

Abstract
The important role of furin in the proteolytic activation of many pathogenic molecules has made this endoprotease a target for the development of potent and selective antiproteolytic agents. Here, we demonstrate the utility of the protein-based inhibitor α1-antitrypsin Portland (α1-PDX) as an antipathogenic agent that can be used prophylactically to block furin-dependent cell killing by Pseudomonas exotoxin A. Biochemical analysis of the specificity of a bacterially expressed His- and FLAG-tagged α1-PDX (α1-PDX/hf) revealed the selectivity of the α1-PDX/hf reactive site loop for furin (Ki, 600 pM) but not for other proprotein convertase family members or other unrelated endoproteases. Kinetic studies show that α1-PDX/hf inhibits furin by a slow tight-binding mechanism characteristic of serpin molecules and functions as a suicide substrate inhibitor. Once bound to furin’s active site, α1-PDX/hf partitions with equal probability to undergo proteolysis by furin at the C-terminal side of the reactive center -Arg355-Ile-Pro-Arg358-↓ or to form a kinetically trapped SDS-stable complex with the enzyme. This partitioning between the complex-forming and proteolytic pathways contributes to the ability of α1-PDX/hf to differentially inhibit members of the proprotein convertase family. Finally, we propose a structural model of the α1-PDX-reactive site loop that explains the high degree of enzyme selectivity of this serpin and which can be used to generate small molecule furin inhibitors.
The recent discovery of a novel family of mammalian subtilisin-related proprotein convertases (PCs) that reside in the secretory pathway has led to a more detailed understanding of the molecular basis of proprotein maturation (1). Members of the PC family identified to date include furin, PACE-4, PC2, PC1/3 (hereafter termed PC3), PC4, PC5/6A and B (hereafter termed PC6A and PC6B), and LPC/PC7/PC8 (hereafter termed PC7) (2).
One PC, furin, trafficks through the trans-Golgi network/endosomal system where it cleaves a wide range of proproteins at the consensus sequence -Arg-Xaa-Lys/Arg-Arg-↓ (3). Mutational analyses showed that the minimal consensus sequence required for furin processing is -Arg-Xaa-Xaa-Arg-↓ (4). By contrast, PC2 and PC3 reside in the regulated secretory pathway of neuroendocrine cells where they process prohormones at -Lys/Arg-Arg- sites (1, 5). The roles of the remaining PCs have yet to be determined.
In addition to endogenous proproteins, many pathogens require furin to become active (3). For example, several viral coat proteins including HIV type 1 (HIV-1) gp160, Newcastle-disease virus Fo, measles virus Fo, and human cytomegalovirus glycoprotein B, require processing by furin in the biosynthetic pathway to generate infectious virions (3). Similarly, cleavage of many bacterial toxins by furin at the plasma membrane (e.g., anthrax toxin) and/or in endosomal compartments [e.g., Pseudomonas exotoxin A (PEA), and diphtheria and Shiga toxins] is required for their cytotoxicity (6).
The discovery that a diverse group of viral and bacterial pathogens requires furin for proteolytic activation has fostered the development of furin inhibitors. These include active-site-directed chloromethyl ketone inhibitors (7, 8), reversible peptide inhibitors (9, 10), and several protein-based inhibitors (11–14). The protein-based inhibitors include a recently described native serpin, PI8 (14), which contains two consensus furin sites, -Arg-Asn-Ser-Arg339- and -Arg339-Cys-Ser-Arg342-, in its reactive site domain. Other protein-based inhibitors represent engineered variants of either the turkey ovomucoid third domain (-Ala-Cys-Thr-Leu18- → -Arg-Cys-Lys-Arg18-) (11), α2-macroglobulin (-Gly-Phe-Tyr-Glu686-Ser-Asp- → -Arg-Ser-Lys-Arg686-Ser-Leu-) (13), or α1-antitrypsin (α1-AT). The latter variant, α1-AT Portland (α1-PDX), is distinct in that it contains a single minimal furin consensus motif in its reactive site loop (RSL; Ala355-Ile-Pro-Met358- → -Arg355-Ile-Pro-Arg358-) (12).
α1-PDX is a potent inhibitor of furin (IC50 = 0.6 nM) and when expressed in cells (either by stable or transient transfection), blocks the processing of HIV-1 gp160 and measles virus-Fo and correspondingly inhibits virus spread (12, 15). However, relative to the chymotrypsin superfamily of serine proteases, little is known regarding the mechanism of inhibition of subtilase superfamily members, including furin, by α1-AT or its engineered variants. In addition, although α1-PDX does not inhibit either elastase or thrombin (12), the selectivity of this furin-directed inhibitor for the other PCs has not been established. Finally, whereas genome expression of α1-PDX effectively blocks proprotein maturation (12, 15), it remains to be determined whether the recombinant protein can be used as a therapeutic agent.
Here, we report the mechanism of furin inhibition by α1-PDX and the intrinsic selectivity of α1-PDX for furin but not for other PCs. Furthermore, we show that α1-PDX can be used prophylactically to block cell killing by PEA, a clinically important pathogen gene product. Our model of the α1-PDX-RSL provides a basis for determining the interactions important for substrate binding and enzyme selectivity. How this model may facilitate development of small molecule therapeutics is discussed.
MATERIALS AND METHODS
Materials.
pGlu-Arg-Thr-Lys-Arg-4-methylcoumaryl-7-amide (pERTKR-MCA) was obtained from Peptides International, N-tert-butoxycarbonyl-Asp(benzyl)-Pro-Arg-MCA and decanoyl-Arg-Val-Lys-Arg-CH2Cl (Dec-RVKR-CH2Cl) from Bachem, thrombin (EC 3.4.21.5) from Sigma, and mAb M2 from Kodak.
His-/FLAG-Tagged α1-AT Variants.
The DNA sequence encoding the human α1-AT signal peptide in pBS-AT (12) was replaced with sequences encoding the epitope(FLAG)-tag (underlined) by insertion of the annealed complimentary oligos 5′-CTAGAGGATCCCATGGACTACAAGGACGACGATGACAAGGAA-3′ and 5′-GATCTTCCTTGTCATCGTCGTCCTTGTAGTCCATGGGATCCT-3′. The resulting cDNA was subcloned into pDS56–6His to generate pDS56α1-AT/hf. To generate pDS56α1-PDX/hf, the DNA sequences encoding the RSL were replaced by the complimentary oligos (5′-GGGGCCATGTTTTTAGAGCGCATACCCA-3′ and 5′-GATCTGGGTATGCGCTCTAAAAACATGGCCCCTGCA-3′) coding for Arg (bold) in positions P4 and P1. To generate pDS56α1-PIT/hf, complimentary oligos (5′-GGGGCCATGTTTTTAGAGGCCATACCCA-3′ and 5′-GATCTGGGTATGGCCTCTAAAAACATGGCCCCTGCA-3′) encoding a P1 Arg were used. The resulting ORFs directed cytosolic expression of the recombinant proteins initiating with a Met followed by the His and FLAG tags and the mature sequences of either α1-PDX/hf or α1-PIT/hf (see Fig. 1A).
Figure 1.

Histidine-FLAG-tagged α1-AT variants. (A) Diagram of α1-AT variants including native α1-AT, α1-PIT/hf, and α1-PDX/hf. The His and FLAG sequences that replace the signal sequence as well as the amino acid substitutions within the RSL are shown. (B) RP-HPLC analysis of purified α1-PDX/hf. The chromatogram shows a single prominent A280 peak eluting at 58 min corresponding to purified α1-PDX/hf. (C) Electrospray MS of purified α1-PDX/hf. The measured molecular mass of α1-PDX/hf and α1-PIT/hf (46,719 ± 3 Da and 46,631 ± 3 Da, respectively) was in close agreement (relative error less than 0.02%) with the calculated values for α1-PDX/hf (46,727 Da) and α1-PIT/hf (46,642 Da).
Expression and Purification of α1-PDX/hf.
α1-AT variants were expressed in Escherichia coli strain BL21 transformed with either pDS56α1-PDX/hf or pDS56α1-PIT/hf. Protein expression was induced by addition of 1 mM isopropyl β-d-thiogalactoside, and cultures were grown overnight at 31°C. The cells were washed in metal-chelation chromatography binding buffer (5 mM imidazole/0.5 M NaCl/20 mM Tris⋅Cl, pH 7.9) and disrupted by cavitation (French press, 1,000 psi). The clarified and filtered supernatants containing soluble α1-AT variants were applied to a Ni2+-agarose column (Pharmacia), and bound proteins were eluted with 100 mM EDTA. The eluates were adjusted to 3.5 M NaCl and applied to a phenyl-Sepharose column (Pharmacia). The bound α1-PDX/hf or α1-antitrypsin Pittsburgh (α1-PIT)/hf was eluted with 20 mM Bis-Tris, pH 7.0 and concentrated (4 mg/ml final) by diafiltration [Diaflo membrane, 10-kDa cut-off (Pierce)] in the same buffer. Protein purity and composition were demonstrated by Coomassie blue staining of SDS/PAGE gels, Western blot (using mAb M2), reverse-phase HPLC (RP-HPLC) (Fig. 1B), amino acid analysis, and MS (Fig. 1C). Serpin preparations were stored at 4°C.
Analytical Characterization of α1-AT Variants.
RP-HPLC was performed as described previously (5) by using a Vydac C4 column (25 cm × 0.46 cm). Electrospray MS analysis and amino acid composition of RP-HPLC-purified α1-AT variants were performed by SynPep, Dublin, CA.
Cell Culture.
BSC-40, AtT20, LoVo, and A7 cells were grown as described (15–17). PEA assays were performed as described (17).
Vaccinia Virus Recombinants.
To monitor enzyme expression the FLAG tag was inserted by loop-in mutagenesis using mutagenic primers C terminal to the proregion cleavage site of each PC (except for PC2) as follows: FLAG-tagged (underline) human PACE-4 (hPACE-4/f), 5′-CGAAGGGTGAAGA-GAGACTACAAGGACGACGATGACAAGCAGGTGCGAAGTGAC-3′ (C terminal to Arg149) (18); murine PC3 (mPC3/f), 5′-AGAGAGAAGTAAACGTGACTACAAGGACGACGATGACAAGTCAGTTCAAAAAGACT-3′ (C terminal to Arg110) (19); murine PC6B (mPC6B/f), 5′-AAAAAGAACCAAGAGGGACTACAAGGACGACGATGACAAGGATTATGACCTCAGCC-3′ (C terminal to Arg116) (20). A secreted soluble FLAG-tagged human PC7 (hPC7/f) was generated by inserting the epitope tag C terminal to Arg141 (21) by using the mutagenic primer 5′-CTGCTAAGGCGGGCTAAGCGCGACTACAAGGACGACGATGACAAGTCGGTCCACTTCAACGACC-3′ and a second primer 5′-CCCAACACCCTCAAGTAGGCCTAGCTGGTAGGCTGTTTC-3′ to insert a stop codon (bold) in place of Thr667. Generation of vaccinia virus recombinants expressing each of the PCs was performed as described previously (22). The human fur713t/f (hereafter termed hfurin/f) and human PC2 (hPC2) constructs were described previously (5, 22).
Expression of Recombinant PCs.
Each PC was produced by infecting cultured cells with the corresponding vaccinia virus recombinant (5 pfu/cell) and collecting the secreted/shed enzyme after 16 hr at 37°C in a serum- and phenol red-free defined medium (MCDB202) (4). hFurin/f, mPC3/f, mPC6B/f, and hPC7/f were expressed in BSC-40 cells. hPACE-4 was expressed in LoVo cells, and hPC2 was expressed in AtT-20 cells. Conditioned medium containing each enzyme preparation was clarified (5,000 × g 10 min), concentrated [Biomax filter, 30-kDa cut-off (Millipore)], and stored at −70°C. Each enzyme preparation was enzymatically pure based on the absence of PC activity in medium from replicate cultures infected with wild-type vaccinia virus (F.J. and G.T., data not shown).
Enzyme Assays.
The enzyme assay data were obtained by using a FluoroMax-2 spectrofluorometer equipped with a 96-well plate reader (Instrument SA, Edison, NJ) using excitation/emission wavelengths of 370/460 nm to measure released AMC (7-amino-4-methylcoumarin). Thrombin assays were performed using N-tert-butoxycarbonyl-Asp(benzyl)-Pro-Arg-MCA as a fluorogenic substrate (12) whereas pERTKR-MCA was used for each of the PCs (8). Furin, PC6B, PC7, and PACE-4 assays were performed in 100 mM Hepes, pH 7.5, containing 0.5% Triton X-100 and 1 mM CaCl2. PC3 and PC2 assays were performed as described (8, 23). Values for Km and Vmax were determined as described (8) whereas E0 and Ki were obtained by fitting the data (v and I) to the equation: v = SA(E0 − 0.5{(E0 + I + Ki) − [(E0 + I + Ki)2 − 4E0I]]1/2}) (SA, specific activity; E0, enzyme concentration, and I, inhibitor concentration) by nonlinear regression (ENZFITTER, Elsevier-Biosoft, Cambridge, UK) (24).
Determination of the RSL Cleavage Site by MS.
α1-PDX/hf (100 μg) was incubated with furin for 1 hr at 25°C. Stoichiometries were chosen so that the RSL cleavage products were generated in sufficient concentration to be detected by MS. Reactions were stopped with EDTA (5 mM final), and the samples were separated by RP-HPLC as described above and subjected to matrix-assisted laser desorption ionization (MALDI)-MS. Positive ion MALDI-time of flight spectra were obtained by using a Voyager Elite Biospectrometry Research Station (PerSeptive Biosystems, Framingham, MA). Samples were dissolved in 0.1% aqueous trifluoroacetic acid and analyzed by using a matrix of sinapinic acid.
RESULTS
Expression of His-/FLAG-Tagged α1-AT Variants.
A rigorous determination of (i) the mechanism by which α1-PDX inhibits furin, (ii) the enzyme specificity of α1-PDX, and (iii) the ability of recombinant α1-PDX to function prophylactically requires preparative amounts of the recombinant protein. To generate sufficient amounts of α1-PDX for these analyses, we expressed this serpin in the cytosol of bacteria. Using PCR mutagenesis, the α1-PDX signal sequence was replaced with an initiator methionine followed by histidine (His) and epitope-FLAG tags (Fig. 1A). The His tag in the resultant construct, α1-PDX/hf, allowed rapid purification of the recombinant protein whereas the FLAG tag was used to assay for α1-PDX/hf by immunodetection. To control for the effect of the His/FLAG tags on serpin activity, the same mutagenesis was performed on α1-PIT (to generate α1-PIT/hf), a naturally occurring α1-AT variant, which inhibits thrombin and differs from α1-PDX by a single residue at position P4 in the RSL (-Ala-Ile-Pro-Arg358-). Plasmids expressing either α1-PDX/hf or α1-PIT/hf were transformed into E. coli, and protein expression was induced by the addition of isopropyl β-d-thiogalactoside at 31°C. After cell disruption, the α1-AT variants were purified by Ni2+ binding- and hydrophobic interaction-chromatography. This procedure yielded recombinant serpins that were essentially pure and intact, as determined by Coomassie blue staining (data not shown) and Western blot analysis (Fig. 2 C and D). The purity and molecular composition of each preparation were confirmed by analytical RP-HPLC (Fig. 1B), amino acid analysis (data not shown), and electrospray MS (Fig. 1C).
Figure 2.

Inhibition of furin by α1-AT variants. Progress curves showing the effect of α1-PDX/hf (A) or α1-PIT/hf (B) on furin activity. Furin/f (5.4 nM) was incubated with pERTKR-MCA (100 μM) at room temperature in the absence (•) or presence (○) of 6.1 (top), 7.7, 15, 21, 31, 61, 123, or 184 (bottom) nM α1-AT variant. Data are representative of at least three independent experiments. SDS/PAGE analysis of reactions of furin/f with α1-PDX/hf (C) or α1-PIT/hf (D). Increasing concentrations of α1-AT variants (134, 268, 670, and 1,340 nM) were incubated with or without furin/f (5.4 nM) for 1 hr at room temperature. Enzyme activity was stopped with EDTA (5 mM final) followed by addition of SDS-SB (containing 2% SDS and 20 mM DTT). Samples were heated (95°C), resolved by SDS/PAGE (10% gel), and transferred to nitrocellulose and FLAG-tagged proteins detected by Western blot using mAb M2.
Mechanism of Furin Inhibition by α1-PDX/hf.
The time- and concentration-dependent inactivation of furin by α1-PDX/hf was examined. Characteristic of other serpin-enzyme interactions (25, 26), the inhibition of furin by α1-PDX/hf obeyed slow-binding inhibition kinetics, as indicated by the biphasic plots, where maximal inhibition was achieved more rapidly with increasing concentrations of α1-PDX/hf (Fig. 2A). The serpin-enzyme complexes were kinetically trapped (i.e., slow off-rate) because no furin activity was liberated when the assays were extended for up to 6 hr (data not shown). By contrast, α1-PIT/hf was unable to inhibit furin at any concentration tested (Fig. 2B), although it potently inhibited thrombin (Table 1). Nonlinear regression analysis of the data in Fig. 2A showed that the initial velocity (v0) was inversely proportional to the concentration of α1-PDX/hf. This finding suggests that the inhibition of furin by α1-PDX/hf follows a two-step mechanism whereby rapid formation of a loose α1-PDX/hf⋅furin complex is followed by a slow isomerization to the tightly bound product (illustrated by the asymptotic component of each curve).
Table 1.
Kinetic and stoichiometric parameters for the proprotein convertases
| Enzyme | Coumarinamide substrate
|
Peptide-based inhibitor
|
Protein-based inhibitor
|
||
|---|---|---|---|---|---|
| pERTKR-MCA Km, μM | Dec-RVKR-CH2Cl Ki, nM | α1-PIT/hf
|
α1-PDX/hf
|
||
| Ki, nM | Ki, nM | SI | |||
| Furin | 4.7 | 2.0 | >500 | 1.4 | 2¶ |
| Furin/f† | 3.2 | 0.60 | >500 | 0.60§ | 2¶ |
| PC6B/f† | 0.41 | 0.11 | >500 | 2.3§ | 8¶ |
| PC3/f† | 25 | 2.0 | >500 | 260§ | 40¶ |
| PC2 | 54 | 0.36 | >500 | 1,000 | nd |
| PACE-4/f† | 6.3 | 3.6 | >500 | >5,000 | nd |
| PC7/f† | 19 | 0.12 | >500 | >5,000 | nd |
| Thrombin | 12‡ | nd | 0.05 | >5,000 | nd |
Assays were performed in duplicate. Reported values are the mean of three independent experiments (SEM <15%). All PCs were active-site titrated with Dec-RVKR-CH2Cl. Calculations assume α1-PDX/hf, α1-PIT/hf, and Dec-RVKR-CH2Cl are fully active. SI values were determined as described in legend to Fig. 4. nd, not determined.
Molecular mass determined by Western blot using mAb M2: Furin/f (86 kDa); PC6B/f [144 kDa (major band) and 60 kDa (minor band)]; PC3/f [86 kDa (major band) and 67 kDa (minor band)]; PACE-4/f (102 kDa); PC7/f (79 kDa).
Boc-D(Bz)-PR-MCA.
EI* complex determined by Western blot; α1-PDX/hf⋅furin/f (160 kDa); α1-PDX/hf⋅PC6B/f (two bands corresponding to the 144- and 60-kDa forms); α1-PDX/hf⋅PC3/f (two bands corresponding to the 86- and 67-kDa forms).
Cleaved α1-PDX/hf (I*) detected on incubation with the respective enzyme.
The stability of the α1-PDX/hf⋅furin complex was examined by assessing its resistance to SDS denaturation. Increasing amounts of α1-PDX/hf or α1-PIT/hf were incubated in the presence or absence of FLAG-tagged soluble furin (furin/f). After incubation to generate serpin-enzyme complexes, residual furin activity was inhibited by addition of EDTA, and the reaction mixtures were analyzed by Western blot (Fig. 2 C and D). In agreement with the predicted mass of α1-PDX/hf, incubation of this serpin in the absence of furin showed a single 47-kDa protein (Fig. 2C, lanes 2, 4, 6, and 8). However, coincubation resulted in a shift of furin/f and α1-PDX/hf to a single high Mr band (≈160 kDa) corresponding to the kinetically trapped, SDS-resistant α1-PDX/hf⋅furin/f complex (Fig. 2C, lanes 1, 3, 5, and 7). By contrast, coincubation of α1-PIT/hf with furin/f failed to generate an SDS-stable complex (Fig. 2D).
The temporal relationship between α1-PDX/hf⋅furin complex formation and inhibition of furin activity also was examined (Fig. 3). The rate of formation (50% complete at ≈5 sec and ≈100% complete by 2 min) was coincident with the loss of furin activity. In addition, a less-abundant cleaved form of α1-PDX/hf also accumulated in parallel with the SDS-stable complex. Matrix-assisted laser desorption ionization-MS analysis of the products revealed that the reaction between furin and α1-PDX/hf resulted in P1-P1′ bond cleavage of the α1-PDX/hf RSL (-ArgP4-Ile-Pro-ArgP1-↓Ser). The molecular mass observed for the liberated N-terminal fragment (42,652 ± 43 Da) was in agreement with the calculated molecular mass (42,614 Da, relative error 0.09%). Thus, the rate of complex formation, cleavage of the α1-PDX/hf, and loss of enzymatic activity, are tightly coupled.
Figure 3.

Time course of inhibition and complex formation between α1-PDX/hf and furin/f. Replicate tubes containing furin/f (10 nM) and α1-PDX/hf (78 nM) were incubated at room temperature. At each time point pERTKR-MCA (100 μM) was added to one set of tubes to measure residual furin/f activity. Shown is one of three independent experiments (variability < 10%). (Inset) At the indicated times, the replicate reactions were stopped by addition of EDTA (final concentration 5 mM), and samples were processed for Western blot analysis.
Stoichiometry of Inhibition (SI) and Determination of Ki.
After formation of the α1-PDX/hf⋅furin acyl intermediate (EI′), α1-PDX/hf may either be hydrolyzed and released (I*) or it may trap the enzyme in a kinetically stable SDS-resistant complex (EI*) (Figs. 2C and 3). The relative flux of a serpin through these pathways reflects its efficiency as an inhibitor for a given endoprotease and is described as the SI (26);
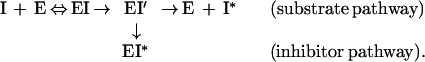 |
Titration experiments were performed to determine the SI between α1-PDX/hf and furin. First, the amount of active enzyme (E0) was determined by titrating furin activity with the active-site-directed irreversible inhibitor Dec-RVKR-CH2Cl. In a parallel analysis, furin activity was titrated with α1-PDX/hf (Fig. 4). Regression analysis of residual furin activity as a function of [I]0:[E]0 showed that approximately 2 mol of α1-PDX/hf are required to inactivate 1 mol of furin, indicating equal flux of this serpin through the inhibitory and hydrolytic pathways (Fig. 4, Inset).
Figure 4.

Tight-binding titration of furin by α1-PDX/hf and Dec-RVKR-CH2Cl. Furin/f (5.4 nM) was incubated with increasing amounts of α1-PDX/hf (○) and Dec-RVKR-CH2Cl (▪) for 30 min at room temperature. pERTKR-MCA (100 μM) was added to determine residual furin activity. Because valid titration curves can be obtained for tight-binding titrants when [E]0/Ki > 2 (24), the Ki of α1-PDX/hf for furin was obtained by fitting the data to an equation for equilibrium binding (24). This analysis revealed a value of Ki = 0.60 nM and a value of [E]0 = 10 nM. Importantly, the calculated ratio of [E]0/Ki = 17 confirmed the validity of the Ki value obtained by using α1-PDX/hf as tight-binding titrants of furin. (Inset) The data shown were used to determine the stoichiometry of the reaction between α1-PDX/hf and furin. The abscissa shows the inhibitor/enzyme ratio using a value for active furin ([E]0 = 5.4 nM) determined by titration using the active-site-directed irreversible inhibitor Dec-RVKR-CH2Cl. The data are averages of duplicate samples and are representative of three independent experiments.
The inhibition of furin by α1-PDX/hf showed that α1-PDX/hf is a tight-binding titrant of furin (Fig. 4). Indeed, α1-PDX/hf fulfilled several criteria of an active site titrant including its stability (Fig. 2A), its formation of a kinetically trapped complex with furin (Fig. 2C), and its rapid combination with the furin active site (Fig. 3).
Intrinsic Specificity of α1-PDX/hf.
The potent inhibition of furin by α1-PDX/hf coupled with its inability to inhibit either elastase or thrombin (Table 1 and ref. 12) suggests this serpin is selective for furin. Therefore, we evaluated the intrinsic specificity of α1-PDX/hf by quantifying its inhibitory properties against the other PCs. To generate active preparations of the other PCs, vaccinia virus recombinants were constructed that expressed FLAG-tagged variants (inserted C terminal to the propeptide cleavage site) of PACE-4, PC3, PC6B, and PC7. Detection of the expressed enzymes was facilitated by the inserted FLAG tag. Because proPC2 activation is disrupted by insertion of the FLAG tag (unpublished results), a nonepitope-tagged PC2 construct was used. Vaccinia virus recombinants encoding each PC then were used to express the enzymes as described in Materials and Methods.
Initially, the Km for hydrolysis of the synthetic peptide substrate pERTKR-MCA was determined for each PC (Table 1). Whereas the PCs display unique cleavage site specificity with proprotein substrates (1), this small fluorogenic substrate can be hydrolyzed by each member of the family. The similar Kms for native- and epitope-tagged furin and PC3 constructs (Table 1 and ref. 8) demonstrate the lack of effect on enzyme expression and activity of this epitope-tag inserted C terminal to the propeptide cleavage site.
The selectivity and effectiveness of α1-PDX/hf for each PC then was compared with α1-PIT/hf and Dec-RVKR-CH2Cl by determining the Ki values (Table 1). Using these criteria, the PCs exhibited similar reactivity toward the peptide inhibitor (Ki = 0.11–3.6 nM). Thus, like peptide substrates, this irreversible peptidyl inhibitor does not discriminate between the PCs. In contrast, α1-PDX/hf displayed a striking selectivity between the PCs. α1-PDX/hf potently inhibited both furin/f (Ki = 0.60 nM) and PC6B/f (Ki = 2.3 nM). However, the SI for PC6B/f was markedly higher (4-fold), indicating that it partitions through the hydrolytic pathway more frequently than furin. Also, α1-PDX/hf inhibits PC3/f approximately 400-fold less effectively than it does furin (Ki = 260 nM) and partitioning greatly favors the hydrolytic pathway relative to the formation of the SDS-stable complex (SI = 40). Furthermore, α1-PDX is a weak competitive inhibitor of PC2 (Ki = 1 μM), suggesting it is unable to form an acyl intermediate. Neither PACE-4/f nor PC7/f are inhibited by α1-PDX at the concentrations tested.
Inhibition of PEA Cytotoxicity by Exogenous α1-PDX/hf.
The selective inhibition of furin by α1-PDX/hf combined with the inherent stability of the α1-PDX/hf⋅furin complex (EI*) suggested that this serpin could be used to inhibit the processing of furin substrates in vivo. Therefore, we investigated the capacity of α1-PDX/hf to prevent the furin-dependent cytotoxicity of PEA. The 67-kDa protoxin requires cleavage within endosomes by furin at the consensus site -ArgP4-Gln-Pro-ArgP1-↓ to become cytotoxic (6). The excised 37-kDa C-terminal fragment translocates into the cytosol where it ADP ribosylates elongation factor 2 to inhibit protein synthesis (6).
Replicate plates of cells pretreated with increasing concentrations of α1-PDX/hf or α1-PIT/hf were incubated with 10 nM PEA. The furin-dependent activation of PEA was monitored by measuring the rate of protein synthesis. Whereas control cells were completely sensitive to PEA, cells pretreated with α1-PDX/hf showed a concentration-dependent protection against toxin activation, with 10 μM α1-PDX/hf completely blocking the effect of the toxin. By contrast, at the same concentrations, α1-PIT/hf was unable to protect the cells from PEA (Fig. 5).
Figure 5.

Exogenous α1-PDX/hf neutralizes PEA-induced cytotoxicity in cultured cells. Replicate cultures of A7 melanoma cells were incubated in the absence or presence of increasing concentrations of α1-PIT/hf (open bars) or α1-PDX/hf (filled bars) for 2 hr at 37°C. Subsequently, PEA (10 nM final) was added, and the cultures were incubated at 37°C for 6 hr. Cells then were incubated with [35S]-Met/Cys for an additional 30 min at 37°C and harvested, and [35S]-Met/Cys incorporation into cellular proteins was determined by scintillation counting of the trichloroacetic acid precipitates. Data shown are representative of three assays (error bars represent standard error).
DISCUSSION
The broad and important role of furin in the proteolytic maturation of proprotein substrates (3), including the activation of many pathogen molecules [e.g., viral envelope glycoproteins (7) and bacterial toxins (6)], has made this endoprotease a target for the development of potent and selective inhibitors. Here, we report the biochemical characterization of α1-PDX/hf, an engineered α1-AT variant containing the minimal consensus furin cleavage site (-ArgP4-Xaa-Xaa-ArgP1-) in its RSL. We show that preparative amounts of the active His- and FLAG-tagged α1-AT variants can be expressed in bacteria and readily purified to homogeneity. Kinetic studies showed α1-PDX/hf inhibits furin by a slow tight-binding mechanism characteristic of serpin molecules and operates as a suicide substrate inhibitor (26). Once bound to furin’s active site, α1-PDX/hf is partitioned with equal probability to undergo proteolysis by furin at the C-terminal side of the -ArgP4-Ile-Pro-ArgP1-↓ site (I*) or to form a kinetically trapped SDS-stable complex (EI*) with the enzyme. Analysis of the specificity of α1-PDX/hf showed it is selective for furin and, to a lesser extent PC6B, but not for other PC family members. Finally, the utility of α1-PDX/hf to serve as a potential therapeutic agent was demonstrated by its ability to block furin-dependent cell killing by PEA.
Our results show that α1-PDX inhibits members of the PC family of mammalian subtilisins by the same mechanism used by α1-AT to inhibit chymotrypsin family members. The ability of α1-PDX to form SDS-stable complexes with furin, PC6B, and PC3, but not PC2, PACE-4, or PC7, partially illustrates the enzyme selectivity of α1-PDX. Unlike chymotrypsin family enzymes, however, the SI between α1-AT and subtilase superfamily members is highly variable (Table 1 and ref. 27). Thus, although α1-PDX has a similar Ki for both furin and PC6B, the SI is strikingly different; four times as much α1-PDX is required to inhibit PC6B at saturating concentrations compared with furin. The highly selective inhibition of furin by α1-PDX compared with the other PCs illustrates that the structure of the reactive center of each PC is unique, implying a different substrate specificity for each family member.
Compared with previously reported furin inhibitors, the enzyme specificity of α1-PDX is highly selective. For example, peptidyl chloromethyl ketones show little selectivity for any PC member (Table 1). Furthermore, their cytotoxicity and the unstable nature of the chloromethane group preclude their usefulness as therapeutic antiproteolytic agents. Similarly, both PI8 and the furin-directed α2-macroglobulin variant show very little enzyme specificity. Both proteins inhibit many proteases in addition to furin greatly limiting their use as therapeutics. Thus, the unique specificity of the α1-PDX/hf-RSL reported here demonstrates a major advantage of the α1-AT scaffold for the design of furin-selective inhibitors.
The solved structure of α1-AT shows that, unlike other serpins, its RSL is stabilized by direct interactions with the core of the protein (28). Based on this information, we have generated an energy-minimized model of α1-PDX (Fig. 6). Ideal for furin, the P1 and P4 residues are predicted to be unnecessary for stabilization of the RSL, and instead they face the solvent where they can interact with the target enzyme’s active site. Importantly, our model of the α1-PDX/hf-RSL provides a basis for determining the interactions important for substrate binding and selectivity between the different PCs. This model also can be used to construct a three-dimensional template with which to develop combinatorial libraries for small molecule furin inhibitors or to synthesize conformationally constrained peptoids (29).
Figure 6.

Model structure of α1-PDX. The global energy-minimum structure was compiled by using the sybyl version 6.3 software (Tripos, St. Louis) based on the atomic coordinates reported for α1-AT (28). The reactive loop P15-P′5 (Gly344–Glu363) is depicted in red. Introduction of the double Arg substitution (Arg358 and Arg355) has negligible effect on the overall conformation because both side groups face the solvent (hence the furin active site). The RSL is stabilized by direct interactions with the core of the protein. These include hydrogen bonding between the P5 Glu (Glu354) and Arg196 as well as hydrophobic interactions between the P3 Ile (Ile356) and P7 Phe (Phe352) and also between the prolines at positions P2, P3′, and P4′ (in addition to the rigidity contributed by these residues).
Our results conflict with those of Benjannet et al. (30), who reported, using overexpression methods, that (i) α1-PDX is a competitive inhibitor of furin and (ii) α1-PDX is not a selective but rather a general inhibitor of the PCs in the constitutive secretory pathway. However, because excessive amounts of enzyme interfere with detection of the serpin-enzyme complex (27), the methodology used by Benjannet et al. may have precluded determination of the inhibitory mechanism. Furthermore, it is likely that overexpression methods limit the ability to dissect a complex issue such as the inherent selectivity of serpin-based inhibition of proteinases (26). For example, overexpression of α1-PIT (a poor inhibitor of furin, see Table 1 and ref. 12) in COS cells inhibits furin (31).
The inherent selectivity and stability of α1-PDX suggest its broad applicability as a therapeutic agent. Indeed, the observation that α1-PDX/hf blocks furin-dependent cell killing by PEA demonstrates its therapeutic potential. Pseudomonas aeruginosa is a clinically important pathogen constituting a major complication in burn patients and people afflicted with cystic fibrosis (32). Animal studies show the contribution of PEA to pathogen virulence (33). Indeed, the exotoxin A gene is amplified in Pseudomonal colonies isolated from the lungs of cystic fibrosis patients, suggesting that it may contribute to the complications of that disease (34). Studies examining the potential of α1-PDX in the treatment of Pseudomonal infections and other pathogens that use furin to become virulent are in progress.
Acknowledgments
We thank C. Lipps for plasmid construction. We thank J. Hicks C. Haskell-Luevano, R. Brennan, L. Boismenu, J. Kedit, and members of the Thomas lab for helpful discussions and reading of the manuscript. We thank M. Gillespie for the PC7 cDNA, K. Nakayama for the PC6B cDNA, S. Smeekens for the PACE-4 cDNA, and R. Draper for the PEA. This work was supported by National Institutes of Health grants (G.T.) and Hedral Therapeutics. F.J. is supported by a Medical Research Council (Canada) postdoctoral fellowship. G.L. is supported by National Research Service Award Fellowship DK09394–02.
ABBREVIATIONS
- α1-PDX
α1-antitrypsin Portland
- α1-PDX/hf
His- and FLAG-tagged α1-PDX
- RSL
reactive site loop
- PC
proprotein convertase
- PEA
Pseudomonas exotoxin A
- α1-AT
α1-antitrypsin
- pERTKR-MCA
pGlu-Arg-Thr-Lys-Arg-4-methylcoumaryl-7-amide
- Dec-RVKR-CH2Cl
decanoyl-Arg-Val-Lys-Arg-CH2Cl
- α1-PIT
α1-antitrypsin Pittsburgh
- RP-HPLC
reverse-phase HPLC
- SI
stoichiometry of inhibition
References
- 1.Rouille Y, Duguay S J, Lund K, Furuta M, Gong Q, Lipkind G, Oliva A A J, Chan S J, Steiner D F. Front Neuroendocrinol. 1995;16:322–361. doi: 10.1006/frne.1995.1012. [DOI] [PubMed] [Google Scholar]
- 2.Seidah N G, Chrétien M. Curr Opin Biotechnol. 1997;8:602–607. doi: 10.1016/s0958-1669(97)80036-5. [DOI] [PubMed] [Google Scholar]
- 3.Nakayama K. Biochem J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molloy S S, Bresnahan P A, Leppla S H, Klimpel K R, Thomas G. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 5.Thomas L, Leduc R, Thorne B A, Smeekens S P, Steiner D F, Thomas G. Proc Natl Acad Sci USA. 1991;88:5297–5301. doi: 10.1073/pnas.88.12.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordon V M, Klimpel K R, Arora N, Henderson M A, Leppla S H. Infect Immun. 1995;63:82–87. doi: 10.1128/iai.63.1.82-87.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garten W, Hallenberger S, Ortmann D, Schafer W, Vey M, Angliker H, Shaw E, Klenk H D. Biochimie. 1994;76:217–225. doi: 10.1016/0300-9084(94)90149-x. [DOI] [PubMed] [Google Scholar]
- 8.Jean F, Boudreault A, Basak A, Seidah N G, Lazure C. J Biol Chem. 1995;270:19225–19231. doi: 10.1074/jbc.270.33.19225. [DOI] [PubMed] [Google Scholar]
- 9.Jean F, Basak A, DiMaio J, Seidah N G, Lazure C. Biochem J. 1995;307:689–695. doi: 10.1042/bj3070689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angliker H. J Med Chem. 1995;38:4014–4018. doi: 10.1021/jm00020a016. [DOI] [PubMed] [Google Scholar]
- 11.Lu W, Zhang W, Molloy S S, Thomas G, Ryan K, Chiang Y, Anderson S, Laskowski M., Jr J Biol Chem. 1993;268:14583–14585. [PubMed] [Google Scholar]
- 12.Anderson E D, Thomas L, Hayflick J S, Thomas G. J Biol Chem. 1993;268:24887–24891. [PubMed] [Google Scholar]
- 13.Van Rompaey L, Ayoubi T, Van De Ven W, Marynen P. Biochem J. 1997;326:507–514. doi: 10.1042/bj3260507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahlen J R, Jean F, Thomas G, Foster D C, Kisiel W. J Biol Chem. 1998;273:1851–1854. doi: 10.1074/jbc.273.4.1851. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe M, Hirano A, Stenglein S, Nelson J, Thomas G, Wong T C. J Virol. 1995;69:3206–3210. doi: 10.1128/jvi.69.5.3206-3210.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bresnahan P A, Leduc R, Thomas L, Thorner J, Gibson H L, Brake A J, Barr P J, Thomas G. J Cell Biol. 1990;111:2851–2859. doi: 10.1083/jcb.111.6.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu G, Thomas L, Warren R A, Enns C A, Cunningham C C, Hartwig J H, Thomas G. J Cell Biol. 1997;139:1719–1733. doi: 10.1083/jcb.139.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiefer M C, Tucker J E, Joh R, Landsberg K E, Saltman D, Barr P J. DNA Cell Biol. 1991;10:757–769. doi: 10.1089/dna.1991.10.757. [DOI] [PubMed] [Google Scholar]
- 19.Smeekens S P, Auruch A S, LaMendola J, Chan S J, Steiner D F. Proc Natl Acad Sci USA. 1991;88:340–344. doi: 10.1073/pnas.88.2.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakagawa T, Murakami K, Nakayama K. FEBS Lett. 1993;327:165–171. doi: 10.1016/0014-5793(93)80163-o. [DOI] [PubMed] [Google Scholar]
- 21.Bruzzaniti A, Goodge K, Jay P, Taviaux S A, Lam M H, Berta P, Martin T J, Moseley J M, Gillespie M T. Biochem J. 1996;314:727–731. doi: 10.1042/bj3140727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molloy S S, Thomas L, VanSlyke J K, Stenberg P E, Thomas G. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamango N S, Zhu X, Lindberg I. Arch Biochem Biophys. 1996;330:238–250. doi: 10.1006/abbi.1996.0249. [DOI] [PubMed] [Google Scholar]
- 24.Knight C G. Methods Enzymol. 1995;248:85–101. doi: 10.1016/0076-6879(95)48008-0. [DOI] [PubMed] [Google Scholar]
- 25.Travis J, Guzdek A, Potempa J, Watorek W. Biol Chem Hoppe Seyler. 1990;371:3–11. [PubMed] [Google Scholar]
- 26.Wright H T. BioEssays. 1996;18:453–464. doi: 10.1002/bies.950180607. [DOI] [PubMed] [Google Scholar]
- 27.Komiyama T, Gron H, Pemberton P A, Salvesen G S. Protein Sci. 1996;5:874–882. doi: 10.1002/pro.5560050509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elliott P R, Lomas D A, Carrell R W, Abrahams J P. Nat Struct Biol. 1996;3:676–681. doi: 10.1038/nsb0896-676. [DOI] [PubMed] [Google Scholar]
- 29.Dolle R E. Mol Divers. 1997;2:223–236. doi: 10.1007/BF01715638. [DOI] [PubMed] [Google Scholar]
- 30.Benjannet S, Savaria D, Laslop A, Munzer J S, Chrétien M, Marcinkiewicz M, Seidah N G. J Biol Chem. 1997;272:26210–26218. doi: 10.1074/jbc.272.42.26210. [DOI] [PubMed] [Google Scholar]
- 31.Wasley L C, Rehemtulla A, Bristol J A, Kaufman R J. J Biol Chem. 1993;268:8458–8465. [PubMed] [Google Scholar]
- 32.Bodey G P, Bolivar R, Fainstein V, Jadeja L. Rev Infect Dis. 1983;5:279–313. doi: 10.1093/clinids/5.2.279. [DOI] [PubMed] [Google Scholar]
- 33.Miyazaki S, Matsumoto T, Tateda K, Ohno A, Yamaguchi K. J Med Microbiol. 1995;43:169–175. doi: 10.1099/00222615-43-3-169. [DOI] [PubMed] [Google Scholar]
- 34.Hoiby N, Koch C. Thorax. 1990;45:881–884. doi: 10.1136/thx.45.11.881. [DOI] [PMC free article] [PubMed] [Google Scholar]