

Abstract
Adenosine protects neurons during hypoxia by inhibiting excitatory synaptic transmission and preventing NMDA receptor activation. Using an adeno-associated viral (AAV) vector containing Cre recombinase, we have focally deleted adenosine A1 receptors in specific hippocampal regions of adult mice. Recently, we found that deletion of A1 receptors in the CA1 area blocks the postsynaptic responses to adenosine in CA1 pyramidal neurons, and deletion of A1 receptors in CA3 neurons abolishes the presynaptic effects of adenosine on the Schaffer collateral input [J Neurosci 23 (2003) 5762]. In the current study, we used this technique to delete A1 receptors focally from CA3 neurons to investigate whether presynaptic A1 receptors protect synaptic transmission from hypoxia. We studied the effects of prolonged (1 h) hypoxia on the evoked field excitatory postsynaptic potentials (fEPSPs) in the CA1 region using in vitro slices. Focal deletion of the presynaptic A1 receptors on the Schaffer collateral input slowed the depression of the fEPSPs in response to hypoxia and impaired the recovery of the fEPSPs after hypoxia. Delayed responses to hypoxia linearly correlated with impaired recovery. These findings provide direct evidence that the neuroprotective role of adenosine during hypoxia depends on the rapid inhibition of synaptic transmission by the activation of presynaptic A1 receptors.
Keywords: CA1, Schaffer collaterals, electrophysiology, AAV, Cre recombinase, inducible knock-out mice
Extracellular adenosine levels in the CNS increase after head injury, seizures, hypoxia, hypoglycemia and ischemia (Winn et al., 1979, 1981; Zhu and Krnjevic, 1993; Headrick et al., 1994; Dunwiddie, 1999). Neuronal damage in these conditions is primarily caused by excessive release of glutamate (Simon et al., 1984; Obrenovitch and Urenjak, 1997; Sattler et al., 2000), and adenosine protects neurons by activating A1 receptors that reduce glutamate release and NMDA receptor activation (Fredholm, 1997; Sweeney, 1997; de Mendonca et al., 2000). Accordingly, protection against hypoxia and ischemia can be achieved by increasing extracellular levels of adenosine through inhibition of adenosine degradation or reuptake (Gidday et al., 1995; Miller et al., 1996; Jiang et al., 1997). Conversely, adenosine receptor antagonists or increased breakdown of extracellular adenosine exacerbates neuronal loss from hypoxia and ischemia (Sweeney, 1997; de Mendonca et al., 2000).
In the CA1 region, hypoxia and ischemia disrupt synaptic transmission, protein synthesis, maintenance of ATP levels, cytoskeletal integrity, and neuronal morphology (Lipton, 1999; Wang et al., 1999). However, hypoxia also induces release of adenosine (Dale et al., 2000; Frenguelli et al., 2003) which rapidly depresses synaptic transmission and neuronal firing (Lipton and Whittingham, 1979; Fowler, 1989; Gribkoff et al., 1990). This inhibition of neuronal activity prevents glutamatergic excitotoxicity, allowing full recovery from hypoxia (Sebastiao et al., 2001). These effects are primarily mediated by adenosine A1 receptors because A1 receptor antagonists or constitutive lack of A1 receptors prevents the full recovery of synaptic activity after hypoxia (Fowler, 1989; Gribkoff et al., 1990; Johansson et al., 2001). Adenosine may protect neurons from hypoxia by preventing NMDA receptor activation (Sebastiao et al., 2001), but it remains unknown whether this protection is mediated by activation of presynaptic A1 receptors that inhibit glutamate release.
Pharmacological tools cannot distinguish between pre-and postsynaptic A1 receptors, but we recently developed a method to focally delete A1 receptors in CA1 and CA3 neurons. Injection of an adeno-associated viral (AAV) vector containing Cre recombinase (AAV-Cre) into the brains of mice with loxP sites flanking the major coding exon for the A1 receptor disrupts the A1 gene in adult mice (Scammell et al., 2003). This focal deletion of A1 receptors from the CA1 neurons blocks their postsynaptic responses to adenosine. In contrast, deletion of A1 receptors from the CA3 neurons abolishes the presynaptic effects of adenosine on the Schaffer collateral input (Scammell et al., 2003). In the current study, we used the AAV-Cre technique to investigate whether presynaptic A1 receptors protect synaptic transmission from hypoxia.
EXPERIMENTAL PROCEDURES
Animals and microinjections
Inducible A1 receptor knock-out mice were produced using homologous recombination to introduce loxP sites around the major coding exon of the adenosine A1 receptor gene (Scammell et al., 2003). Cre recombinase can then be introduced to delete the sequence between the loxP sites, producing a truncated and non-functional A1 receptor gene.
Mice were housed in a pathogen-free barrier facility maintained at 21.5–22.5 °C with lights on at 7:00 A.M. and off at 7:00 P.M. Mice had food and water available ad libitum. The Institutional Animal Care and Use Committee and The Committee on Microbiologic Safety of Harvard Medical School approved all procedures in accordance with international guidelines. All efforts were made to minimize the number of animals used and their suffering. Ten adult, male mice weighing 25–35 g were anesthetized with a bolus of chloral hydrate (450 mg/kg i.p.), and 1 μl AAV-Cre was ster-eotaxically microinjected into the right dorsal hippocampus (1.9 mm behind bregma, 2.1 mm lateral, and 2.1 mm below the dural surface). As a control, 1 μl of an AAV expressing green fluorescent protein (AAV-GFP) was injected into the left dorsal hippocampus. To minimize tissue injury, each AAV was injected slowly over 1 h, using a pressure-injection system (Picospritzer II; General Valve, Fairfield, NJ, USA) and glass pipettes with a 10- to 20-μm diameter tip (Scammell et al., 1998).
Slice preparation and extracellular field recordings
Two to 4 weeks after the AAV injections, in vitro slices were prepared from inducible A1 receptor knock-out mice. Under isoflurane anesthesia, mice were decapitated and the brain rapidly removed and placed in cold artificial cerebrospinal fluid (ACSF) containing (in mM): 128 NaCl, 3 KCl, 0.5 NaH2PO4, 1 MgCl2, 1.5 CaCl2, 23.5 NaHCO3, and 10 glucose, equilibrated with 95% O2 and 5% CO2 (pH 7.35, 315–320 mOsm). Coronal forebrain slices containing the hippocampus (400 μm thick) were cut using a vibrating microtome (VT1000; Leica, Bannockburn, IL, USA) while maintained in ice cold oxygenated ACSF. Slices were hemisected and kept at 22 °C in oxygenated ACSF for 1 h before the recording started.
Field excitatory postsynaptic potentials (fEPSPs) were recorded from the CA1 stratum radiatum (Fig. 1A) with glass electrodes filled with 2 M NaCl (2–4 MΩ) using a Multiclamp 700A amplifier (Axon Instruments, Foster City, CA, USA). Signals were filtered at 1–2 kHz and digitized at 40 kHz with Digidata 1200 hardware and pClamp 8.2 software (Axon Instruments). Using a concentric stimulating electrode (FHC, Bowdoinham, ME, USA) placed in the stratum radiatum adjacent to the CA2 field (Fig. 1A), the Schaffer collateral fibers were stimulated every 20 s with single, constant current pulses of 0.2 ms duration. Stimulation pulses were delivered with a constant-current source (Iso-flex; A.M.P.I. Jerusalem, Israel), triggered by Clampex software (Axon Instruments), and stimulus strength was adjusted to give approximately 75% of maximum fEPSP amplitude ranging between 0.5 and 2 mV. Slices were recorded submerged and perfused (6 ml/min) with ACSF (Pearson et al., 2001) maintained at 32 °C using a temperature controller (TC-344B; Warner Instruments, Hamden, CT, USA). In every experiment, adenosine effects were measured initially, and then after the complete washout of adenosine, the response to 1 h hypoxia was tested. Hypoxia was induced using ACSF pre-equilibrated with 95% N2 and 5% CO2, and each slice received only one exposure to hypoxia. Stock solutions of the adenosine A1 receptor antagonist 8-cyclopentyltheophylline (CPT) were prepared in dimethyl sulfoxide (DMSO); the final concentration of DMSO in the ACSF was <0.025%.
Fig. 1.

The A1 receptor antagonist CPT impairs the responses to hypoxia. (A) Electrode positions for the Schaffer collateral stimulations (S) and fEPSPs recording (R). Scale bar=200 μm. (B) Effects of 1 h hypoxia on fEPSPs in the absence (B1) and the presence of 1 μM CPT (B2). Each trace is the average of four consecutive fEPSPs recorded during 1-min periods: just before hypoxia starts (Baseline), after 3 min hypoxia, at the end of the 1 h hypoxia and at the end of the 30 min recovery. (C) Hypoxia quickly and reversibly depresses fEPSP slopes in control conditions (n=4), but in the presence of the A1 receptor antagonist CPT (n=6), hypoxia-induced depression of the fEPSPs occurs at a slower rate and the recovery from hypoxia is impaired. In the graph the fEPSP slopes are represented every 1 min and are expressed as the mean±S.E.M.; 100% is the average of fEPSP slopes during baseline (10 min preceding hypoxia).
In situ hybridization
In situ hybridization for A1 receptor mRNA was performed as previously described (Scammell et al., 2003). After electrophysiologic recordings, slices were fixed overnight in 4% paraformaldehyde and equilibrated in 20% sucrose with DEPC. To ensure that the sections were cut parallel to the slice we first cut a flat surface in the frozen embedding medium. We then laid the slice on a microscope slide and flipped the slide to attach the slice to the flat embedding medium. Sections were then cut at 30 μm, mounted on slides, and hybridized with an antisense riboprobe directed against bases 436–900 of the rat A1 receptor cDNA sequence (Reppert et al., 1991). Deletion of A1 receptors from the CA3 region was confirmed using film autoradiography and emulsions.
Data analysis and statistics
Evoked field potentials were quantified as the slope of the fEPSP measured between 10 and 90% of the fEPSP peak amplitude. Only data from slices with histologically confirmed deletion of A1 receptors limited to the CA3 area and with less than 20% fEPSP inhibition by adenosine were used for the analysis of the responses to hypoxia. Only one slice from each animal met these two criteria and in every case this was the slice showing the most extended CA3 deletion. Onset of the hypoxia-mediated depression of the fEPSPs was estimated by fitting the normalized data during the 1 h hypoxia with single exponential functions with a nonlinear least-squares fitting method using IGOR Pro 4.0 (Wave-Metrics, Lake Oswego, OR, USA). Since the fEPSP depression in response to hypoxia depends on both adenosine-dependent and adenosine-independent mechanisms, to calculate the time constant of the adenosine-mediated effect we assumed that adenosine-dependent and -independent effects are compounded (fEPSPslope = e−1/τ1 · e−1/τ2. Therefore the effective inverse decay time constant, measured in control condition can be expressed as 1/τe = 1/τ1 + 1/τ2. Data are presented as means ± S.E.M. and statistical significance was established by unpaired two-tailed t-tests.
RESULTS
Hypoxia in the presence of A1 receptor antagonists
Hypoxia completely depressed the hippocampal fEPSPs within 5 min in slices from wild-type mice. After 1 h of hypoxia, reoxygenation produced a complete recovery of the fEPSPs (Fig. 1). In the presence of the A1 receptor antagonist CPT (1 μM), the fEPSPs was depressed more slowly by hypoxia than in control conditions (Fig. 1). Specifically, the time constant for the depression of the fEPSPs was 10.3±1.5 min (n=6) in the presence of CPT compared with 1.1±0.1 min (n=4) in control ACSF (P=0.0015, unpaired t-test). CPT also impaired the recovery of synaptic transmission (Fig. 1): 30 min after the return to normoxia, the fEPSP slopes recovered to 107±17% of baseline in control ACSF but only to 45.3±9.2% in the presence of CPT (P=0.026, unpaired t-test).
Hypoxia in slices with deletion of presynaptic A1 receptors
To determine whether these neuroprotective effects of adenosine were presynaptic, we focally deleted A1 receptors in the CA3 neurons. Ten inducible receptor knock-out mice A1 were injected with AAV-Cre into the CA3 region to delete A1 receptors and injected contralaterally with AAV-GFP as a control. In situ hybridization confirmed that focal deletion of A1 receptors was restricted to the CA3 area in five animals.
In slices lacking A1 receptor mRNA only in CA3, the response to adenosine was markedly reduced, and the fEPSPs were more vulnerable to hypoxia (Fig. 2). Application of adenosine (50 μM) inhibited the Schaffer collateral fEPSPs by only 14.4±3.0% (n=4) on the AAV-Cre injection side, compared with an inhibition of 57.7±4.5% (n=5) on the AAV-GFP injection side (P=0.002, unpaired t -test). In addition, focal deletion of A1 receptors in the CA3 region slowed the response to hypoxia, and after 1 h of hypoxia, reoxygenation did not elicit a full recovery of synaptic transmission. The time constant of the hypoxia-mediated depression of fEPSPs was 8.5±1.1 min (n=4) in the AAV-Cre injection side compared with 1.1±0.2 min (n=5) in the AAV-GFP injection side (P=0.005, unpaired t-test). Thirty minutes after the return to normoxia, the fEPSP slopes recovered to 63.8±7.7% in the slices with deletion of receptors in CA3 compared with the full A1 recovery (103±7%) in slices from the AAV-GFP injection side (P=0.008, unpaired t-test).
Fig. 2.
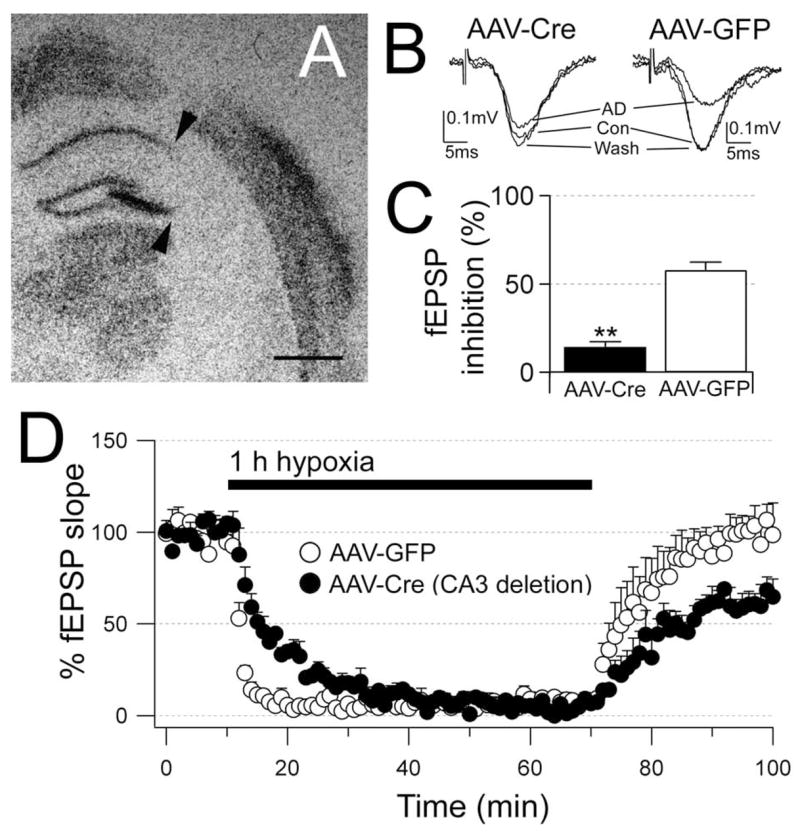
Focal deletion of the presynaptic A1 receptors impairs the responses to adenosine and hypoxia. (A) In situ hybridization for adenosine A1 receptor mRNA shows deletion of receptors in most of CA3 (arrows). (B) In the same slice, the response of the CA1 fEPSPs to adenosine (50 μM) is markedly reduced (AAV-Cre) compared with the response on the opposite side (AAV-GFP). Each trace is the average of six consecutive fEPSPs. (C) The average response to adenosine in slices with deletion of CA3 A1 receptors (n=4) is significantly reduced compared with the AAV-GFP injection side (n=5). (D) Hypoxia-induced depression of the fEPSPs occurs at a slower rate and the recovery from hypoxia is impaired in slices with focal deletion of the CA3 A1 receptors (n=4) compared with the response in slices from the AAV-GFP injection side (n=5). In the graph the fEPSP slopes are represented every 60 s and are expressed as the mean±S.E.M.; 100% is the average of fEPSP slopes during baseline (10 min preceding hypoxia). Significance by unpaired t-tests P<0.01 (**). Scale bar=1 mm.
Across all these experiments, delay in the onset of the hypoxia-mediated inhibition of the fEPSP strongly inversely correlated with the recovery of the fEPSP after the hypoxia (r=0.819; n=19; F=34.7; P<0.0001; Fig. 3).
Fig. 3.

Incomplete recovery occurs when the response to hypoxia is delayed. (A, B) Application of A1 receptor antagonists and focal deletion of the presynaptic A1 receptors on the Schaffer collateral input delays hypoxia-mediated fEPSP depression and impairs recovery from hypoxia. The two graphs compare the response to hypoxia in slices from wild-type mice in normal ACSF and in the presence of CPT (1 μM) and in inducible A1 receptor knock-out mice in slices from the AAV-GFP-injected side and from the AAV-Cre-injected side. (C) Delay in the onset of the hypoxia-mediated fEPSP depression inversely correlates with lack of full recovery from hypoxia. Summary of all experiments: wild-type mice are represented with triangles with recordings in normal ACSF (open triangles; n=4) and in the presence of CPT (filled triangles; n=6); inducible receptor knock-out mice are A1 represented with circles with the recordings from the AAV-Cre-injected side (filled circles; n=4) and from the AAV-GFP-injected side (open circles; n=5). The onset of the fEPSP depression is determined by best fit with single exponential functions. Values are expressed as mean±S.E.M.; 100% is the average of fEPSP slopes during baseline (10 min preceding hypoxia). Significance by unpaired t-tests P<0.01 (**); P<0.05 (*).
DISCUSSION
We found that focal deletion of the presynaptic A1 receptors on the Schaffer collaterals delayed the depression of the CA1 fEPSP in response to hypoxia and impaired the recovery of the fEPSP from prolonged hypoxia, demonstrating that protection of the synaptic inputs during hypoxia requires activation of presynaptic receptors. In A1 addition, we found that delays in responding to hypoxia linearly correlated with lack of recovery, suggesting that full recovery from hypoxia requires rapid depression of synaptic signaling.
Technical comments
AAV-Cre robustly induces recombination in neurons without altering their electrophysiological properties (Ehrengruber et al., 2001; Scammell et al., 2003), but this method has some limitations. First, because this method depends on stereotaxic injections, the region of recombination can vary, but this variability provides useful anatomic controls. Using small volumes of AAV-Cre we achieved relatively specific deletion of the A1receptors in the CA3 area (Fig. 2A). Secondly, these injections induced recombination in most neurons, but the small residual responses to adenosine (Fig. 2B, C) indicate that some A1 receptors persist. Using in situ hybridization, we previously showed that about 90% of the neurons at the AAV-Cre injection site undergo recombination (Scammell et al., 2003). However, even partial deletion of the A1 receptors in the CA3 area worsens the neuronal injury from prolonged hypoxia.
The half life of A1 receptors is 21 h (Hettinger et al., 1998) and AAV-Cre-mediated recombination is complete within 7 days (Kaspar et al., 2002). We recorded hippocampal activity 2 to 4 weeks after the injection of AAV-Cre, and we thus anticipate that A1 receptor proteins were maximally deleted from CA3 neuron and terminals.
Adenosine-mediated fEPSP depression
During hypoxia, release of adenosine (Fowler, 1989; Gribkoff et al., 1990; Frenguelli et al., 2003) is not the only mechanism that depresses synaptic transmission (de Mendonca and Ribeiro, 1997; Coelho et al., 2000). Hypoxia can depress the fEPSPs even when the adenosine receptors are blocked with an A1 antagonist (Fowler, 1989; Latini et al., 1999; Sebastiao et al., 2001) or in A1 receptor knock-out mice (Johansson et al., 2001), although this depression occurs more slowly and, after prolonged hypoxia, the recovery of the fEPSP is incomplete (Johansson et al., 2001; Sebastiao et al., 2001). Consistent with these studies, we found that in the presence of CPT as well as in animals with focal deletion of the presynaptic A1 receptors, the rate of the fEPSP depression in response to hypoxia is slower and the recovery after 1 h of hypoxia is impaired. Using the compound model for adenosine-dependent and -independent effects (see Experimental Procedures) and the time constants of the fEPSP depression measured in control conditions (1.1 min) and in the presence of CPT (adenosine-independent time constant; 10.3 min) we estimate that the time constant of the adenosine-mediated depression of fEPSPs is 1.2 min. This suggests that the adenosine-independent mechanisms do not significantly affect the initial depression of the fEPSPs. Indeed, the contribution of the adenosine-independent mechanisms such as the activation of the muscarinic M2 and the GIII metabotropic glutamatergic receptors, that operate at slower rate, is only apparent when adenosine A1 receptors are blocked (de Mendonca and Ribeiro, 1997; Coelho et al., 2000). Adenosine-independent mechanisms may contribute, along with adenosine, to the inhibition of fEPSPs later on, helping maintain depression of glutamatergic transmission when the A1 receptors have become down-regulated (Onodera and Kogure, 1990; Frenguelli et al., 2003).
Presynaptic A1 receptors protect synaptic transmission from hypoxia
Previous studies have shown that blockage of either NMDA receptors or synaptic transmission facilitates the recovery of synaptic activity after hypoxia (Boening et al., 1989; Fowler and Li, 1998; Sebastiao et al., 2001). It remains unknown whether the protection is mediated by activation of presynaptic A1 receptors that inhibit glutamate release or by activation of postsynaptic A1 receptors that reduce the activation of the NMDA receptors. Several pieces of evidence now suggest that it is a presynaptic mechanism. For example, under hypoxia, when the fEPSPs are abolished, the postsynaptic response to glutamate is preserved (Hershkowitz et al., 1993), the frequency of miniature EPSCs is reduced, but the amplitude is unchanged (Hershkowitz et al., 1993), and ultimately, under hypoxia, when the fEPSPs are not yet abolished, paired-pulse facilitation is increased (Coelho et al., 2000). In the current work, we present direct evidence that presynaptic A1 receptors protect synaptic input from hypoxia.
These experiments focused on the role of presynaptic A1 receptors, but postsynaptic receptors may also be protective by inhibiting NMDA receptors (Greene and Haas, 1985; de Mendonca et al., 1995; Costenla et al., 1999). Thus, focal deletion of A1 receptors from the CA1 neurons would allow one to test the necessity of postsynaptic A1 receptors. Alternatively, one could create recombinant mice with loxP sites flanking a transcriptional blocking sequence upstream of the A1 receptor gene. These mice would have the phenotype of A1 receptor knock-out mice, but injection of AAV-Cre in the CA3 and CA1 regions could define whether the expression of pre- or postsynaptic receptor is sufficient to rescue the knock-out A1 phenotype.
Acknowledgments
This study was supported by the USPHS grants HS33987 and HL60292 and a Sleep Medicine Education and Research Foundation Young Investigator Award. The authors wish to thank Dr. Nancy L. Chamberlin and Dr. Matthew P. Anderson for helpful discussions, Quan H. Ha and Mihn T. Ha for providing excellent technical assistance.
Abbreviations
- AAV
adeno-associated virus
- ACSF
artificial cerebro-spinal fluid
- CPT
8-cyclopentyltheophylline
- DMSO
dimethyl sulfoxide
- fEPSPs
field excitatory postsynaptic potentials
- GFP
green fluorescent protein
References
- Boening JA, Kass IS, Cottrell JE, Chambers G. The effect of blocking sodium influx on anoxic damage in the rat hippocampal slice. Neuroscience. 1989;33:263–268. doi: 10.1016/0306-4522(89)90205-4. [DOI] [PubMed] [Google Scholar]
- Coelho JE, de Mendonca A, Ribeiro JA. Presynaptic inhibitory receptors mediate the depression of synaptic transmission upon hypoxia in rat hippocampal slices. Brain Res. 2000;869:158–165. doi: 10.1016/s0006-8993(00)02381-7. [DOI] [PubMed] [Google Scholar]
- Costenla AR, De Mendonca A, Sebastiao A, Ribeiro JA. An adenosine analogue inhibits NMDA receptor-mediated responses in bipolar cells of the rat retina. Exp Eye Res. 1999;68:367–370. doi: 10.1006/exer.1998.0645. [DOI] [PubMed] [Google Scholar]
- Dale N, Pearson T, Frenguelli BG. Direct measurement of adenosine release during hypoxia in the CA1 region of the rat hippocampal slice. J Physiol. 2000;526(Pt 1):143–155. doi: 10.1111/j.1469-7793.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendonca A, Ribeiro JA. Contribution of metabotropic glutamate receptors to the depression of excitatory postsynaptic potentials during hypoxia. Neuroreport. 1997;8:3667–3671. doi: 10.1097/00001756-199712010-00003. [DOI] [PubMed] [Google Scholar]
- de Mendonca A, Sebastiao AM, Ribeiro JA. Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport. 1995;6:1097–1100. doi: 10.1097/00001756-199505300-00006. [DOI] [PubMed] [Google Scholar]
- de Mendonca A, Sebastiao AM, Ribeiro JA. Adenosine: does it have a neuroprotective role after all? Brain Res Brain Res Rev. 2000;33:258–274. doi: 10.1016/s0165-0173(00)00033-3. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV. Adenosine and suppression of seizures. Adv Neurol. 1999;79:1001–1010. [PubMed] [Google Scholar]
- Ehrengruber MU, Hennou S, Bueler H, Naim HY, Deglon N, Lundstrom K. Gene transfer into neurons from hippocampal slices: comparison of recombinant Semliki Forest virus, adenovirus, adeno-associated virus, lentivirus, and measles virus. Mol Cell Neurosci. 2001;17:855–871. doi: 10.1006/mcne.2001.0982. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists delay hypoxia-induced depression of neuronal activity in hippocampal brain slice. Brain Res. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- Fowler JC, Li Y. Contributions of Na+ flux and the anoxic depolarization to adenosine 5′-triphosphate levels in hypoxic/hypoglycemic rat hippocampal slices. Neuroscience. 1998;83:717–722. doi: 10.1016/s0306-4522(97)00460-0. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Adenosine and neuroprotection. Int Rev Neurobiol. 1997;40:259–280. [PubMed] [Google Scholar]
- Frenguelli BG, Llaudet E, Dale N. High-resolution real-time recording with microelectrode biosensors reveals novel aspects of adenosine release during hypoxia in rat hippocampal slices. J Neurochem. 2003;86:1506–1515. doi: 10.1046/j.1471-4159.2003.01957.x. [DOI] [PubMed] [Google Scholar]
- Gidday JM, Fitzgibbons JC, Shah AR, Kraujalis MJ, Park TS. Reduction in cerebral ischemic injury in the newborn rat by potentiation of endogenous adenosine. Pediatr Res. 1995;38:306–311. doi: 10.1203/00006450-199509000-00006. [DOI] [PubMed] [Google Scholar]
- Greene RW, Haas HL. Adenosine actions on CA1 pyramidal neurones in rat hippocampal slices. J Physiol. 1985;366:119–127. doi: 10.1113/jphysiol.1985.sp015788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff VK, Bauman LA, VanderMaelen CP. The adenosine antagonist 8-cyclopentyltheophylline reduces the depression of hippocampal neuronal responses during hypoxia. Brain Res. 1990;512:353–357. doi: 10.1016/0006-8993(90)90648-U. [DOI] [PubMed] [Google Scholar]
- Headrick JP, Bendall MR, Faden AI, Vink R. Dissociation of adenosine levels from bioenergetic state in experimental brain trauma: potential role in secondary injury. J Cereb Blood Flow Metab. 1994;14:853–861. doi: 10.1038/jcbfm.1994.107. [DOI] [PubMed] [Google Scholar]
- Hershkowitz N, Katchman AN, Veregge S. Site of synaptic depression during hypoxia: a patch-clamp analysis. J Neurophysiol. 1993;69:432–441. doi: 10.1152/jn.1993.69.2.432. [DOI] [PubMed] [Google Scholar]
- Hettinger BD, Leid M, Murray TF. Cyclopentyladenosine-induced homologous down-regulation of A1 adenosine receptors (A1AR) in intact neurons is accompanied by receptor sequestration but not a reduction in A1AR mRNA expression or G protein alpha-subunit content. J Neurochem. 1998;71:221–230. doi: 10.1046/j.1471-4159.1998.71010221.x. [DOI] [PubMed] [Google Scholar]
- Jiang N, Kowaluk EA, Lee CH, Mazdiyasni H, Chopp M. Adenosine kinase inhibition protects brain against transient focal ischemia in rats. Eur J Pharmacol. 1997;320:131–137. doi: 10.1016/s0014-2999(96)00905-3. [DOI] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci USA. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar BK, Vissel B, Bengoechea T, Crone S, Randolph-Moore L, Muller R, Brandon EP, Schaffer D, Verma IM, Lee KF, Heinemann SF, Gage FH. Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc Natl Acad Sci USA. 2002;99:2320–2325. doi: 10.1073/pnas.042678699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latini S, Bordoni F, Pedata F, Corradetti R. Extracellular adenosine concentrations during in vitro ischaemia in rat hippocampal slices. Br J Pharmacol. 1999;127:729–739. doi: 10.1038/sj.bjp.0702591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lipton P, Whittingham TS. The effect of hypoxia on evoked potentials in the in vitro hippocampus. J Physiol. 1979;287:427–438. doi: 10.1113/jphysiol.1979.sp012668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LP, Jelovich LA, Yao L, DaRe J, Ugarkar B, Foster AC. Pre- and peristroke treatment with the adenosine kinase inhibitor, 5′-deoxyiodotubercidin, significantly reduces infarct volume after temporary occlusion of the middle cerebral artery in rats. Neurosci Lett. 1996;220:73–76. doi: 10.1016/s0304-3940(96)13234-1. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Urenjak J. Altered glutamatergic transmission in neurological disorders: from high extracellular glutamate to excessive synaptic efficacy. Prog Neurobiol. 1997;51:39–87. doi: 10.1016/s0301-0082(96)00049-4. [DOI] [PubMed] [Google Scholar]
- Onodera H, Kogure K. Calcium antagonist, adenosine A1, and muscarinic bindings in rat hippocampus after transient ischemia. Stroke. 1990;21:771–776. doi: 10.1161/01.str.21.5.771. [DOI] [PubMed] [Google Scholar]
- Pearson T, Nuritova F, Caldwell D, Dale N, Frenguelli BG. A depletable pool of adenosine in area CA1 of the rat hippocampus. J Neurosci. 2001;21:2298–2307. doi: 10.1523/JNEUROSCI.21-07-02298.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE, Arrigoni E, Thompson MA, Ronan PJ, Saper CB, Greene RW. Focal deletion of the adenosine A1 receptor in adult mice using an adeno-associated viral vector. J Neurosci. 2003;23:5762–5770. doi: 10.1523/JNEUROSCI.23-13-05762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE, Griffin JD, Elmquist JK, Saper CB. Microinjection of a cyclooxygenase inhibitor into the anteroventral preoptic region attenuates LPS fever. Am J Physiol. 1998;274:R783–789. doi: 10.1152/ajpregu.1998.274.3.R783. [DOI] [PubMed] [Google Scholar]
- Sebastiao AM, de Mendonca A, Moreira T, Ribeiro JA. Activation of synaptic NMDA receptors by action potential-dependent release of transmitter during hypoxia impairs recovery of synaptic transmission on reoxygenation. J Neurosci. 2001;21:8564–8571. doi: 10.1523/JNEUROSCI.21-21-08564.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- Sweeney MI. Neuroprotective effects of adenosine in cerebral ischemia: window of opportunity. Neurosci Biobehav Rev. 1997;21:207–217. doi: 10.1016/s0149-7634(96)00011-5. [DOI] [PubMed] [Google Scholar]
- Wang T, Raley-Susman KM, Wang J, Chambers G, Cottrell JE, Kass IS. Thiopental attenuates hypoxic changes of electrophysiology, biochemistry, and morphology in rat hippocampal slice CA1 pyramidal cells. Stroke. 1999;30:2400–2407. doi: 10.1161/01.str.30.11.2400. [DOI] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine production in the rat during 60 seconds of ischemia. Circ Res. 1979;45:486–492. doi: 10.1161/01.res.45.4.486. [DOI] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. Am J Physiol. 1981;241:H235–242. doi: 10.1152/ajpheart.1981.241.2.H235. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Krnjevic K. Adenosine release is a major cause of failure of synaptic transmission during hypoglycaemia in rat hippocampal slices. Neurosci Lett. 1993;155:128–131. doi: 10.1016/0304-3940(93)90689-i. [DOI] [PubMed] [Google Scholar]