997. pp. 779–799. [Google Scholar]
- Radoshitzky S, Abraham J, Spiropoulou C, Kuhn J, Nguyen D, Wenhui L, Nagel J, Schmidt P, Nunberg J, Andrews N, Farzan M, Choe H. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrisi M, Lotti L, Belleudi F, Gradini R, Salcini A, Confalonieri S, Pelicci P, De Fiore P. Eps15 is recruited to the plasma membrane upon epidermal growth factor receptor activation and localizes to components of the endocytic pathway during receptor internalization. Molecular Biology of the Cell. 1997;10:417–434. doi: 10.1091/mbc.10.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]