

Abstract
Purpose
To determine the pharmacokinetics, tissue, and blood distribution of rapamycin PEG-block-poly(ε-caprolactone) (PEG-b-PCL) micelle formulations with and without the addition of α-tocopherol compared to control rapamycin in Tween 80/PEG 400/N,N-dimethylacetamide (DMA) (7:64:29).
Methods
Rapamycin was incorporated at 10% w/w into PEG-b-PCL micelles (5:10 kDa) using a solvent extraction technique. The co-incorporation of 2:1 α-tocopherol:PEG-b-PCL was also studied. Rapamycin was quantified utilizing LC/MS in a Waters XTerra MS C18 column with 32-desmethoxyrapamycin as the internal standard. Male Sprague Dawley rats (N = 4 per group; ~200 g) were cannulated via the left jugular and dosed intravenously (IV) with the rapamycin control and micelle formulations (10 mg/kg, 1:9 ratio for rapamycin to PEG-b-PCL). For tissue distribution 24 h after IV dosing, whole blood, plasma, red blood cells, and all the representative tissues were collected. The tissues were rapidly frozen under liquid nitrogen and ground to a fine powder. The rapamycin concentrations in plasma and red blood cells were utilized to determine the blood distribution (partition coefficient between plasma and red blood cells). For the determination of the pharmacokinetic parameters, blood, plasma, and urine samples were collected over 48 h. The pharmacokinetic parameters were calculated using WinNonlin® (Version 5.1) software.
Results
Rapamycin concentrations were considerably less in brain after administration of both micelle formulations compared to a rapamycin in the Tween 80/PEG 400/DMA control group. There was a 2-fold and 1.6-fold increase in the plasma fraction for rapamycin micelles with and without α-tocopherol. There was a decrease in volume of distribution for both formulations, an increase in AUC, a decrease in clearance, and increase in half life respectively for rapamycin in PEG-b-PCL + α-tocopherol micelles and in PEG-b-PCL micelles. There was no mortality with the micelle formulations compared to 60% mortality with rapamycin in Tween 80/PEG 400/DMA.
Conclusions
The decreased distribution into the brain of rapamycin in PEG-b-PCL micelles may ameliorate rapamycin neurotoxicity. Both micelle formulations increase rapamycin distribution in plasma, which could facilitate access into solid tumors. The micellar delivery systems of rapamycin impart in vivo controlled release, resulting in altered disposition, and dramatically reduced mortality.
Keywords: Rapamycin, Nanocarrier, Poly(ethylene glycol)-b-poly(ε-caprolactone), Polymer micelle, Pharmacokinetics, Delivery
Introduction
Rapamycin (Sirolimus) (Fig. 1a) is a large (MW 914 g/mol) lipophilic (X log PO/W 5.77) carboxylic lactone-lactam macrolide antibiotic that was first isolated from the soil bacteria Streptomyces hygroscopicus from the soil of the Vai Atari region of Rapa Nui (Easter Island, Chile) more than 25 years ago [39]. It was originally developed as an antifungal agent [10, 25], but posterior investigations reported its potent antiproliferative and immunosuppressive effects in vitro [14, 27, 32, 34] and in vivo [25, 28, 46] allowing it to be currently utilized as conjunction therapy with cyclosporine A in kidney and liver transplant recipients [31]. Further investigations also revealed that rapamycin has a unique anti-tumor mechanism through the binding with FKBP12 and the inhibition of mammalian target of rapamycin (mTOR), which is a central regulator of proliferation, apoptosis [5, 11, 37, 41], and cell growth by inducing cell-cycle arrest in the G1 phase [7]. These latter pharmaco-logical properties have allowed rapamycin to be recognized recently as a possible chemotherapeutic agent against many solid tumor types including breast [21], colon [15], prostate [50], and renal cell carcinomas [38] with a typical IC50 < 50 nM.
Fig. 1.
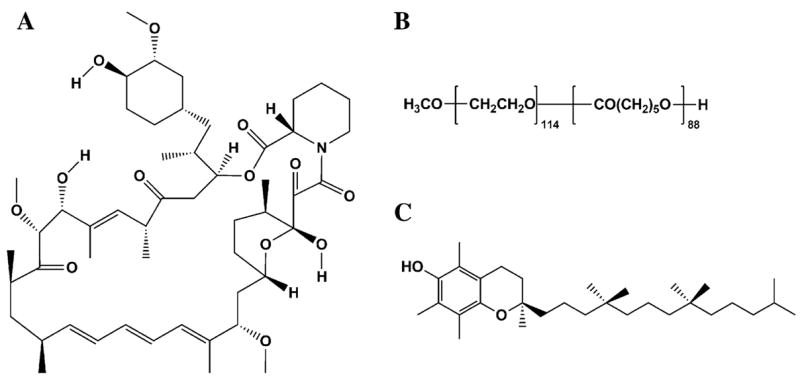
Chemical structures of rapamycin (a), PEG-b-PCL (b), and α-tocopherol (c)
Even though rapamycin offers promising pharmacological activities, it faces two major pharmacokinetic limitations. First, the clinical development of rapamycin has been hindered by its poorly water soluble (2.6 μg/ml) behavior [34, 40] making previous attempts to develop intravenous (IV) formulations difficult [43] but still allowing the development of the currently employed oral solution and tablet forms [30]. However, rapamycin has a low oral bioavailability (<15%) [35, 45, 49] limiting the development of tablet formulations except for low-dosage treatments such as immunosuppression in renal and liver transplant recipients [31]. Furthermore, this low bioavailability has been related to rapamycin sensitivity to gastric acid, partial intestinal absorption, and mainly first-pass hepatic metabolism (less than 3% excreted in urine) [24]. The second limitation is the strong partition of rapamycin to erythrocytes, cells, and tissue with red blood cell to plasma ratios of 36/1 or plasma to red blood cell ratio (Kd) of ca. 0.05, which may hinder accessibility into solid tumor sites [45, 46, 49]. Rapamycin exhibits a preferential distribution into red blood cells (94.5%) compared to plasma (3.1%), lymphocytes (1.01%) and granulocytes (1.0%) [30, 48]. Furthermore, this strong uptake and sequestration of rapamycin into red blood cells (RBC) can be partially explained by the high content of immunophillins [26] and lipoproteins [23] in RBC. The strong partition into cells and tissues can be attributed to two factors. The lipophilic nature of rapamycin allows it to be widely distributed into lipid membranes in the body as well as inducing its extensive apparent volume of distribution (5.6–16.7 l/kg) [17, 30, 49] of rapamycin. The extensive tissue distribution of rapamycin has been confirmed by studies performed in rats where tissue to blood partition coefficients (Kp) were reported to be more than 40 [35]. However, these partitioning determinations have not been performed in humans and the relationship between the concentration of rapamycin in tissues and the effictive and toxic effects of rapamycin is currently unknown. A single report demonstrated that rapamycin induces toxic effects on brain cell metabolism in vitro [42], but these observations have not yet been reported clinically. Therefore, a better understanding of the tissue distribution of rapamycin is warranted to try to establish a plausible relationship between its toxic effects and biodistribution. Furthermore, this extensive partitioning into tissues provides a relatively long half-life of more than 5 h up to 87 h [9, 22, 24] that permit once-a-day dosing, a delay in reaching steady-state concentrations [24, 51], a wide inter- and intra-patient variability in drug clearance (0.090–0.339 l/h kg) [9, 17, 24] and a less than optimal correlation between blood or plasma concentrations and dose [30].
It is because of these limitations that different approaches have been taken to improve the formulation and delivery of rapamycin. One of these approaches has been the development of Temsirolimus (CCI-779), a water-soluble rapamycin ester that has demonstrated promise in early Phase-I trials [1]. However, CCI-779 still has limited solubility in water, ~120 μg/ml, requiring the use of ethanol as a co-solvent [36] for IV formulations [33]. Moreover, phase-I trials have demonstrated that the CCI-779 prodrug is rapidly hydrolyzed by plasma esterases back into rapamycin and can thus redistribute and partition into blood erythrocytes [36, 49], which may reduce accumulation into solid tumor sites. Even though, phases I and II clinical studies have reported significant changes in the pharmacokinetic profile (5-fold increase in Cmax, 5-fold decrease in tmax, 3-fold decrease in t1/2, and 1-fold decrease in AUC) [20, 36], high inter-patient variability and mild to moderate side effects such as neurotropenia, thrombocytopenia, diarrhea, manic-depressive syndrome, and diarrhea were observed [20, 36]. Furthermore, some of the side effects were exacerbated compared to the observed side effects after rapamycin administration: headache and abdominal pain [16, 44], skin disorders, nocturnal calf cramps, and muscle aching [18]. Therefore, the necessity to explore other formulation alternatives to improve the pharmacokinetic and biodistribution profile, diminish the preferential partition of rapamycin into erythrocytes, and reduce some of the toxic side-effects is warranted.
The therapeutic utility of rapamycin could be optimized by novel nanoformulated PEG-b-poly(ε-caprolactone) micelle delivery into solid tumors. These nanoparticle polymeric micelle delivery systems may optimally accumulate at solid tumors by the enhanced permeability and retention (EPR) effect. Therefore, in the present studies, we optimized the incorporation of rapamycin into PEG-b-PCL micelles, which we have previously reported to effectively encapsulate rapamycin [19]. Rapamycin loaded PEG-b-PCL micelles were prepared using a scalable solvent-extraction technique that resulted in high drug loading, good stability, and extended release [19]. The present study has the objective to determine the pharmacokinetics, tissue and blood distribution, and assess any observable toxicities after administration of rapamycin PEG-block-poly(ε-caprolactone) (PEG-b-PCL) (Fig. 1b) micelle formulations with and without the addition of α-tocopherol (Fig. 1c) compared to control rapamycin in Tween 80/PEG 400/N,N-dimethylacetamide (DMA) (7:64:29).
Materials and methods
Rapamycin formulation in PEG-b-PCL micelles
Rapamycin loaded PEG-b-PCL micelles were prepared by a solvent extraction technique as previously described [19]. Poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) (5,000:10,500, Mw/Mn 1.18, JCS Biopolytech, Toronto, Ontario) and rapamycin (LC Laboratories, Woburn, MA) were dissolved in a minimum volume of acetone and added drop-wise to vigorously stirred ddH2O using a syringe pump. The organic solvent was removed by stirring under an air purge. After removing the organic solvent, PEG-b-PCL micelle solutions were made up to 5% w/v dextrose and passed through a 0.22-μm polyestersulfone filter to remove unincorporated drug and enable sterilization. Solutions were protected from light and stored at 4°C, protected from freezing, until administered to rats. Micelles containing α-tocopherol (Sigma Chemical Co., St. Louis, MO, USA) were made in the same manner, with the addition of 10:1 α-tocopherol:PEG-b-PCL (mol: mol) to the organic phase [19]. In a typical experiment, 12.5 mg rapamycin and 112.5 mg PEG-b-PCL (1:9 w/w) were dissolved in 1 ml of acetone and added (50 μl/min) to 6 ml of ddH2O. The resulting clear solution was air purged to ca. 3 ml and made up to 6.25 ml (2 mg/ml rapamycin final). The drug solution was made isotonic with 300 mg dextrose (5% w/v) and then filter-sterilized. All rapamycin and PEG-b-PCL formulations in this study, with and without tocopherol, were 1:9 w/w rapamycin to PEG-b-PCL, i.e. 1 mg/kg rapamycin and 9 mg/kg PEG-b-PCL.
The incorporation of rapamycin into PEG-b-PCL micelles was verified by equivalent retention times in ultraviolet (UV) and refractive index (RI) chromatographs from gel permeation chromatography. PEG-b-PCL micelles were injected on an OHpak SB-806M GPC column (20 μl injections, 0.5 mM PEG-b-PCL, 0.75 ml/min of 10 mM HEPES eluent, pH 7.0, 30°C) (Shodex, Kawasaki, Japan) and detected by RI and UV absorbance (277 nm). Rapamycin loading into PEG-b-PCL micelles was quantitatively determined by reverse-phase HPLC (Ace 3-μm C18 4.6 × 50 mm) using a 50 mM acetic acid—MeOH gradient (25:75—0:100, 50°C, 277 nm detection). Hydrodynamic diameters of PEG-b-PCL micelles were determined by dynamic light scattering (DLS) (NICOMP 380 ZLS, Particle Sizing Systems, Santa Barbara, CA). Data were analyzed by intensity-weighted Gaussian distribution fitting (NICOMP Version 1.76). Measurements were made for a minimum of 15 min or at least 1 × 106 counts in channel 1.
A control formulation for IV administration of rapamycin was made by dissolving rapamycin in DMA (1% w/v) and adding to a solution of PEG 400 and Tween 80 (final 29:64:7 DMA:PEG:Tween), based on the rodent pharmacokinetics and distribution studies by Kahan et al. [6, 35]. In a typical experiment, 15 mg rapamycin was completely dissolved in 1.5 ml DMA, and added to a premixed solution of 3.375 ml PEG 400 and 0.375 ml Tween 80. The resulting mixture was vortexed until clear and passed through a 0.45-μm PTFE syringe filter (final 2.8 mg/ml rapamycin). The concentration of rapamycin was verified by reverse-phase HPLC as above.
Measurement of rapamycin concentrations in blood, plasma, urine, and tissue samples by LC/MS
The protocol previously described by Annesley and Clayton [4] was slightly modified. For our purpose, 100 μl of whole blood, plasma, calibrator or control, or 100 mg of tissue was added in a regular polypropylene microcentrifuge tube. Then, 250 μl of deionized water, 250 μl of aqueous 0.1 M zinc sulfate, and 500 μl methanol containing the internal standard (32-desmethoxyrapamycin) were added. The mixture was vortexed for 30 s, and the tubes were left at room temperature for 4 min. Then, the tubes were centrifuged for 4 min, and the colorless supernatant was injected into the LC/MS system. The analyses were carried out on an Agilent 1100 Series LC/MSD. In the positive-ion mode the monitored single plot transitions (m/z) were: rapamycin 936→937, and 32-desmethoxyrapamycin 816→906. Separation was performed with a Waters XTerra MS C18 2.1 × 100 mm column maintained at 40°C. The injection volume was 20 μl with a flow rate of 0.4 ml/min. The mobile phases were: (A) 2 mM ammonium acetate and 0.1% formic acid in water, (B) 2 mM ammonium acetate and 0.1% formic acid in methanol, and (C) acetonitrile. The gradient program was 50% A and 50% B for 0.10 min, followed by an immediate change to 10% A and 90% B at 0.11 min. At 1.80 min, the mobile phase was increased from 90% B to 100% B, and at 6.10 min, the mobile phase was changed to 100% C to clean the column. At 9.10 min, the mobile phase reverted to 50% A and 50% B. Rapamycin eluted at ca. 4.46 min and 32-desmethoxyrapamycin eluted at ca. 5.50 min. For quantification in tissues, concentration ranges of 0.001–10 μg/ml were employed in the calibration samples. For blood, plasma, and urine, the calibration samples were prepared at a concentration of 0.001–50 μg/ml. For all runs, quality control samples were incorporated to ensure the integrity of the results. Standard curves were linear (R2 = 0.999) and bias and precision data were less than 10%. The recovery was determined by enriching rat blood, plasma, urine, and tissue extract samples with rapamycin (0.05, 0.5, and 5 μg/ml), and comparing the peak areas of the extracted samples to the areas obtained for injections of drug in saline (corrected for volume or dilution). It was observed that the recoveries for the different biological matrices ranged from 94 to 97%, which are comparable results to previous studies [4, 13].
Surgical procedures
Male Sprague-Dawley rats (200–240 g) were obtained from Simonsen Labs (Gilroy, CA, USA) and given food (Purina Rat Chow 5001) and water ad libitum in our animal facility for at least 3 days before use. Rats were housed in temperature-controlled rooms with a 12 h light/dark cycle. The day before the pharmacokinetic experiment the right jugular veins of the rats were catherized with sterile silastic cannula (Dow Corning, Midland, MI, USA) under halothane (Sigma Chemical Co. St. Louis, MO, USA) anesthesia. This involved exposure of the vessel prior to cannula insertion. After cannulation, the Intramedic PE-50 polyethylene tubing (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) connected to the cannula was exteriorized through the dorsal skin. The cannula was flushed with 0.9% saline. The animals were transferred to metabolic cages and were fasted overnight. Animal use protocols were approved by The Institutional Animal Care and Use Committee at Washington State University, in accordance with “Principles of laboratory animal care” (NIH publication No. 85-23, revised 1985).
Pharmacokinetic study
To assess the effect of formulation on the pharmacokinetic parameters, rats were cannulated as described above and dosed intravenously with rapamycin (10 mg/kg) dissolved in DMA, PEG, and Tween 80 (control formulation); in PEG-b-PCL formulation (1 mg/kg rapamycin and 9 mg/kg polymer); or in PEG-b-PCL co-incorporated with α-tocopherol (PEG-b-PCL + α-tocopherol formulation) (n = 4 for each treatment group). The rats were given between 0.5 and 1 ml of each formulation as IV bolus within 5 min. After dosing, serial blood samples (~0.30 ml) were collected from the cannula at 0, 1, and 30 min, then 1, 2, 4, 6, 12, 24, and 48 h after IV administration, and the cannula flushed with 0.9% saline. After dosing and after each serial blood sampling, blinded observers were present to record any visible behavior, bleeding, or overall appearance of the animal as signs of acute toxicity. Each blood sample was divided into one 0.1 ml fraction and one 0.2 ml fraction, the first fraction (0.1 ml) was collected into regular polypropylene microcentrifuge tubes and labeled as blood sample and stored at −70°C until further LC/MS analysis. The second fraction (0.2 ml) was collected in heparanized tubes (Monoject, Mansfield MA) and following centrifugation, the plasma and red blood cell (RBC) fractions were collected and stored at −70°C until analyzed. Urine samples were also collected at 0, 2, 6, 12, 24, 24, and 48 h following IV administration and were stored at −70°C until analyzed.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed using data from individual rats for which the mean and standard error of the mean (SEM) were calculated for each group. The elimination rate constant (KE) was estimated by linear regression of the blood or plasma concentrations in the log-linear terminal phase. In order to estimate the blood or plasma concentrations (C0) immediately after rapamycin IV dosing, a two-compartmental model was fitted to the plasma or whole blood concentration versus time data using WinNonlin® software (Version 5.1). The estimated C0 was then used with the actual measured blood or plasma concentrations to determine the area under the blood or plasma concentration–time curve (AUC). The AUC0–∞ was calculated using the combined log-linear trapezoidal rule for data from time of dosing to the last measured concentration, plus the quotient of the last measured concentration divided by KE. Non-compartmental pharmacokinetic methods were used to calculate mean residence time (MRT by dividing AUMC0-∞ by AUC0-∞), clearance (CL by dividing dose by AUC0-∞) and volume of distribution (Vdβ by dividing CL by KE). Based on the cumulative urinary excretion, the fraction excreted in urine (fe by dividing the total cumulative amount of rapamycin excreted in urine (ΣXu) by the dose), renal clearance (CLr by multiplying fe by CL), hepatic clearance (CLhepatic by subtracting CLr from CL), extraction ratio (ER by dividing CLhepatic by hepatic flow (Q)). The mean hepatic blood flow (Q) is approximately 3.22 lL/h kg [12]. Using the hematocrit in rat [12] of 0.48, this yields a mean hepatic plasma flow of 1.74 l/h kg. Therefore, depending on plasma or blood the correct hepatic flow (Q) needs to be employed. The estimated bioavailability could be calculated utilizing two methods: either by subtracting the ER from 1 or by using the relationships F = Q/(Q + CLint) and CLhepatic = (Q × CLint)/(Q + CLint), been CLint the intrinsic clearance.
Biodistribution study
To assess the effect of formulation on the tissue distribution of rapamycin, rats were cannulated and intravenously administered with rapamycin dissolved in control formulation; in PEG-b-PCL formulation; or in PEG-b-PCL + α-tocopherol formulation at the same dose (10 mg/kg) as pharmacokinetic studies. At 24 h after formulation injection, each animal (n = 4 for each group) was anaesthetized and exsanguinated by cardiac puncture. Brain, lungs, heart, liver, spleen, kidneys, urinary bladder, muscle as well as samples of blood and plasma were collected. Tissue samples were blotted with paper towel, washed in ice-cold saline, bottled to remove excess fluid, weighed and rapidly frozen in liquid nitrogen, pulverized to a fine powder with a mortar and pestle under liquid nitrogen and stored at −70°C until assessed for rapamycin concentration by LC/MS analysis. Tissue to plasma concentration ratios (Kp) were calculated by dividing rapamycin concentration in each tissue by rapamycin concentration in plasma for individual animals in the biodistribution studies.
Blood distribution
To assess the effect of formulation on the blood distribution (partition coefficient between plasma and red blood cells) of rapamycin, rats were cannulated and intravenously administered with rapamycin dissolved in DMA, PEG, and Tween 80 (control formulation); in PEG-b-PCL formulation; or in PEG-b-PCL + α-tocopherol formulation at the same dose (10 mg/kg) as pharmacokinetic studies. A total of 24 rats were used for this experiment: four rats per time-point, two time-points (1 min and 24 h) were collected, and three formulations were dosed. One minute after dosing, the corresponding rats were anaesthetized and exsanguinated by the cannula. At 24 h post-dose, the same procedure was followed. After each collection, the blood samples were collected in heparanized tubes (Monoject, Mansfield, MA) and following centrifugation, the plasma and red blood cell (RBC) fractions were collected and stored at −70°C until LC/MS analysis.
Data analysis
Compiled data were presented as mean and standard error of the mean (mean ± SEM). Where possible, the data were analyzed for statistical significance using NCSS Statistical and Power Analysis software (NCSS, Kaysville, UT). Student’s t-test was employed for unpaired samples with a value of P < 0.05 being considered statistically significant.
Results
Formulation of rapamycin in PEG-b-PCL micelles
The solubilization of rapamycin in PEG-b-PCL micelles was verified by GPC analysis as previously described [19]. PEG-b-PCL micelles have low UV absorbance at 276 nm, the absorbance maxima of rapamycin. GPC chromatography reported rapamycin coeluted with PEG-b-PCL micelles as indicated by overlapping UV and RI chromatographs (data not shown). The resulting PEG-b-PCL micelles solubilized 10% w/w rapamycin, with a loading efficiency of ca. 90%, greater than the 50% previously reported. The increase in loading efficiency may be attributed to relatively smaller filtration and measurement loses at larger scales utilized in this study and the use of higher quality, less polydisperse PEG-b-PCL, which we observed generally results in greater micelle formation and less aggregated material. Yamamoto et al. reported that micelles from polymers with narrower polydispersity are generally more stable [47].
PEG-b-PCL micelles solubilizing 10% w/w rapamycin were 37 nm in diameter. Incorporation of α-tocopherol increased the micelle hydrodynamic diameter to 101 nm. Nanocarriers in this size range are expected to have long circulation times and extravasate in the leaky vasculature of tumors [2].
Pharmacokinetics of polymeric micellar rapamycin after intravenous administration
Linearity in the standard curves was demonstrated for the blood and plasma samples over the concentration range studied, and chromatograms were free of interference from endogenous components. The blood and plasma rapamycin concentration versus time profiles observed for the three formulations were similar (Figs. 2, 3, respectively). A rapid decline in concentrations, representing a distribution phase, was observed for the first 6 h in blood and 12 h in plasma after dosing for the three formulations. A slight increase in rapamycin concentration was evident at 6 h on a semi-log concentration time profile only after the administration of control formulation, indicating the possibility of enterohepatic recycling. This was followed by an elimination phase characterized with a t1/2 of 12–16 h for blood and 11–22 h for plasma. Rapamycin in PEG-b-PCL yielded a significantly higher t1/2 in plasma compared to rapamycin control (Table 2).
Fig. 2.
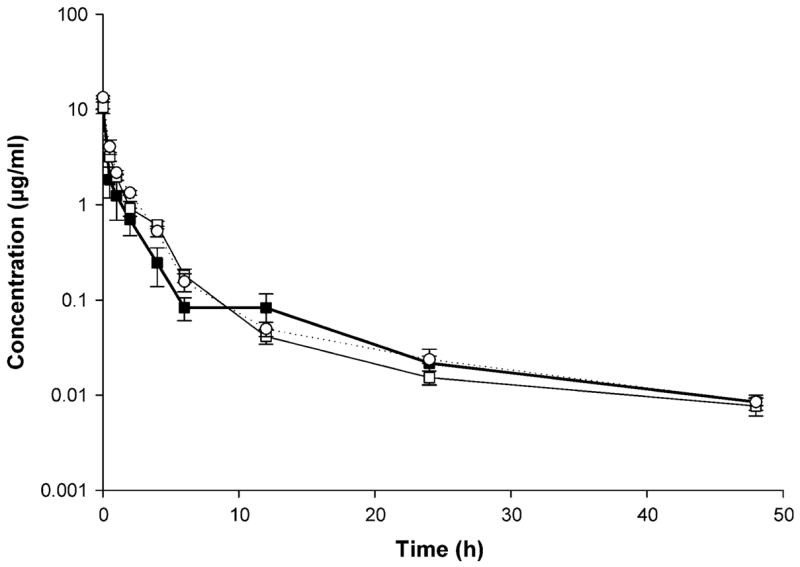
Rapamycin concentration–time profile in blood after IV administration of control rapamycin (filled square), rapamycin in PEG-b-PCL (open square), and rapamycin in PEG-b-PCL + α-tocopherol formulation (open circle) (10 mg/kg) to rats (mean ± SEM, n = 4 per group)
Fig. 3.
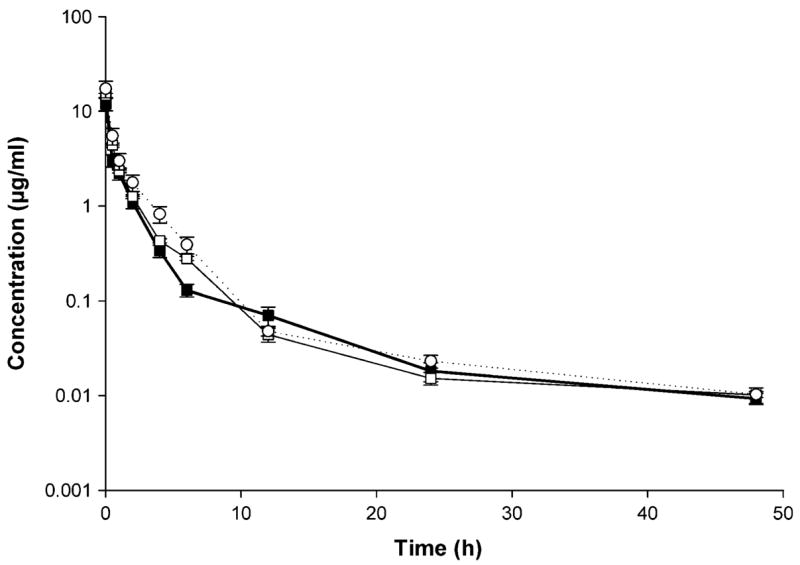
Rapamycin concentration–time profile in plasma after IV administration of control rapamycin (filled square), rapamycin in PEG-b-PCL (open square), and rapamycin in PEG-b-PCL + α-tocopherol formulation (open circle) (10 mg/kg) to rats (mean ± SEM, n = 4 per group)
Table 2.
Pharmacokinetics of control rapamycin, rapamycin in PEG-b-PCL, and rapamycin in PEG-b-PCL + α-tocopherol in plasma after IV administration in rats (mean ± SEM, n = 4)
| Pharmacokinetic parameter | Rapamycin control | Rapamycin in PEG-b-PCL | Rapamycin in PEG-b-PCL + α-tocopherol |
|---|---|---|---|
| AUCinf (μg h/ml) | 9.443 ± 0.972 | 11.552 ± 1.163 | 15.313 ± 2.670 |
| AUC0-48h (μg h/ml) | 9.289 ± 1.003 | 11.205 ± 1.069 | 15.056 ± 2.646 |
| Vdβ (l/kg) | 17.635 ± 2.891 | 27.247 ± 2.444 | 16.247 ± 0.624 |
| CLrenal (l/h kg) | 0.038 ± 0.003 | 0.018 ± 0.006a | 0.013 ± 0.001a |
| CLhepatic (l/h kg) | 1.032 ± 0.108 | 0.856 ± 0.082 | 0.661 ± 0.120a,b |
| CLtot (l/h kg) | 1.070 ± 0.110 | 0.874 ± 0.088 | 0.674 ± 0.119 |
| KE (h−1) | 0.061 ± 0.004 | 0.032 ± 0.001a | 0.041 ± 0.006a |
| t1/2 (h) plasma | 11.346 ± 0.704 | 21.616 ± 0.239a | 17.124 ± 2.378 |
| t1/2 (h) urine | 10.184 ± 0.024 | 11.232 ± 0.054a | 20.919 ± 0.037a,b |
| MRT (h) | 4.673 ± 1.382 | 5.288 ± 0.652 | 4.319 ± 0.269 |
| Extraction ratio (ER) | 0.593 ± 0.062 | 0.492 ± 0.047a | 0.380 ± 0.069a |
| Estimated bioavailability (%) | 40.682 ± 6.184 | 61.841 ± 13.681a | 61.990 ± 6.906a |
Denotes statistical significant difference (P < 0.05) between control and formulation
Denotes statistical significant difference (P < 0.05) between both formulations
Non-compartmental analysis of the blood and plasma concentrations showed a significant change in some pharmacokinetic parameters of rapamycin in both polymeric micelles compared to control formulation (Tables 1, 2). PEG-b-PCL micelles with the co-incorporation of α-tocopherol provided higher AUCinf values in blood (57% increase) compared to control rapamycin formulation. The micelle carrier with the co-incorporation of α-tocopherol decreased the total clearance (CLtot) in blood (34%) compared to rapamycin control. The volume of distribution at the elimination phase (Vdβ) in blood was found to be similar for the control and both formulations. Even though these changes in pharmacokinetic parameters, the reported mean residence time (MRT) for both formulations is not significantly different than the control formulation.
Table 1.
Pharmacokinetics of control rapamycin, rapamycin in PEG-b-PCL, and rapamycin in PEG-b-PCL + α-tocopherol in blood after IV administration in rats (mean ± SEM, n = 4) between both formulations
| Pharmacokinetic parameter | Rapamycin control | Rapamycin in PEG-b-PCL | Rapamycin in PEG-b-PCL + α-tocopherol |
|---|---|---|---|
| AUCinf (μg h/ml) | 7.584 ± 0.887 | 8.556 ± 0.248 | 11.929 ± 0.412a,b |
| AUC0-48h (μg h/ml) | 7.294 ± 0.992 | 8.378 ± 0.204 | 11.751 ± 0.399a,b |
| Vdβ (l/kg) | 23.993 ± 3.466 | 26.696 ± 1.302 | 17.745 ± 1.272b |
| CLrenal (l/h kg) | 0.053 ± 0.012 | 0.022 ± 0.002a | 0.012 ± 0.003a,b |
| CLhepatic (l/h kg) | 1.284 ± 0.012 | 1.150 ± 0.035 | 0.828 ± 0.027a,b |
| CLtot (l/h kg) | 1.337 ± 0.156 | 1.172 ± 0.034 | 0.840 ± 0.028a,b |
| KE (h−1) | 0.056 ± 0.001 | 0.044 ± 0.002a | 0.048 ± 0.03 |
| t1/2 (h) blood | 12.396 ± 0.346 | 15.820 ± 0.843 | 14.626 ± 0.812 |
| t1/2 (h) urine | 10.296 ± 0.014 | 10.647 ± 0.061a | 21.393 ± 0.026a,b |
| MRT (h) | 8.770 ± 3.120 | 4.833 ± 0.780 | 4.304 ± 0.355 |
| Extraction ratio (ER) | 0.399 ± 0.045 | 0.357 ± 0.011 | 0.257 ± 0.008a,b |
| Estimated bioavailability (%) | 60.123 ± 3.169 | 64.293 ± 1.092 | 74.297 ± 0.836a,b |
Denotes statistical significant difference (P < 0.05) between control and formulation
Denotes statistical significant difference (P < 0.05)
The mean hepatic blood flow (Q) is approximately 3.22 l/h kg [12]. Using the hematocrit in rat [12] of 0.48, this yields a mean hepatic plasma flow of 1.74 l/h kg. While the CLtot values in blood (0.8–1.3 l/h kg) are lower (<50%) than the mean hepatic blood flow (3.22 l/h kg), the CLtot values in plasma (0.7–1.1 l/h kg) are closer to the mean hepatic plasma flow (1.74 l/h kg). This can be observed by the changes in extraction ratio (ER). In the case of PEG-b-PCL + α-tocopherol micellar formulation there is a significant reduction in CLtot in blood that correlates with lower ER values and higher estimated bioavailability (F). These observations indicate that α-tocopherol induces a more sustained release of rapamycin out of the micelles in vivo (higher AUC) followed by lower degree of distribution (lower Vdβ), and faster elimination (lower t1/2), followed by an increase in the amount of available rapamycin to exert pharmacological activity in blood.
The terminal urine half-life of rapamycin significantly increased in both micelle formulations; the PEG-b-PCL formulation increased the half-life by 6–11% while the PEG-b-PCL + α-tocopherol formulation increased the half-life by more than 2-fold. Three plots are commonly employed to estimate the half-life of a compound. The amount remaining to be excreted (ARE) plot is often used to determine the accumulation of drug in urine over time. The rate of excretion plot is used in order to establish how rapidly a drug is eliminated. The third plot commonly employed in determining half-life is the blood, serum, or plasma concentration versus time plot. Furthermore, pharmacokinetic theory suggests that the slopes of the amount remaining to be excreted (ARE) plot [log (ΣXuinf − ΣXu) = log (keX0/KE) − KE/2.303t], the rate of excretion plot [log (dXu/dt) = log (keX0) − KE/2.303t], and the serum concentration versus time plot [log C = log C0 − KE/2.303t] must be parallel because they share the same slope (KEt/2.303) and first order elimination rate constant (KE). The half-life should theoretically be the same as t1/2 = 0.693/KE. When plotted on the same graph, the three plots are parallel, in this case the ARE plot, rate plot, blood, and plasma concentrations were plotted together (Fig. 4a–c). This indicates that the elimination rates of these three matrices, namely blood, plasma, and urine, are similar. Based on the CLrenal values can be observed that rapamycin is excreted via non-renal routes and that the micelle formulation results in amount of rapamycin cleared by the kidneys.
Fig. 4.
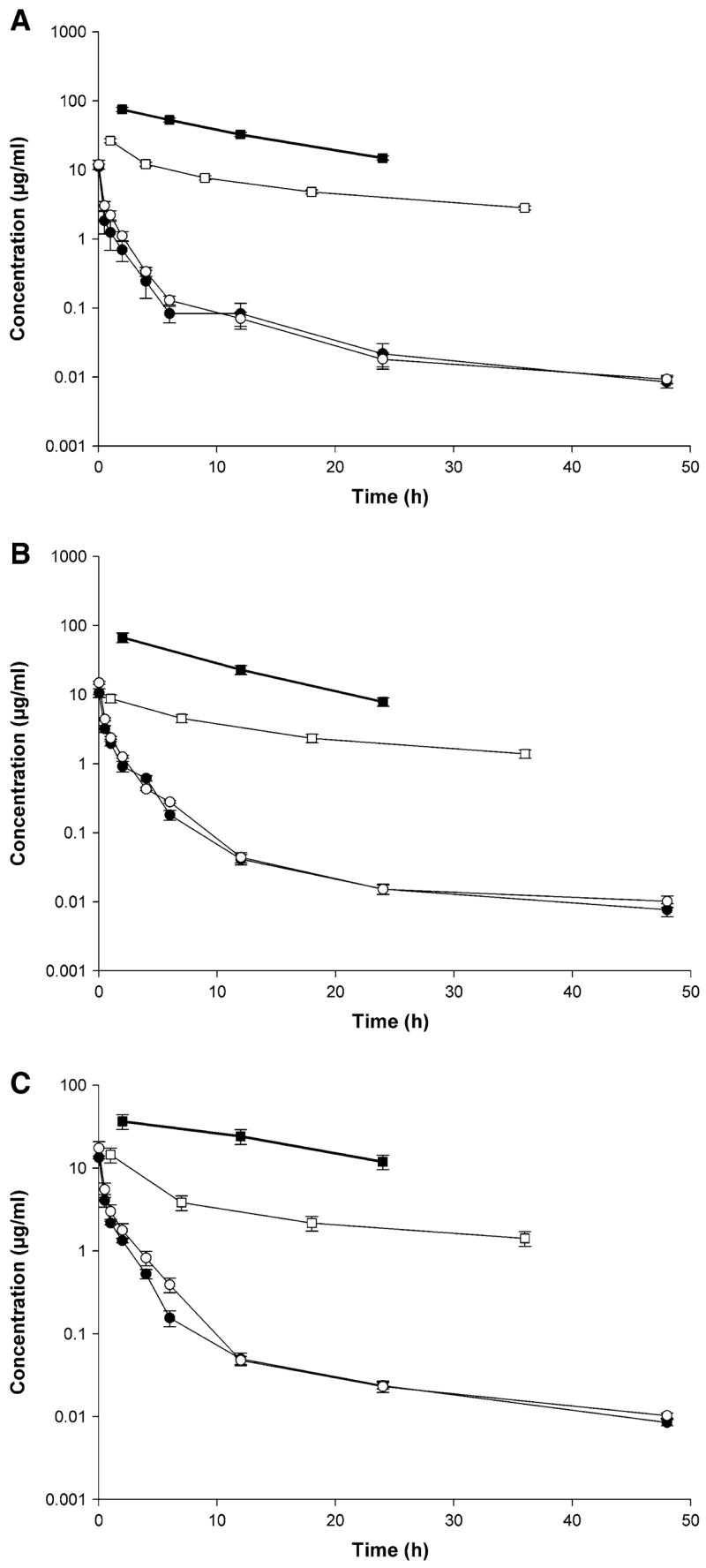
Rapamycin amount remaining to be excreted in urine (ARE) plot (filled square), rate of urinary excretion plot (open square), concentration-time profile in blood (filled circle) and plasma plots (open circle) after IV administration of a) control rapamycin, b) rapamycin in PEG-b-PCL, and c) rapamycin in PEG-b-PCL + α-tocopherol formulation (10 mg/kg) to rats (mean ± SEM, n = 4 per group)
Observations performed by blinded observers reported that immediately between 2 h post-IV dosing of control formulation the rats presented nose bleeding, disorientation, heavy breathing, slight decrease in response to sound, and 60% of the control subjects died within 24 h post-dose. The animals that received the PEG-b-PCL or PEG-b-PCL + α-tocopherol formulations only presented slight nasal bleeding and 100% of the animals survived until the end of the experiment (72 h post-dose).
Biodistribution
As described in the pharmacokinetics study, marked distribution and elimination phases were noted in blood and plasma (Figs. 2, 3). Quantifiable amounts of rapamycin were observed in all assayed tissues. The order in concentrations from highest to lowest for the rapamycin control formulation was lung > kidney > spleen > liver > urinary > bladder > heart > brain > muscle > plasma > blood (Table 3). The corresponding order for PEG-b-PCL micelle formulation was lung > spleen > kidney > liver > urinary > bladder > heart > muscle > brain > plasma > blood, and for PEG-b-PCL + α-tocopherol the order was urinary bladder > kidney > lung > liver > spleen > heart > muscle > brain > p lasma > blood. The Kp values for all the tissues were between 20 and 40, which correlates with previous observations [35]. However, significantly lower Kp values (5–15) were observed in muscle and brain of animals dosed with both micelle formulations compared to control formulation. Rapamycin in PEG-b-PCL reported a statistical significant decrease in concentration in blood by 27% and brain by 75%. Rapamycin in PEG-b-PCL + α-tocopherol resulted in a statistically significant decreased in spleen (25%) and brain (75%) concentrations, while the concentration significantly increased by 69% in urinary bladder (Table 3).
Table 3.
Mean concentrations (μg/g of tissue) of rapamycin in rat tissues (and tissue to plasma rations—Kd values—in parenthesis) measured at 24 h post IV administration of control rapamycin, rapamycin in PEG-b-PCL, and rapamycin in PEG-b-PCL + α-tocopherol in plasma after IV administration (mean ± SEM, n = 4)
| Tissue | Rapamycin control | Rapamycin in PEG-b-PCL | Rapamycin in PEG-b-PCL + α-tocopherol |
|---|---|---|---|
| Plasma | 0.020 ± 0.002 | 0.015 ± 0.002 | 0.020 ± 0.001 |
| Blood | 0.018 ± 0.004 (0.989 ± 0.011) | 0.013 ± 0.001a (0.739 ± 0.052)a | 0.018 ± 0.002b (0.828 ± 0.022)a |
| Liver | 0.663 ± 0.076 (33.659 ± 7.210) | 0.534 ± 0.021 (35.143 ± 1.386) | 0.528 ± 0.022 (23.865 ± 1.959)a,b |
| Kidney | 0.704 ± 0.106 (35.870 ± 8.907) | 0.553 ± 0.016 (32.513 ± 2.831) | 0.664 ± 0.005 (28.217 ± 1.657) |
| Spleen | 0.668 ± 0.074 41.708 ± 0.687) | 0.556 ± 0.018 (32.425 ± 5.362)a | 0.499 ± 0.011a,b (22.330 ± 1.529)a,b |
| Brain | 0.300 ± 0.067 (18.391 ± 7.889) | 0.075 ± 0.005a (5.172 ± 0.636)a | 0.076 ± 0.004a (3.357 ± 0.164)a,b |
| Muscle | 0.173 ± 0.019 (9.804 ± 1.319) | 0.174 ± 0.023 (12.851 ± 0.599) | 0.153 ± 0.013 (6.131 ± 0.094)a,b |
| Lung | 0.721 ± 0.105 (44.839 ± 0.834) | 0.710 ± 0.036 (39.645 ± 3.911) | 0.568 ± 0.014b (25.793 ± 2.919)a,b |
| Heart | 0.445 ± 0.034 (27.919 ± 1.430) | 0.382 ± 0.004 (22.498 ± 2.409)a | 0.362 ± 0.005 (16.997 ± 1.098)a,b |
| Urinary bladder | 0.529 ± 0.019 (30.000 ± 2.942) | 0.482 ± 0.027 (31.741 ± 1.782) | 0.892 ± 0.080a,b (38.562 ± 8.195) |
Denotes statistical significant difference (P < 0.05) between control and formulation
Denotes statistical significant difference (P < 0.05) between both formulations
The tissue to plasma concentration ratios (Kp) of rapamycin were measured 24 h post-IV administration of control and both PEG-b-PCL micelle formulations reporting quite different profiles (Fig. 5). Rapamycin in PEG-b-PCL formulation showed significant lower Kp values in blood, spleen, brain, and heart compared to control formulation. Similarly, rapamycin in PEG-b-PCL + α-tocopherol showed significant lower Kp values in blood, liver, spleen, brain, muscle, lung, and heart (Table 3).
Fig. 5.
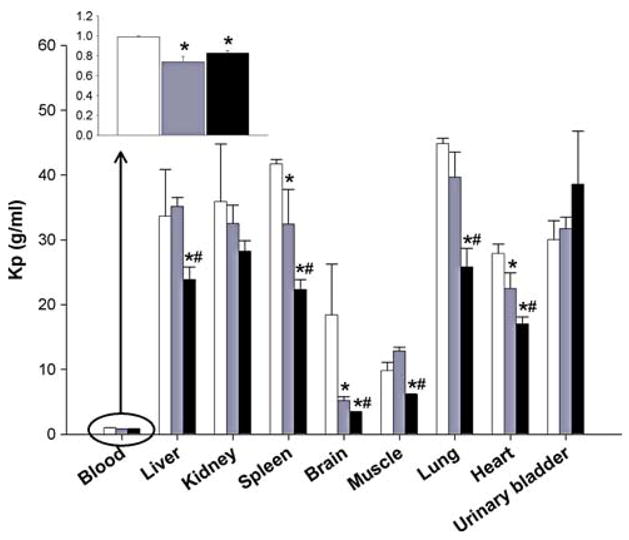
Tissue to plasma ratios (Kp) measured at 24 h post-IV administration of control rapamycin (open square), rapamycin in PEG-b-PCL (dotted rectangle), and rapamycin in PEG-b-PCL + α-tocopherol formulation (filled square) (10 mg/kg) to rats (mean ± SEM, n = 4 per group). *Denotes statistical significant difference (P < 0.05) between control and formulation. #Denotes statistical significant difference (P < 0.05) between both formulations
Blood distribution
The partition coefficient (Kd) representing the ratio of plasma to red blood cells concentrations were calculated at 1 min (Fig. 6a) and 24 h (Fig. 6b) after IV dosing of the different rapamycin formulations. At 1 min post-dose, the formulations without and with the co-incorporation of α-tocopherol have a reported increase in Kd of 1.39-fold and 1.98-fold respectively compared to control formulation. The same pattern was observed at 24 h post-dosing, the Kd increasing 1.31-fold and 2.11-fold for the PEG-b-PCL formulations without and with the co-incorporation of α-tocopherol as compared to control formulation. This indicates the micelle formulations are changing the distribution of rapamycin in the blood, reducing the binding affinity of the drug to RBC as expressed by the higher Kd values. This would allow higher amounts of rapamycin circulating in the plasma that would be readily accessible to solid tumors. Finally, the co-incorporation of α-tocopherol provided higher Kd values indicating a higher distribution into the plasma.
Fig. 6.
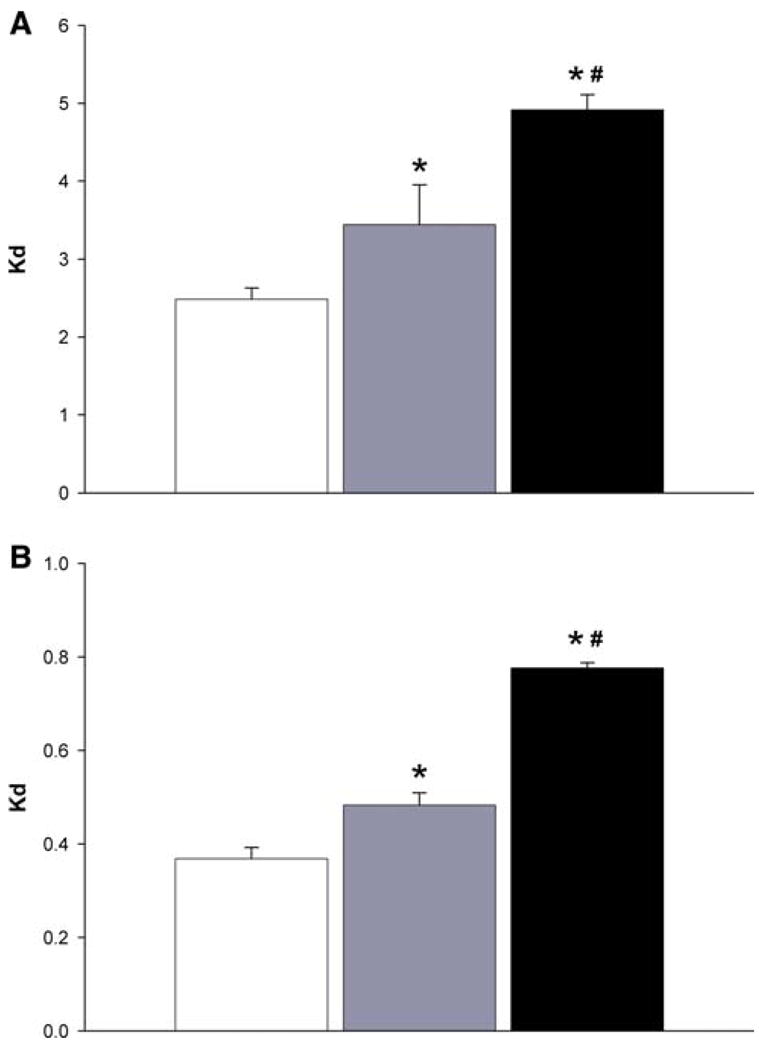
Plasma/RBC ratios (Kd) of rapamycin control formulation (open square), rapamycin in PEG-b-PCL (dotted rectangle), and rapa-mycin in PEG-b-PCL + α-tocopherol formulation (filled square) at a) 1 min after IV administration. (n = 4, mean ± SEM) and at b) 24 h after IV administration. (n = 4, mean ± SEM). *Denotes statistical significant difference (P < 0.05) between control and formulation. #Denotes statistical significant difference (P < 0.05) between both formulations
Discussion
Different clinical studies have demonstrated treatment limiting toxicities after administration of rapamycin, some of those include headaches, asthenia, mucositis, nausea, hypertriglyceridemia, thrombocytopenia, hypercholesterolemia, elevated transaminases, hyperglycemia [20], and mild dermatological toxicities [36]. Therefore, the objective of this study was to develop novel delivery systems that can act as an effective alternative to DMA, PEG, and Tween 80, Cremophor, or ethanol for rapamycin solubilization, control the rate of release, its disposition, increase its plasma distribution and ultimately make it safer for human administration by providing a more effective carrier that could enable the use of lower doses.
Rapamycin pharmacokinetics have been studied extensively in different species including rat, monkey, rabbit, and human. These studies have characterized rapamycin to be a drug with a relatively long half-life of more than 5 hours, with volume of distribution values that indicates a substantial proportion of the drug residing extravascularly [8, 22, 45, 52]. Rapamycin is a lipophilic compound with a partition coefficient (X log PO/W) of 5.77 and is highly distributed into the tissue as evidenced by the high volume of distribution value. In addition, rapamycin is highly extracted as suggested by its clearance values.
A PEG-b-PCL micelle delivery system for rapamycin was developed because of the biocompatibility of the two blocks and the thermodynamic stability of the micellar structure. The micelles increased the aqueous solubility of rapamycin to several mg/ml for PEG-b-PCL and PEG-b-PCL + α-tocopherol micelles, which is much higher than water solubility of rapamycin (2.6 μg/ml) [34, 40].
For carriers with long circulating properties with sufficient stability, a higher plasma concentration is hypothesized with long circulating properties [3]. The PEG-b-PCL micelle formulation with the co-incorporation of α-tocopherol demonstrated significant differences in pharmacokinetic parameters from the rapamycin control formulation (Tables 1, 2). The significant change in AUC in blood indicates a higher degree of in vivo stability of the PEG-b-PCL + α-tocopherol micelle formulations. The solubilization of rapamycin in PEG-b-PCL + α-tocopherol micelles led to a change in biological fate characterized by an increase in AUC and a reduction in CLtot for the solubilized drug. Although not as dramatic, These findings correlate with the results of the pharmacokinetics studies by Aliabadi et al. [3], who recently reported augmented disposition of cyclosporine loaded PEG-b-PCL micelles in rats. Although clearly each drug and carrier may have a unique degree of alteration or augmentation in plasma or tissue concentrations, our observations also indicate an increase in estimated bioavailability after rapamycin was solubilized in micelles with the co-incorporation of α-tocopherol. The co-incorporation of α-tocopherol seems to further increase the sustained release and bioavailability of rapamycin.
We observed that the micelle formulation increased the Kd values indicating a higher partitioning of rapamycin into plasma. Rapamycin binds to FKBP (FK506 binding protein) in red blood cells [21] allowing for low Kd values of 0.05 that has been reported to affect the low clearance values of rapamycin out of the red blood cells [21]. Our results indicate that the encapsulation of rapamycin increase the Kd values significantly and that rapamycin is more rapidly cleared from plasma than RBC due to the observed lower Kd values at 24 h compared to the higher Kd values one minute after administration. This change in blood distribution would provide higher levels of rapamycin in plasma that could be readily accessible to solid tumors.
In the biodistribution study, the decrease in blood rapamycin concentrations seemed to coincide with a decrease in brain rapamycin concentrations for both formulations. Lower rapamycin concentrations were detected in spleen and lung, while higher concentrations were detected in urinary bladder. In general, formulation in PEG-b-PCL micelles led to no significant change in the concentrations of rapamycin except for blood, brain, spleen, lung, and urinary bladder. The uptake by the brain was dramatically reduced, which is of note because rapamycin has been associated with the induction of neurotoxicity [42]. Liu et al. recently examined the distribution of PEG-b-PCL micelles in vivo [29]. Brain distribution was not reported in that study, but it is expected the blood–brain barrier would not be permeable to large molecules (>500 MW) such as micelles. Further studies are underway to determine if the micelle formulation of rapamycin might actually translate into reduced rapamycin neurotoxicity. The trend towards accumulation of rapamycin in urinary bladder after administration of rapamycin in PEG-b-PCL + α-tocopherol formulation is an interesting observation that also requires further mechanistic clarification.
The Kp value for rapamycin were lowered in blood, spleen, brain, and heart for both formulations, and additionally the Kp was lowered in liver, spleen, muscle, and lung by the PEG-b-PCL + α-tocopherol formulation. The high Kp value after 24 h indicates the distribution of rapamycin into tissues, which can be explained by the low concentrations in plasma and reflects the rapid clearance of rapamycin from plasma.
Conclusions
The PEG-b-PCL polymeric micelle system with the co-incorporation of α-tocopherol allowed for the uptake, protection, and retention of the water insoluble drug rapamycin by increasing the area under the curve, at the same time reducing the total clearance while stabilizing the micellar structure in several biological matrices. The PEG-b-PCL polymeric micelle carrier with and without the co-incorporation of α-tocopherol facilitated an increase in the distribution of rapamycin in plasma rather than the preferential binding of rapamycin for red blood cells by increasing the Kd between 1.3 and 2.1-fold compared to a control formulation with significantly less mortality. Based on our results it seems that the PEG-b-PCL micelles with co-incorporation of α-tocopherol might be of the appropriate size and core/shell properties to escape the uptake by the reticu-loendothelial system and circulate for longer periods preferentially distributing in plasma versus RBCs. Further disposition studies of PEG-b-PCL polymeric micelle systems are ongoing with a variety of other hydrophobic drugs.
Acknowledgments
This research was supported by NIH grant AI-43346-08 and generous grants from Hoffman-La Roche Inc. and Wisconsin Alumni Research Fund. MLF was partially supported by a PhRMA post-doctoral fellowship.
Contributor Information
Jaime A. Yáñez, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Pullman, WA 99164, USA
M. Laird Forrest, Department of Pharmaceutical Chemistry, University of Kansas, Lawrence, KS 66047, USA.
Yusuke Ohgami, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Pullman, WA 99164, USA.
Glen S. Kwon, Division of Pharmaceutical Sciences, School of Pharmacy, University of Wisconsin, Madison, WI 53705, USA, e-mail: gskwon@pharmacy.wisc.edu
Neal M. Davies, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Pullman, WA 99164, USA, e-mail: ndavies@wsu.edu
References
- 1.Temsirolimus: CCI 779, CCI-779, cell cycle inhibitor-779. Drugs R D. 2004;5:363–367. doi: 10.2165/00126839-200405060-00011. [DOI] [PubMed] [Google Scholar]
- 2.Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92:1343–1355. doi: 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- 3.Aliabadi HM, Brocks DR, Lavasanifar A. Polymeric micelles for the solubilization and delivery of cyclosporine A: pharmacokinetics and biodistribution. Biomaterials. 2005;26:7251–7259. doi: 10.1016/j.biomaterials.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 4.Annesley TM, Clayton L. Simple extraction protocol for analysis of immunosuppressant drugs in whole blood. Clin Chem. 2004;50:1845–1848. doi: 10.1373/clinchem.2004.037416. [DOI] [PubMed] [Google Scholar]
- 5.Aoki M, Blazek E, Vogt PK. A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA. 2001;98:136–141. doi: 10.1073/pnas.011528498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai S, Stepkowski SM, Kahan BD, Brunner LJ. Metabolic interaction between cyclosporine and sirolimus. Transplantation. 2004;77:1507–1512. doi: 10.1097/01.tp.0000128372.09220.b5. [DOI] [PubMed] [Google Scholar]
- 7.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 8.Brattstrom C, Sawe J, Jansson B, Lonnebo A, Nordin J, Zimmerman JJ, Burke JT, Groth CG. Pharmacokinetics and safety of single oral doses of sirolimus (rapamycin) in healthy male volunteers. Ther Drug Monit. 2000;22:537–544. doi: 10.1097/00007691-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Brattstrom C, Sawe J, Tyden G, Herlenius G, Claesson K, Zimmerman J, Groth CG. Kinetics and dynamics of single oral doses of sirolimus in sixteen renal transplant recipients. Ther Drug Monit. 1997;19:397–406. doi: 10.1097/00007691-199708000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Calne RY. The rejection of renal homografts. Inhibition in dogs by 6-mercaptopurine. Lancet. 1960;1:417–418. doi: 10.1016/s0140-6736(60)90343-3. [DOI] [PubMed] [Google Scholar]
- 11.Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): pro- and anti-apoptotic. Cell Death Differ. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- 12.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 13.Deters M, Kirchner G, Resch K, Kaever V. Simultaneous quantification of sirolimus, everolimus, tacrolimus and cyclosporine by liquid chromatography-mass spectrometry (LC-MS) Clin Chem Lab Med. 2002;40:285–292. doi: 10.1515/CCLM.2002.045. [DOI] [PubMed] [Google Scholar]
- 14.Dumont FJ, Staruch MJ, Koprak SL, Melino MR, Sigal NH. Distinct mechanisms of suppression of murine T cell activation by the related macrolides FK-506 and rapamycin. J Immunol. 1990;144:251–258. [PubMed] [Google Scholar]
- 15.Eng CP, Sehgal SN, Vezina C. Activity of rapamycin (AY-22,989) against transplanted tumors. J Antibiot (Tokyo) 1984;37:1231–1237. doi: 10.7164/antibiotics.37.1231. [DOI] [PubMed] [Google Scholar]
- 16.Ettenger RB, Grimm EM. Safety and efficacy of TOR inhibitors in pediatric renal transplant recipients. Am J Kidney Dis. 2001;38:S22–S28. doi: 10.1053/ajkd.2001.27838. [DOI] [PubMed] [Google Scholar]
- 17.Ferron GM, Mishina EV, Zimmerman JJ, Jusko WJ. Population pharmacokinetics of sirolimus in kidney transplant patients. Clin Pharmacol Ther. 1997;61:416–428. doi: 10.1016/S0009-9236(97)90192-2. [DOI] [PubMed] [Google Scholar]
- 18.Finsterer J, Kanzler M, Weinberger A. Sirolimus myopathy. Transplantation. 2003;76:1773–1774. doi: 10.1097/01.TP.0000090752.85709.9F. [DOI] [PubMed] [Google Scholar]
- 19.Forrest ML, Won CY, Malick AW, Kwon GS. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. J Control Release. 2006;110:370–377. doi: 10.1016/j.jconrel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Hidalgo M, Buckner JC, Erlichman C, Pollack MS, Boni JP, Dukart G, Marshall B, Speicher L, Moore L, Rowinsky EK. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12:5755–5763. doi: 10.1158/1078-0432.CCR-06-0118. [DOI] [PubMed] [Google Scholar]
- 21.Ho S, Clipstone N, Timmermann L, Northrop J, Graef I, Fiorentino D, Nourse J, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Clin Immunol Immunopathol. 1996;80:S40–S45. doi: 10.1006/clin.1996.0140. [DOI] [PubMed] [Google Scholar]
- 22.Honcharik N, Fryer J, Yatscoff R. Pharmacokinetics of rapamycin: single-dose studies in the rabbit. Ther Drug Monit. 1992;14:475–478. [PubMed] [Google Scholar]
- 23.Hoogeveen RC, Ballantyne CM, Pownall HJ, Opekun AR, Hachey DL, Jaffe JS, Oppermann S, Kahan BD, Morrisett JD. Effect of sirolimus on the metabolism of apoB100- containing lipoproteins in renal transplant patients. Transplantation. 2001;72:1244–1250. doi: 10.1097/00007890-200110150-00011. [DOI] [PubMed] [Google Scholar]
- 24.Kahan BD. Update on pharmacokinetic/pharmacodynamic studies with FTY720 and sirolimus. Ther Drug Monit. 2002;24:47–52. doi: 10.1097/00007691-200202000-00009. [DOI] [PubMed] [Google Scholar]
- 25.Kahan BD, Gibbons S, Tejpal N, Stepkowski SM, Chou TC. Synergistic interactions of cyclosporine and rapamycin to inhibit immune performances of normal human peripheral blood lymphocytes in vitro. Transplantation. 1991;51:232–239. doi: 10.1097/00007890-199101000-00038. [DOI] [PubMed] [Google Scholar]
- 26.Kay JE, Sampare-Kwateng E, Geraghty F, Morgan GY. Uptake of FK 506 by lymphocytes and erythrocytes. Transplant Proc. 1991;23:2760–2762. [PubMed] [Google Scholar]
- 27.Kimball PM, Kerman RH, Kahan BD. Production of synergistic but nonidentical mechanisms of immunosuppression by rapamycin and cyclosporine. Transplantation. 1991;51:486–490. doi: 10.1097/00007890-199102000-00041. [DOI] [PubMed] [Google Scholar]
- 28.Knight R, Ferraresso M, Serino F, Katz S, Lewis R, Kahan BD. Low-dose rapamycin potentiates the effects of subtherapeutic doses of cyclosporine to prolong renal allograft survival in the mongrel canine model. Transplantation. 1993;55:947–949. [PubMed] [Google Scholar]
- 29.Liu J, Zeng F, Allen C. In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur J Pharm Biopharm. 2006 doi: 10.1016/j.ejpb.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 30.Mahalati K, Kahan BD. Clinical pharmacokinetics of sirolimus. Clin Pharmacokinet. 2001;40:573–585. doi: 10.2165/00003088-200140080-00002. [DOI] [PubMed] [Google Scholar]
- 31.Maramattom BV, Wijdicks EF. Sirolimus may not cause neurotoxicity in kidney and liver transplant recipients. Neurology. 2004;63:1958–1959. doi: 10.1212/01.wnl.0000144351.63740.81. [DOI] [PubMed] [Google Scholar]
- 32.Metcalfe SM, Richards FM. Cyclosporine, FK506, and rapamycin. Some effects on early activation events in serum-free, mitogen-stimulated mouse spleen cells. Transplantation. 1990;49:798–802. [PubMed] [Google Scholar]
- 33.Montaguti P, Melloni E, Cavalletti E. Acute intravenous toxicity of dimethyl sulfoxide, polyethylene glycol 400, dimethyl-formamide, absolute ethanol, and benzyl alcohol in inbred mouse strains. Arzneimittelforschung. 1994;44:566–570. [PubMed] [Google Scholar]
- 34.Morris RE. In vivo immunopharmacology of the macrolides FK 506 and rapamycin: toward the era of rational immunosuppressive drug discovery, development, and use. Transplant Proc. 1991;23:2722–2724. [PubMed] [Google Scholar]
- 35.Napoli KL, Wang ME, Stepkowski SM, Kahan BD. Distribution of sirolimus in rat tissue. Clin Biochem. 1997;30:135–142. doi: 10.1016/s0009-9120(96)00157-9. [DOI] [PubMed] [Google Scholar]
- 36.Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, Leister C, Korth-Bradley J, Hanauske A, Armand JP. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22:2336–2347. doi: 10.1200/JCO.2004.08.116. [DOI] [PubMed] [Google Scholar]
- 37.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 38.Seeliger H, Guba M, Koehl GE, Doenecke A, Steinbauer M, Bruns CJ, Wagner C, Frank E, Jauch KW, Geissler EK. Blockage of 2-deoxy-D-ribose-induced angiogenesis with rapamycin counteracts a thymidine phosphorylase-based escape mechanism available for colon cancer under 5-fluorouracil therapy. Clin Cancer Res. 2004;10:1843–1852. doi: 10.1158/1078-0432.ccr-1176-3. [DOI] [PubMed] [Google Scholar]
- 39.Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc. 2003;35:7S–14S. doi: 10.1016/s0041-1345(03)00211-2. [DOI] [PubMed] [Google Scholar]
- 40.Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22, 989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo) 1975;28:727–732. doi: 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]
- 41.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- 42.Serkova N, Christians U, Flogel U, Pfeuffer J, Leibfritz D. Assessment of the mechanism of astrocyte swelling induced by the macrolide immunosuppressant sirolimus using multinuclear nuclear magnetic resonance spectroscopy. Chem Res Toxicol. 1997;10:1359–1363. doi: 10.1021/tx970071k. [DOI] [PubMed] [Google Scholar]
- 43.Simamora P, Alvarez JM, Yalkowsky SH. Solubilization of rapamycin. Int J Pharm. 2001;213:25–29. doi: 10.1016/s0378-5173(00)00617-7. [DOI] [PubMed] [Google Scholar]
- 44.Tejani A, Alexander S, Ettenger R, Lerner G, Zimmerman J, Kohaut E, Briscoe DM. Safety and pharmacokinetics of ascending single doses of sirolimus (Rapamune, rapamycin) in pediatric patients with stable chronic renal failure undergoing dialysis. Pediatr Transplant. 2004;8:151–160. doi: 10.1046/j.1399-3046.2003.00137.x. [DOI] [PubMed] [Google Scholar]
- 45.Trepanier DJ, Gallant H, Legatt DF, Yatscoff RW. Rapamycin: distribution, pharmacokinetics and therapeutic range investigations: an update. Clin Biochem. 1998;31:345–351. doi: 10.1016/s0009-9120(98)00048-4. [DOI] [PubMed] [Google Scholar]
- 46.Tu Y, Stepkowski SM, Chou TC, Kahan BD. The synergistic effects of cyclosporine, sirolimus, and brequinar on heart allograft survival in mice. Transplantation. 1995;59:177–183. [PubMed] [Google Scholar]
- 47.Yamamoto Y, Nagasaki Y, Kato Y, Sugiyama Y, Kataoka K. Long-circulating poly(ethylene glycol)-poly(D,L-lactide) block copolymer micelles with modulated surface charge. J Control Release. 2001;77:27–38. doi: 10.1016/s0168-3659(01)00451-5. [DOI] [PubMed] [Google Scholar]
- 48.Yatscoff R, LeGatt D, Keenan R, Chackowsky P. Blood distribution of rapamycin. Transplantation. 1993;56:1202–1206. doi: 10.1097/00007890-199311000-00029. [DOI] [PubMed] [Google Scholar]
- 49.Yatscoff RW, Wang P, Chan K, Hicks D, Zimmerman J. Rapamycin: distribution, pharmacokinetics, and therapeutic range investigations. Ther Drug Monit. 1995;17:666–671. doi: 10.1097/00007691-199512000-00020. [DOI] [PubMed] [Google Scholar]
- 50.Yu K, Toral-Barza L, Discafani C, Zhang WG, Skotnicki J, Frost P, Gibbons JJ. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer. 2001;8:249–258. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 51.Zimmerman JJ, Kahan BD. Pharmacokinetics of sirolimus in stable renal transplant patients after multiple oral dose administration. J Clin Pharmacol. 1997;37:405–415. doi: 10.1002/j.1552-4604.1997.tb04318.x. [DOI] [PubMed] [Google Scholar]
- 52.Zimmerman JJ, Lasseter KC, Lim HK, Harper D, Dilzer SC, Parker V, Matschke K. Pharmacokinetics of sirolimus (rapamycin) in subjects with mild to moderate hepatic impairment. J Clin Pharmacol. 2005;45:1368–1372. doi: 10.1177/0091270005281350. [DOI] [PubMed] [Google Scholar]