

Abstract
Many molecular diagnostic laboratories have evolved from research laboratories, initially performing low numbers of homebrew assays, but many laboratories now perform more kit-based assays, with ever increasing test volumes. One such assay is assessment of bone marrow transplantation engraftment. Allogeneic bone marrow transplantation is performed primarily in the treatment of hematological malignancies. Monitoring of engraftment was traditionally evaluated using minisatellites (variable number tandem repeats) and Southern blotting, but most laboratories now use Food and Drug Administration-cleared microsatellite (short tandem repeats) kits to assess the extent of engraftment. With the increase in equipment reliability, the use of kit-based assays, and the desire to provide the highest quality clinical data, we began applying traditional clinical pathology quality control tools to the molecular diagnostics laboratory. In this study, we demonstrate this approach using a microsatellite-based bone marrow engraftment assay. We analyzed control samples (pure and mixed) for two different microsatellites to establish quality control parameters and constructed Levey-Jennings charts to monitor both the precision and accuracy of this assay. By incorporating these tools into an overall quality assurance program, a laboratory can identify systematic errors and perform corrective actions before actual assay failure, thereby improving the quality of patient care.
The clinical laboratory has long functioned as a dynamic environment in which novel diagnostic tests are developed from new technologies. Molecular diagnostics combines laboratory medicine with molecular biology and has significantly evolved over the last several decades, benefiting from advances in technology as well as discoveries in the field of molecular genetics. Molecular diagnostics laboratories have generally evolved from research laboratories, and are generally considered to perform high-complexity esoteric tests. The volume of research work in areas such as molecular oncology has translated into a growing list of diagnostic tests for the service laboratory.
Allogeneic bone marrow transplantation is performed for the treatment of many hematological malignancies as well as other diseases, such as leukemia, severe aplastic anemia, myelodysplasia, and severe combined immunodeficiency disease.1 The success of engraftment has long been evaluated using many different techniques, including cytogenetic markers and red blood cell phenotypes to assess chimerism between the donor and recipient, as well as the extent of sustained engraftment.2,3 In light of the discovery and characterization of polymorphic DNA loci that are capable of distinguishing individuals, most laboratories have transitioned to variable number tandem repeats (also called minisatellites) and Southern blotting, although these methods produce only a semiquantitative assessment of engraftment.4,5 More recently, Scharf et al6 proposed a rapid, nonradioactive, quantitative analysis technique to determine the extent of engraftment by analyzing microsatellites amplified with fluorescent primers and resolved on a commercially available DNA sequencer. Although engraftment analysis using single nucleotide polymorphisms (SNPs) and real-time PCR has been demonstrated,7 the overwhelming majority of laboratories continue to use microsatellites, which are also currently used as the standard in the forensic community.8
The standard practice of quality control (QC) procedures has long been used in clinical laboratories. Although QC tools have been applied to high-volume, molecular diagnostic virology testing, they have not been commonly used for lower-volume, molecular diagnostic assays. Tools such as statistical QC techniques can evaluate the results of a work process and identify when they exceed the expected variation under stable routine operations. In a health care laboratory, controls are analyzed to monitor the variation inherent in the testing process. Results from these control samples are expected to fall within certain statistical limits, eg, ∼95% within the mean plus or minus 2 SD. These data are then used to construct control charts (eg, Levey-Jennings charts) to display the variation between these known samples over time.9 Control rules, such as those established by Dr. James O. Westgard (Westgard rules), are used to identify control values that exceed expected variations and determine the acceptability of the performance of the assay and the reporting of patient results.10
Since the purpose of implementing QC is to monitor the performance of the entire analytical process, it is important to choose control materials with appropriate concentrations. Quality requirements in the form of medically important changes (or clinical decision intervals) and total analytical errors (allowable total errors) can readily be used with available QC planning tools. These requirements include the number of materials necessary to monitor the critical medical decision levels and working range of the method, as well as the type of material that will best simulate a true patient sample and be analyzed in the same fashion as actual patient samples. It is important to collect measurements that will characterize the actual performance of the method under each laboratory's own operating conditions. The implementation of the QC materials offers an approach to truly assess the performance of the overall analytical process, including quantitative analysis, after the assay is completed.
In this study, we report the application of QC practices to bone marrow engraftment assessment in a molecular diagnostics laboratory. In the initial evaluation study, the mean and SD were calculated based on data from 20 runs to establish the basic QC parameters. Twenty samples are typically needed to establish sufficiently narrow standard deviations, because if too few controls are included in establishing the ±2 SD range, it will be artificially wide, and runs that should be rejected might pass QC.11 After assessing method performance from the initial evaluation studies, we monitored real-time QC data using these established parameters in the second phase. This QC application has provided a tool to monitor the overall testing performance, including precision and accuracy, of bone marrow engraftment analysis.
Materials and Methods
General Approach
Results of bone marrow engraftment can vary anywhere from 100% donor to 100% recipient. In planning for QC analysis, typically two levels of QC materials at concentrations close to the medical decision levels are included with each analytical run. In full myeloablative bone marrow transplantation for leukemia, small amounts of residual recipient DNA may represent residual leukemic cells, warranting additional treatment, such as donor lymphocyte infusion.
For this assay, we report a formal limit of detection of 5% since we require that minimum peak heights for apparently pure (100% donor or 100% recipient) results be at least 2000 relative fluorescence units (RFU). Since we set the threshold of the instrument at 75 RFU, we can meet this formal limit of detection (75/2000 = 3.75%). In reality, peak heights are in the 4000 to 6000 range, and since we can detect small peaks in the electropherogram that are below 75 RFU, the actual limit of detection is in the 1 to 4% range. Because the true limit of detection is specific to the actual peak height of that particular locus for that patient sample, out of convenience, we do not quote a specific precise limit of detection value for each patient sample.
Since medically relevant decisions are made at these levels, and in light of our quoted limit of detection, we chose control mixes at known chimerism ratios of 95% “donor” (95%D/5%R) and 100% “donor” (100%D/0%R). These QC materials are prepared from the DNA isolated from two different individuals [arbitrarily designated as “donor” (D) and “recipient” (R)]. The quantification of the assay results is determined by two informative alleles of the two individuals.
DNA Isolation
DNA was extracted from peripheral blood (from control individuals) using DNA blood kits (QIAGEN, Valencia, CA), according to the manufacturer's instructions. After determining the DNA concentration (NanoDrop, Wilmington, DE), two master stocks were prepared using the two different individuals' samples [arbitrarily designated “donor” (D) and “recipient” (R)]: 95%D/5%R (w/w) and 100%D/0%R (w/w). These two master stocks were used as quality control materials and included in every single run along with patient samples. Subsequently, the working stock was prepared using the master stock at a final concentration of 2 ng/μL, aliquoted in a final volume of 50 μl, and stored at −20°C (not frost-free). The working stock should last one month and the master stock should last at least four years under these conditions.
Bone Marrow Engraftment Analysis
Extracted DNA from patients and quality control materials were used as the templates for amplification using the AmpFlSTR Profiler PCR Amplification Kit (Applied Biosystems, Foster City, CA). This is a multiplex microsatellite PCR kit originally designed for forensic purposes. It contains locus-specific, fluorophore-labeled (5-FAM, JOE, and NED) and unlabeled primers to amplify the following microsatellite loci: D3S1358, vWA (von Willebrand factor), FGA (fibrinogen α-chain), THO1, TPOX, CSF1PO, D5S818, D13S317, D7S820, and the gender marker, amelogenin. The 25-μL PCR mixture contains 9.55 μl of AmpFlSTR PCR mix, 5 μl of AmpFlSTR Profiler® primer set, 0.45 μl of AmpliTaq Gold DNA polymerase (5 units/μL), 9 μl of H2O (molecular biology reagent grade; Sigma-Aldrich, St. Louis, MO), and 1 μl of DNA template (2 ng/μL). The reaction mixtures were subjected to a denaturation cycle at 95°C for 11 minutes, followed by 28 cycles of 94°C for 1 minute, 59°C for 1 minute, and 72°C for 1 minute, and a final extension of 60°C for 45 minutes in a thermocycler (GeneAmp PCR System 9700) (Applied Biosystems). A final hold at 4°C was used until samples were removed. The 95%D/5%R, 100%D, and no-template controls were also amplified along with the patient samples. Following PCR amplification, 1 μl of the PCR product was diluted in 8.5 μl of deionized formamide (Hi-Di formamide) and 0.5 μl of GeneScan-500 [ROX] internal lane size standards (both from Applied Biosystems). After denaturation at 95°C for 2 minutes and placement on ice for 2 minutes, the samples were analyzed with an ABI PRISM 3100 Genetic Analyzer using Data Collection v3.0, POP-4 polymer, a 36-cm capillary array, dye set F, run module genescan36_pop4, 45 seconds initial injection time, and GS500 profiler.gsp analysis module (all from Applied Biosystems).
After data collection and processing, the peak height and area of a fluorescent signal were displayed in both electropherogram and tabular data. The electropherogram is a chromatographic display with fluorescence intensity (peak height) indicated as relative fluorescence units on the y axis. After the internal size standard peaks are identified, GeneScan (version 3.7) constructs a standard curve relating the size of the molecule to time, and the electropherogram is displayed with the number of bases on the x axis. The peak height of a fluorophore is directly dependent on the emission energy of the fluorophore and the number of molecules present (the amount of the PCR product DNA). Peaks of all heights within the analysis range (relative fluorescence units: 2000 to 7000) are displayed on the electropherogram, but those peaks below the peak amplitude threshold (typically set to 75 RFU) are not listed in tabular form.
Determining the percentage of the “recipient” at an informative locus provides a quantitative measurement of the degree of mixed chimerism. An informative locus consists of an allele that the other person does not share. Furthermore, it cannot be in the stutter position of the other person if a low percentage is being determined. Once selected, the informative loci are used to evaluate the percentage of engraftment after bone marrow transplantation. The instrument also provides the capability of simultaneous detection of four differently colored fluorescent dyes. The ability to distinguish PCR products by both molecular weight and fluorophore allows one to distinguish the 10 different loci included in this kit on a single capillary. One can selectively analyze the data from each locus independently of the other loci that may be detected in the same sample, although one must be aware of the phenomenon of bleeding from one color into another, especially with offscale peaks.
In our setting, the informative loci selected for the two control individuals are FGA and D13S317, which are amplified with blue and yellow fluorescent-labeled primers, respectively. For the FGA locus, the “donor” has alleles of 220 and 232 bases, whereas the “recipient” is a 236-base homozygote. For the D13S317 locus, the “donor” is a 221-base homozygote, and the “recipient” has alleles of 209 and 221 bases. The percentage of “recipient” in the chimeric samples is calculated to determine the degree of engraftment in the posttransplant sample as follows:
where H is peak height, D is a “donor” allele, R designates a “recipient” allele, and D/R is a shared allele. If an allele is shared, only the informative alleles (ie, alleles that distinguish “donor” from “recipient”) are used in the numerator of the calculation. The final reported clinical result is determined by the mean of the calculation results from the two loci.
Results
In the initial phase of this study, we retrospectively analyzed control samples using informative loci to provide baseline values to generate Levey-Jennings plots. As expected, the 100%D/0%R control never showed evidence of individual R's alleles, and thus it was not analyzed further. The minimum requirement for data collection in the initial study is to analyze the control materials to obtain a minimum of 20 measurements over at least a 10-day period. In our study, data from 20 runs (over 1 month) for the 95%D/5%R were used to establish the mean and the SD to construct the Levey-Jennings chart (Table 1). The accuracy of the entire assay process was assessed by bias and the precision was evaluated by %CV. Since it was assumed that the 95%D/5%R control contained exactly 5% of the individual R's DNA, it was noted that there was a small positive bias of the FGA (mean 5.2%) and D13S317 (mean 6.4%) loci by 4% and 28%, respectively. Based on the mean and SD of the initial portion of the study, the %CV values of FGA and D13S317 at 5% are 19.2 and 28.1, respectively.
Table 1.
QC Parameters Established from the Initial Phase of the Study (n = 20)
| Informative locus | FGA | D13S317 |
|---|---|---|
| Mean (%) | 5.2 | 6.4 |
| SD (%) | 1.0 | 1.8 |
| %CV | 19.2 | 28.1 |
In the second phase of this study, we used the Levey-Jennings chart established in the initial phase to monitor the subsequent performance of the assay for 50 new consecutive runs (Figure 1). The data are presented with days on the x axis, and mean plus or minus 3 SDs on the y axis. According to the Westgard multi-rule QC procedure, one control measurement exceeding 2 SDs (12S rule) is used as the warning rule to trigger careful inspection of the control data and the application of the other rules.10 If there are no other violations, the run can be accepted and patient results released. There were three such 12S warnings (Figure 1, circles). For FGA, these occurred on days 10 and 18, and for D13S317, it occurred on day 9. There were no days in which both controls (FGA and D13S317) were outside 2 SDs, and in each case, subsequent control values were well within 2 SDs; thus, the 22S rule was never violated. No other commonly used Westgard rules, such as when one control sample is outside 3 SDs (13S rule), were violated.
Figure 1.
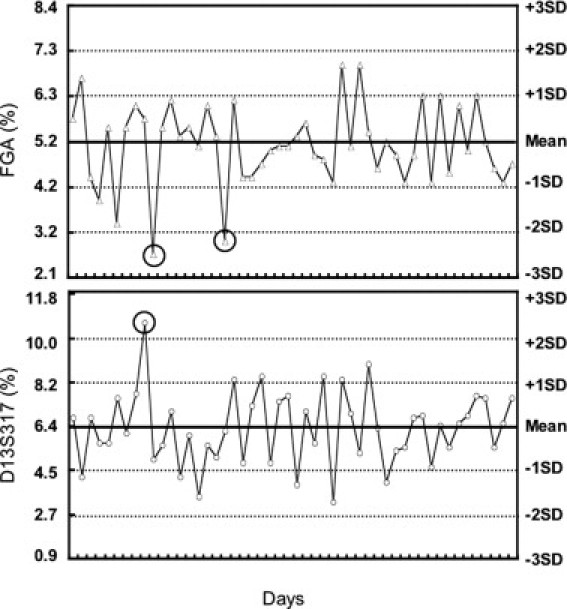
Follow-up QC monitoring of the 95%Donor/5%Recipient chimeric control on the established Levey-Jennings chart. The percentage of the two informative loci, FGA and D13S317, in the 95%D/5%R chimeric control from 50 consecutive runs was calculated as described (see Materials and Methods). Results were then plotted on a Levey-Jennings chart established from the QC parameters obtained in the initial evaluation study. Tick marks on the x axis represent successive days on which the assay was run. Circles indicate control values that exceeded 2 SDs.
Discussion
Many QC procedures are already used in clinical molecular diagnostic laboratories to ensure acceptable assay performance. For example, a major concern in all clinical molecular diagnostic laboratories is PCR contamination. In addition to assigning separate areas for pre- and post-PCR procedures, no-template controls are always included in each run. Other good laboratory practices, such as using filter tips, semiopen workstations, dedicated pipettes for preparing master mix, separate laboratory coats for pre- and post-PCR areas, and use of uracil N-glycosylase (UNG), are also encouraged.12 Another QC example is the inclusion of the undesignated band in the size standard mix (typically about 264 bases) to ensure that the instrument and software have produced an appropriate standard curve.13 Ideally, a second or third undesignated molecule of different molecular weight could be included in the size standard. Similarly, we acknowledge that it might be desirable to use additional controls with higher amounts of recipient DNA in our BMT engraftment assay.
Other areas within the molecular diagnostic laboratory could benefit from more rigorous quality control. In general, results are becoming more quantitative, especially with the advent of real-time PCR, and therefore statistical process control can be applied. PCR assay could be monitored (especially for the enzyme) with internal controls, using long oligonucleotides, which contain primer-binding sites, and are approximately the same size as the PCR product, but contain a unique probe-binding site.14,15 This control documents that the primers, nucleotide, and polymerase have all been added and function properly, and that the thermocycler is functioning correctly. As discussed, medically relevant external controls should be included within each run. Finally, the migration time of the 246-base control size standard peak could be plotted, in addition to its full-width half-maximum. It is possible that a failing capillary could be detected by a change in these parameters.
In the initial phase of this study, we first established the QC parameters for two microsatellite loci using the 95%D/5%R control. There was a small bias for the D13S317 marker resulting in a mean of 6.4% (28% positive bias). We consider this acceptable given the errors associated with DNA quantification and pipetting. Furthermore, there are intrinsic errors in using microsatellites, in which heterozygote peaks, which should be at a 1:1 molar ratio, are known to vary in peak height (lower molecular weight allele typically having a higher peak height). In our study, the average %CV of these two informative loci on these two individuals is 20 to 30%, which represents the precision of the entire assay process, including the final calculation. We consider this CV to be medically acceptable when decisions to alter treatment are based on major shifts in the degree of chimerism (eg, a patient with 0% recipient initially and changing to 20% recipient in the subsequent sample). This variation helps justify the use of two loci to evaluate controls and patient samples. Evaluating more than one locus for patient samples is also valuable when loss of heterozygosity is present.16 We have demonstrated that with this relatively low CV, routine use of duplicate assays is unnecessary (unpublished data). QC parameters should be recalculated if control samples are changed or when one might expect a systematic change in fundamental assay performance.
We used these initial data to establish Levey-Jennings plots for use in the second phase of the study. We had three warnings resulting from the 12S rule; however, there were no unacceptable runs. Furthermore, the expected result is to get 12S warnings based on the definition of the 2 SDs where approximately 5% of subsequent controls are expected to be outside this range. For these 100 data points, one would anticipate about five such 12S warnings. Although there was an initial positive bias (5.2% and 6.4%), there were no further shifts or trends in the mean value over time in the second phase, and the %CV was relatively consistent (17.6% and 23.8%). Laboratories may wish to employ specific Westgard rules to identify random (such as 13S and R4S rules) and systematic errors (such as 22S rule). An important part of this protocol is to describe when and how many control samples are to be analyzed, where they are to be located in an analytical run, how the control results are to be interpreted, and what to do when they are “out-of-control.” Ideally, Levey-Jennings plots could be incorporated into the instrument software so that significant technologist time is not required to produce them.
When controls fail, patient results are not signed out. It is often helpful to assemble a chart to temporally examine when control values deviate compared with new lots of reagents and controls, in addition to software and instrument changes. If one can identify another instrument at one's institution, mix-and-match experiments can be very useful to determine whether a given problem is an instrument versus a reagent problem.
During subsequent use of these charts, a 22S error occurred in the 5% control where both markers were systematically reporting below 5%. We believe that this was due to cumulative freeze-thawing of the same 5% control aliquot for over one year. Although we do not have a full explanation of exactly how this caused the error, we have corrected the problem by limiting the number of freeze-thaws of a single 5% aliquot to one month (eight freeze-thaws).
In this paper, we have demonstrated the initial application of a classic QC tool to the clinical molecular pathology laboratory. By incorporating these tools into an overall quality assurance program, a laboratory can identify systematic errors and perform corrective actions before actual assay failure.
Footnotes
Funded in part through The Sol Goldman Center for Pancreatic Cancer Research, the Rolfe Foundation, NIH/NCI Early Detection Research Network (CA 115102), and NIH/NCI R21 CA122265 (to J.R.E.).
Current address of S.-L.L.: Beth Israel Deaconess Medical Center, Yamins 309, 330 Brookline Ave, Boston, MA 02215.
References
- 1.Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 2.Thomas ED, Buckner CD, Banaji M, Clift RA, Fefer A, Flournoy N, Goodell BW, Hickman RO, Lerner KG, Neiman PE, Sale GE, Sanders JE, Singer J, Stevens M, Storb R, Weiden PL. One hundred patients with acute leukemia treated by chemotherapy, total body irradiation, and allogeneic marrow transplantation. Blood. 1977;49:511–533. [PubMed] [Google Scholar]
- 3.Roy DC, Tantravahi R, Murray C, Dear K, Gorgone B, Anderson KC, Freedman AS, Nadler LM, Ritz J. Natural history of mixed chimerism after bone marrow transplantation with CD6-depleted allogeneic marrow: a stable equilibrium. Blood. 1990;75:296–304. [PubMed] [Google Scholar]
- 4.Serrano J, Roman J, Sanchez J, Jimenez A, Castillejo JA, Herrera C, Gonzalez MG, Reina L, Rodriguez MC, Alvarez MA, Maldonado J, Torres A. Molecular analysis of lineage-specific chimerism and minimal residual disease by RT-PCR of p210(BCR-ABL) and p190(BCR-ABL) after allogeneic bone marrow transplantation for chronic myeloid leukemia: increasing mixed myeloid chimerism and p190(BCR-ABL) detection precede cytogenetic relapse. Blood. 2000;95:2659–2665. [PubMed] [Google Scholar]
- 5.Mackinnon S, Barnett L, Heller G, O'Reilly RJ. Minimal residual disease is more common in patients who have mixed T-cell chimerism after bone marrow transplantation for chronic myelogenous leukemia. Blood. 1994;83:3409–3416. [PubMed] [Google Scholar]
- 6.Scharf SJ, Smith AG, Hansen JA, McFarland C, Erlich HA. Quantitative determination of bone marrow transplant engraftment using fluorescent polymerase chain reaction primers for human identity markers. Blood. 1995;85:1954–1963. [PubMed] [Google Scholar]
- 7.Oliver DH, Thompson RE, Griffin CA, Eshleman JR. Use of single nucleotide polymorphisms (SNP) and real-time polymerase chain reaction for bone marrow engraftment analysis. J Mol Diagn. 2000;2:202–208. doi: 10.1016/S1525-1578(10)60638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pakstis AJ, Speed WC, Kidd JR, Kidd KK. Candidate SNPs for a universal individual identification panel. Hum Genet. 2007;121:305–317. doi: 10.1007/s00439-007-0342-2. [DOI] [PubMed] [Google Scholar]
- 9.Levey S, Jennings ER. The use of control charts in the clinical laboratory. Am J Clin Pathol. 1950;20:1059–1066. doi: 10.1093/ajcp/20.11_ts.1059. [DOI] [PubMed] [Google Scholar]
- 10.Westgard JO, Barry PL, Hunt MR, Groth T. A multi-rule Shewhart chart for quality control in clinical chemistry. Clin Chem. 1981;27:493–501. [PubMed] [Google Scholar]
- 11.Tholen DW, Kallner A, Kennedy JW, Krouwer JS, Meier K: Evaluation of Precision Performance of Quantitative Measurement Methods. Approved Guideline. Second Edition. EP5–A2. National Committee on Clinical Laboratory Standards 2004, 24: No 25
- 12.Burkardt HJ. Standardization and quality control of PCR analyses. Clin Chem Lab Med. 2000;38:87–91. doi: 10.1515/CCLM.2000.014. [DOI] [PubMed] [Google Scholar]
- 13.Butler JM, McCord BR, Jung JM, Lee JA, Budowle B, Allen RO. Application of dual internal standards for precise sizing of polymerase chain reaction products using capillary electrophoresis. Electrophoresis. 1995;16:974–980. doi: 10.1002/elps.11501601163. [DOI] [PubMed] [Google Scholar]
- 14.Abdulmawjood A, Roth S, Bulte M. Two methods for construction of internal amplification controls for the detection of Escherichia coli O157 by polymerase chain reaction. Mol Cell Probes. 2002;16:335–339. doi: 10.1006/mcpr.2002.0431. [DOI] [PubMed] [Google Scholar]
- 15.Das A, Spackman E, Senne D, Pedersen J, Suarez DL. Development of an internal positive control for rapid diagnosis of avian influenza virus infections by real-time reverse transcription-PCR with lyophilized reagents. J Clin Microbiol. 2006;44:3065–3073. doi: 10.1128/JCM.00639-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou M, Sheldon S, Akel N, Killeen AA. Chromosomal aneuploidy in leukemic blast crisis: a potential source of error in interpretation of bone marrow engraftment analysis by VNTR amplification. Mol Diagn. 1999;4:153–157. doi: 10.1016/s1084-8592(99)80039-3. [DOI] [PubMed] [Google Scholar]