

Abstract
We evaluated the branched-chain DNA (bDNA) assay QuantiGene Reagent System to measure RNA in formalin-fixed, paraffin-embedded (FFPE) tissues. The QuantiGene Reagent System does not require RNA isolation, avoids enzymatic preamplification, and has a simple workflow. Five selected genes were measured by bDNA assay; quantitative polymerase chain reaction (qPCR) was used as a reference method. Mixed-effect statistical models were used to partition the overall variance into components attributable to xenograft, sample, and assay. For FFPE tissues, the coefficients of reliability were significantly higher for the bDNA assay (93–100%) than for qPCR (82.4–95%). Correlations between qPCRFROZEN, the gold standard, and bDNAFFPE ranged from 0.60 to 0.94, similar to those from qPCRFROZEN and qPCRFFPE. Additionally, the sensitivity of the bDNA assay in tissue homogenates was 10-fold higher than in purified RNA. In 9- to 13-year-old blocks with poor RNA quality, the bDNA assay allowed the correct identification of the overexpression of known cancer genes. In conclusion, the QuantiGene Reagent System is considerably more reliable, reproducible, and sensitive than qPCR, providing an alternative method for the measurement of gene expression in FFPE tissues. It also appears to be well suited for the clinical analysis of FFPE tissues with diagnostic or prognostic gene expression biomarker panels for use in patient treatment and management.
Fixation in formalin is the routine procedure to preserve tissue integrity for diagnostic evaluation. However formalin cross-links proteins and nucleic acids, which complicates their quantification.1,2 Through prolonged exposure to formalin and subsequent storage, the quality of RNA is severely compromised.3,4,5 RNA in formalin-fixed and paraffin-embedded (FFPE) tissues is degraded into fragments below 200 bases4;6 and chemically modified.7,8 As a result, the efficiency of RNA extraction and reverse transcription are greatly reduced8 and in general, the measurement of RNA in FFPE tissues by polymerase chain reaction (PCR)-based methods is less reliable than in frozen tissues.
Recent methodological advances for measurement of RNA in FFPE tissues have been achieved through improvements in extraction and amplification of formalin-treated RNA and in the adaptation of a primer design to amplify short RNA fragments4.9 Two methods, cDNA-mediated annealing, selection extension and ligation (DASL)10 and whole transcriptome amplification (WTA),11 facilitate measurements of large gene expression panels that comprise hundreds of genes (DASL) or generate probes for global gene expression array analysis (WTA). While these methods are well suited for gene discovery efforts from FFPE tissues, they have little clinical utility because their reliability on a gene specific basis is not known.
We tested the branched-chain DNA (bDNA) technology QuantiGene Reagent System for quantifying the expression of RNA in formalin-fixed tissues.8 This sandwich nucleic acid hybridization assay provides a unique advantage over existing methods by amplifying the reporter signal and by avoiding enzymatic amplification of the target RNA.12,13,14,15,16,17 In addition, the assay measures RNA directly from tissue homogenates (THs), thereby overcoming errors that are caused by RNA extraction. The probe sets are designed such that shorter fragments of formalin-treated RNA can be measured. In addition, the short complementary sequence of individual capture and detection probes confers specificity in hybridization kinetics that strongly favor the RNA of interest.18 Here we evaluate for a small set of specific genes in FFPE tissues the assay performance of the bDNA assay (QuantiGene Reagent System). In a gene expression panel that consists of a small number of genes it is critical to measure each gene with high accuracy. This requires selecting genes that can be quantified with good precision and optimizing the primer design or probe set for each gene. Here we demonstrate the utility of a collection of prostate xenografts in determining the reliability of measuring single genes. We also compare the assay performance of the QuantiGene Reagent System to qPCR and demonstrate the applicability of the QuantiGene Reagent System for RNA measurements in macrodissected archival clinical tissue specimens.
Materials and Methods
Sample Collection from Xenografts
Ten different xenografts were snap-frozen in liquid nitrogen and stored at −80°C. Four samples were taken from each xenograft, two of which were fixed in formalin, while two others were embedded in OCT. RNA was isolated from each sample and divided after purification for measurements by qPCR or bDNA assays as described below. In each sample, we measured six genes and used one of these (β-actin) for normalization of gene expression.
Sample Preparation from Xenografts and Prostate Tissues
Xenografts (LuCaP 23.8, 49, 58, 69, 70, 73, 77, 93, 35, 35V) were grown by serial passage in immunocompromised BALB/C-nu/nu (athymic) mice (Simonsen Laboratories, Gilroy, CA). Mice were housed at the University of Washington Medical Center and all procedures were approved by the Animal Health and Welfare Committee. Pieces of xenograft tissues were snap-frozen in liquid nitrogen and transferred to −80°C for storage. To generate formalin-fixed tissue samples and to mimic the processing of surgical tissues in the clinical pathology laboratory, two pieces per xenograft were submerged in 10% phosphate-buffered formalin for 18 to 24 hours at room temperature and transferred to 70% ethanol until they were processed in the tissue processor for 8 hours (Tissue Tek; Miles Scientific, Naperville, IL). Two frozen and two deparaffinized formalin-fixed tissue samples for each xenograft (50–100 mg) were homogenized with one 5-mm stainless steel bead (Qiagen, Valencia, CA) in 1 ml of Trizol (Invitrogen, Carlsbad, CA) for frozen tissues, or in the buffer from the Optimum FFPE RNA Isolation Kit (Ambion Diagnostics, Austin, TX) for formalin-fixed tissues. Samples were homogenized in the TissueLyser (Qiagen) at 30 Hz for 3 minutes.
Human Tissues
Patient tissues were collected under an Institutional Review Board-approved protocol. Archival tissue blocks from radical prostatectomies, dating from 1993 to 1998, were cut into 25 sections of 8-μm thickness. Hematoxylin and eosin stain was used to identify areas of normal and cancer epithelium and these were transferred onto the unstained slides. Normal or cancerous tissues were isolated separately by scraping, placed into 1.5-ml microcentrifuge tubes, and processed into tissue homogenates.
Tissue Homogenates
Tissue homogenates were prepared according to the procedure described in the QuantiGene Sample Processing Kit for FFPE Tissues (Panomics, Inc., Fremont, CA). Briefly, 300 μl of homogenizing solution supplemented with 2 μl of proteinase K (50 μg/μl) were incubated with 10 deparaffinized 10-μm sections overnight at 65°C. The next day, the tissue homogenate was separated from debris by brief centrifugation, then transferred to a new tube. The resulting tissue homogenate was frozen at −80°C and stored until further use. Macrodissected tissue scrapings from human normal and prostate tissues were homogenized in half of the recommended volume.
RNA Isolation
RNA in frozen tissue was isolated by Trizol (Invitrogen). The RNA-containing aqueous phase of the Trizol extract was precipitated with an equal amount of 70% ethanol and immediately purified with the RNeasy Mini column (Qiagen). The RNA from fixed tissues was isolated using the Optimum kit (Ambion, Austin, TX) according to the manufacturer's protocol. RNA was eluted from the columns in a final volume of 30 μl.
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
First-strand cDNA was synthesized using 0.5 μg of oligo(dT) and 200 units of Superscript III reverse transcriptase (Invitrogen) on a Mastercycler (Eppendorf, Hamburg, Germany) following the manufacturer's protocol. The cDNA was diluted to a final concentration 10 ng/μl of input RNA. qPCR was performed with SYBR Green fluorophore (Invitrogen) in triplicate using My iQ single-color qPCR detection system (Bio-Rad, Hercules, CA). The optimized PCR conditions consisted of 30 ng of cDNA, 0.5 μmol/L forward and reverse primers, 12.5 μl Platinum SYBR Green qPCR Super-Mix-UDG (Invitrogen), and 0.5 μl of magnesium chloride (50 mmol/L). PCR cycling was performed as follows: 95°C for 10 minutes for one cycle, 95°C for 20 seconds, and 60°C for 45 seconds for 40 cycles. Primers were designed using the OligoPerfect Designer Website (Invitrogen). Primer pairs were designed to span approximately 100 nucleotides and hybridized as closely as possible to the 3′ end of the transcript (polyA tail) (Table 1). Genes were measured in triplicate by qPCR. For comparison to the bDNA assay, cycle values (Ct) were de-logged. β-Actin was used for normalization.
Table 1.
qPCR Primer Sequences
| Gene and accession number | Sequence |
|---|---|
| ACTβ | Forward 5′-TCCCCCAACTTGAGATGTATGAAG-3′ |
| NM_001101 | Reverse 5′-AACTGGTCTCAAGTCAGTGTACAGG-3′ |
| RPL32 | Forward 5′-AGTTCCTGGTCCACAACGTC-3′ |
| NM_000994 | Reverse 5′-CGGTTCTTGGAGGAAACATT-3′ |
| SDHA | Forward 5′-CTGGGAAGGTCACTCTGGAA-3′ |
| NM_004168 | Reverse 5′-CTCATCAGTAGGAGCGAAT-3′ |
| TCEA1 | Forward 5′-CTCCTTTGCTCCCTTTTTCC-3′ |
| NM_006756 | Reverse 5′-GTGGGCCAATTCTTAACACG-3′ |
| RPL3 | Forward 5′-TTCATTGACACCACCTCCAA-3′ |
| NM_000967 | Reverse 5′-CCTTCTTCCTTTGCAATTCG-3′ |
| RPL15 | Forward 5′-CTGGCCAAACAACCCTAAAA-3′ |
| NM_002948 | Reverse 5′-CATGGTGCAAACAGAAATGC-3′ |
| KRT18 | Forward 5′-AACCATCCAAAAGACCACCA-3′ |
| NM_000224 | Reverse 5′-CCTGCTTCTGCTGGCTTAAT-3′ |
| PSA | Forward 5′-AGCAAGGATGGAGCTGAAAA-3′ |
| NM_001648 | Reverse 5′-AAAGGAAGACCCTCCCTCCT-3′ |
bDNA Assay
Standard probe design software was used to design specific oligonucleotide probe sets for target genes for use in QuantiGene 1.0 or QuantiGene 2.0 Reagent Systems (Panomics, Inc.). QuantiGene 1.0 Reagent System gives 45-fold signal amplification, whereas the QuantiGene 2.0 Reagent System gives 400-fold signal amplification. A probe set for a target gene consists of three types of oligonucleotide probes that capture the target RNA to the surface of plate well and hybridize to DNA signal amplification molecules. For each target sequence, the software algorithm identifies one or more continuous regions that can serve as annealing templates for CEs (capture extenders, 5–10 per gene), LEs (label extenders, 10–20 per gene), or BL (blocking probes). QuantiGene1.0 and QuantiGene 2.0 Reagent Systems were performed according to manufacturer's recommended protocols (Panomics, Inc.). Briefly, probe set oligonucleotides (250 fmol CE, 500 fmol BL, and 1000 fmol LE) were mixed with the sample, and the mixture was added to an assay well in a 96-well plate covalently coated with capture probe oligonucleotide (5′-CACTTCACTTTCTTTCCAAGAG-3′). Target RNA was captured during an overnight incubation at 53°C (QuantiGene 1.0) or 55°C (QuantiGene 2.0). Unbound material was removed by three washes with 200 to 300 μl of wash buffer (0.1× standard saline citrate containing 0.3 g/L lithium lauryl sulfate) followed by sequential hybridization of DNA amplifier molecules, then 3′-alkaline phosphatase-conjugated Label Probe oligonucleotides, with three washes after each incubation. After the final wash, the dioxetane alkaline phosphatase substrate Lumiphos Plus (Lumingen Inc., Southfield, MI) was added to the wells, and after a short incubation, luminescent signal was measured in either an LMax (Molecular Devices, Mountain View, CA) or GloRunner (Turner Biosystems, Sunnyvale, CA) microplate luminometer.
Based on the large amount of intra-assay consistency observed during the early phases of the project, duplicate measurements were considered sufficient to measure gene expression in FFPE tissues when using the QuantiGene 1.0 and QuantiGene 2.0 Reagent Systems. The “no template” background values were subtracted from each probe set signal. Values were normalized to the ribosomal protein L19 (RPL19) or β-actin for the cancer gene panel or xenografts, respectively. Additionally, a ratio of normalized values was calculated for evaluating the over expression of genes in cancer versus normal tissues.
Statistical Analysis
Analysis of xenograft data requires decomposing the variability in assay values into component sources of variation. Variability is attributable to xenografts, samples within each xenograft, and replicate measurements within each sample. Components of variance, which differ by gene, sample preparation, as well as assay, estimated from a mixed-model analysis of variance.
Residuals were approximately normally distributed for both qPCR and bDNA assays for log-transformed response. To interpret the qPCR assay values in terms of cycle number, a log base-2 transformation was used. PSA value for one replicate obtained using qPCR applied to frozen tissue was quite discrepant from all other PSA values from the same xenograft (−1.8009 versus −9.9309, −9.5809, −10.7709, −10.9309, −11.2809), and this value was excluded from the analysis.
Assay validity depends on two components of the variance decomposition: 1) assay reliability measured as the ability to obtain consistent results when the assay is repeated on the same sample; and 2) ability to obtain results, which correlate highly with those obtained with a gold standard assay. Assay reliability is obtained as the ratio of biological variance to total variance:
Reliability is high when the assay variance (pure error variance) σA2 represents only a small portion of the total variance. The qPCR assay obtained from frozen tissue is regarded as a gold standard assay to which other assay/sample preparation methods are compared. Correlations between the qPCR assay values and any other assay value were obtained by fitting a multivariate extension mixed model analysis of variance. Variance components and the xenograft correlation structure were obtained using the MIXED procedure in SAS.
Results
To evaluate the performance of the bDNA assay, the main objective is to separate the variability of replicate measurements in the assay, also called measurement error, from the biological variability. In this system, the biological variability is generated by differences in gene expression among xenografts and heterogeneous gene expression within a xenograft and represents a much larger component of variability than the variability attributed to the assay. Table 2 gives proportion of variances for five genes for frozen and formalin-fixed sample preparations. Measurements were performed with qPCR or with the bDNA QuantiGene 1.0 Reagent System. Values for total variability are comparable between the bDNA and qPCR assays, and in both assays, the relative amount of total variance across genes is similar. For formalin-fixed tissues, the reliability coefficients range between 99.3% and 100% for the bDNAFFPE assay and between 82.4% and 95% for the qPCRFFPE assay. For each gene, the reliability of the bDNA assay is significantly higher than of the qPCR assay. The increase in assay reliability in the bDNA assay is statistically significant, since the lower limit confidence interval in the bDNA assay is always higher than the upper-bound qPCR confidence interval. In formalin-fixed tissues, KRT18 and TCEA1 possess the lowest variability among xenografts and the highest sample and assay variability. The decrease in the reliability coefficient for TCEA1 is likely caused by the relatively low level of gene expression, while the complexity of the KRT18 gene locus and splice variants of the KRT18 gene likely account for the decreased reliability of KRT18 measurements.
Table 2.
Components of Variability in Gene Expression Measurements
| Assay | Sample preparation | Gene | V(Xeno)* | V(sample)† | V(assay)‡ | Reliability coefficient§ | Total variance¶ |
|---|---|---|---|---|---|---|---|
| qPCR | Frozen | TCEA1 | 74.70% | 19.30% | 6.00% | 94.0% (76.1, 96.9) | 0.83 |
| RPL3 | 84.60% | 13.60% | 1.80% | 98.2% (93.1, 99.3) | 1.32 | ||
| RPL15 | 78.50% | 18.50% | 3.10% | 96.9% (89.1, 99.4) | 1.95 | ||
| KRT18 | 63.50% | 18.80% | 17.70% | 82.3% (54.9, 93.1) | 0.96 | ||
| PSA | 99.00% | 0.60% | 0.40% | 99.6% (85.2, 99.6) | 25.65 | ||
| qPCR | Formalin-fixed | TCEA1 | 45.90% | 36.50% | 17.60% | 82.4%*** (46.6, 93.6) | 0.74 |
| RPL3 | 62.80% | 32.20% | 5.00% | 95.0%*** (87.9, 97.4) | 1.24 | ||
| RPL15 | 53.50% | 36.80% | 9.70% | 90.3%*** (67.4, 96.6) | 1.85 | ||
| KRT18 | 7.00% | 84.20% | 8.80% | 91.2%* (75.0, 92.3) | 2.3 | ||
| PSA | 66.70% | 26.90% | 6.50% | 93.5%*** (65.7, 98.4) | 4.6 | ||
| bDNA | Frozen | TCEA1 | 86.10% | 13.70% | 0.20% | 99.8%*** (99.5, 99.8) | 0.43 |
| RPL3 | 96.20% | 3.50% | 0.30% | 99.7%*** (99.1, 99.9) | 0.78 | ||
| RPL15 | 95.10% | 4.30% | 0.60% | 99.4%*** (98.4, 99.7) | 0.7 | ||
| KRT18 | 25.20% | 74.40% | 0.50% | 99.5%*** (98.5, 99.9) | 0.57 | ||
| PSA | 98.60% | 1.40% | 0.00% | 100.0%*** (99.9, 100) | 8.15 | ||
| bDNA | Formalin-fixed | TCEA1 | 80.20% | 19.10% | 0.70% | 99.3%*** (95.1, 99.8) | 0.43 |
| RPL3 | 83.60% | 16.30% | 0.10% | 99.9%*** (99.6, 99.9) | 1.23 | ||
| RPL15 | 92.60% | 7.40% | 0.10% | 99.9%*** (99.7, 100) | 1.39 | ||
| KRT18 | 84.30% | 15.10% | 0.60% | 99.4%*** (97.5, 99.8) | 0.56 | ||
| PSA | 97.20% | 2.80% | 0.00% | 100.0%*** (99.8, 100) | 5.33 |
Percent of total variance due to xenograft variation.
Percent of total variance attributable to variability of samples within xenografts.
Percent of total variance attributable to the assay.
Percent of total variance which is attributable to xenograft and sample (biologic variability). Confidence intervals of 95% are shown in parentheses.
Total absolute variance.
P < 0.05 for a two-sided test compared against reliability coefficient of QPCR assay with frozen tissue preparation.
P < 0.001 for a two-sided test compared against reliability coefficient of QPCR assay with frozen tissue preparation.
Table 3 gives the correlation coefficients between qPCRFROZEN and qPCRFFPE and qPCRFROZEN and bDNAFROZEN or FFPE. Using qPCRFROZEN as the criterion measure, with the exception of KRT18, the validity (intermethod reliability) was similar for qPCRFFPE (range, 0.59–0.91) and bDNAFFPE (range, 0.6–0.94). KRT18, which has low biological variability among xenografts, has the lowest correlation, while PSA, which has the highest biological variability, has the highest correlation (see Supplementary Figure 1 at http://jmd.amjpathol.org). The confidence intervals of the correlations are large because we only measured 10 xenografts. However, the confidence intervals for the bDNAFFPE assay only included zero for one of the genes, suggesting a slightly better correlation than for qPCRFFPE. Overall, it appears that the bDNA assay QuantiGene 1.0 Reagent System has a comparable accuracy to the qPCR assay in measuring genes in FFPE tissues, and thus with the exception of KRT18, the bDNA assay demonstrates satisfactory validity for genetic measurements in FFPE tissues. Since precisions of measurements vary among genes, it will be necessary to evaluate each gene separately for inclusion into a gene expression panel for analysis of tissues in a clinical setting.
Table 3.
Correlation between the QuantiGene 1.0 Assay and qPCR
| Gene | Gene ID | ρ* (2qPCRFROZEN, qPCRFFPE)† | ρ(qPCRFROZEN, bDNAFROZEN) | ρ(qPCRFROZEN, bDNAFFPE) |
|---|---|---|---|---|
| TCEA1‡ | 21399 | 0.81 (−0.36, 0.99) | 0.49 (−0.50, 0.95) | 0.60 (−0.80, 0.97) |
| RPL3 | 6122 | 0.80 (−0.07, 0.99) | 0.81 (0.20, 0.99) | 0.81 (0.39, 0.98) |
| RPL15 | 6138 | 0.59 (−0.22, 0.97) | 0.76 (0.16, 0.99) | 0.85 (0.59, 0.99) |
| KRT18 | 3875 | 0.23 (−0.78, 0.94) | 0.38 (−0.75, 0.97) | 0.82 (0.29, 0.98) |
| PSA | 354 | 0.91 (0.81, 0.99) | 0.95 (0.89, 1.00) | 0.94 (0.89, 1.00) |
ρ, correlation coefficient; 95% confidence intervals for the correlation coefficients are indicated in parentheses. Confidence intervals excluding zero are significant at P < 0.05.
PCRFROZEN, qPCR in frozen tissue preparations; PCRFFPE, qPCR in formalin-fixed and paraffin-embedded tissue preparations. bDNAFROZEN assay (QuantiGene 1.0) in frozen tissue preparations, bDNAFFPE assay (QuantiGene 1.0) in formalin-fixed and paraffin-embedded tissue preparations.
TCEA1, transcription elongation factor A1; RPL3, ribosomal protein L3; RPL15, ribosomal protein L15; KRT18, keratin 18; PSA, prostate-specific antigen.
Figure 1.
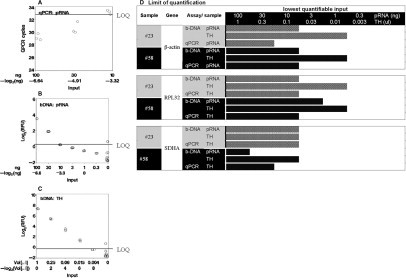
LOQ of the bDNA assay in archival tissue blocks. A–C: Limiting dilution assay with qPCR or QuantiGene 1.0 measurements. β-Actin measurements in purified RNA (pRNA) or TH from sample 23. Measurements of β-actin were conducted by qPCR (A) or bDNAFFPE assay QuantiGene 1.0 (B and C) in a serial dilution of the sample. The starting material is 100 ng of purified RNA or 1 μl of TH, which is the volume from which approximately 100 ng of RNA is extracted. Therefore sample inputs of pRNA and TH and subsequent linear dilution series are comparable. The concentrations of the serial dilution are indicated on the x axis in a log2 scale. The y axes in B and C are log2 relative luminescence units (log2RLU) of data obtained in the bDNA assay to facilitate comparison to qPCR. Thus the amounts of analyte indicated in the x axis are comparable between the three panels. In each panel, the LOQ is indicated by a vertical line. The LOQ in A is at 33 cycles, which is the conventional definition of the LOQ for this PCR machine. In panels 2 and 3, the LOQ is at the inflection point of the linear line, which indicates the point at which further dilution does not result in a proportional decrease in signal intensity. D: Limit of quantification comparing pRNA and TH in the QuantiGene 1.0 Reagent System. Two archival blocks, 23 and 58, were used to generate pRNA or TH. The limit of quantification for β-actin, RPL32, and SDHA was determined for each sample using the limiting dilution approach described above. Dilutions of sample input that provided quantifiable results are shaded in the graph. The end of the shaded bar represents the LOQ for each sample/assay/gene combination.
Figure 1 shows our approach for comparing the sensitivity of the bDNA assay QuantiGene 1.0 Reagent System to the qPCR. Because the bDNA assay does not involve preamplification of RNA as is the case in the qPCR, its sensitivity is entirely based on the signal amplification strength of its detection reagents. Despite this limitation, the limit of quantification (LOQ) was the same for the bDNA QuantiGene 1.0 Reagent System and the qPCR assay. As demonstrated in Figure 1, A and B, the LOQ for measurements of β-actin in purified RNA was at 10 ng of RNA input for both methodologies. Since the bDNA assay has the unique advantage that it does not require RNA purification, we investigated whether measuring RNA inside a tissue homogenate and without prior isolation would possess greater sensitivity. We compared measurements in TH and in RNA extracted from a comparable amount of TH. By measuring the expression of three genes (β-actin, RPL32, SDHA), expressed at different magnitudes, we observed a 10-fold greater LOQ for measurements in TH than purified RNA (Figure 1, compare B, C, and D). For example, the LOQ for β-actin was at 10 ng of sample input for purified RNA, while it was at 1 ng of sample input for TH. Similar results were obtained for the other genes and samples. The LOQ was defined as a cycle number above 33 cycles for qPCR. In the limiting dilution experiment of the bDNA assay, samples were diluted sequentially by a factor of 3. The LOQ was defined as the concentration at which a further dilution did not result in a threefold decline in signal intensity.
Figure 2 demonstrates the application of the bDNA QuantiGene 2.0 Reagent System to measuring a panel of 14 “prostate cancer” genes in archival prostate cancer tissue blocks. Genes were measured in adjacent macrodissected normal and cancerous tissues from the same archival blocks collected 9 to 13 years ago. Before macrodissection, we evaluated whether the RNA in a given tissue block is of sufficient quantity and quality for measurements of 10 genes. This decision was made by first measuring 28S rRNA and 18S rDNA concentrations in TH obtained from three sections (8 μmol/L) of the tissue block (Figure 2A). The amount of 18S rDNA (10-μl sample, 102-fold dilution) is proportional to the number of cells in the TH and was used to normalize the 28S rRNA measurement. However, the qualification of a tissue block was based only on the 28S rRNA measurement. The threshold for an acceptable quality of RNA in a tissue block was defined as a quantifiable amount of 28S rRNA in a 10-μl sample from a 105-fold dilution of the original TH. Blocks that met this requirement were further sectioned and TH prepared from macrodissected areas of cancer and normal. The dissected areas ranged between 0.25 and 1.5 cm2 per slide solubilized in 150 μl of tissue homogenization solution. Sample 44 had the smallest amount of tissue and the poorest RNA quality, but nevertheless provided measurements for 13 of 14 genes (Figure 2B).
Figure 2.
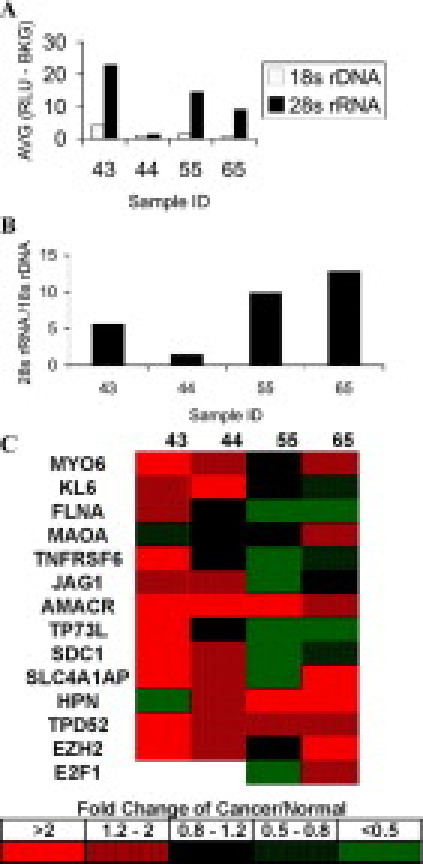
Relative gene expression in cancerous versus normal prostate epithelium. A and B: Assessment of RNA quality and amount. A: Three sections from archival tissue blocks were solubilized in tissue homogenizing solution. Measurements of 28S rRNA (10 μl) were performed with the QuantiGene 1.0 Reagent System in a 105-fold dilution of the original sample. Measurements of 18S rDNA (10 μl, 102-fold dilution) are proportional to cell number in the sample and were conducted with the QuantiGene 1.0 Reagent System in the same TH. B: The amount of measurable 28S rRNA was normalized to 18S rDNA for comparison of RNA quality between samples. Samples that provided 28S rRNA measurements above the limit of detection in the bDNA assay, defined as 3 SD above background, were used for gene expression measurements. C: Measurement of a cancer gene panel with the QuantiGene 2.0 Reagent System. Cancer and normal tissues from the same blocks were macrodissected and dissolved in homogenizing solution. A panel of 14 prostate cancer genes were measured in cancer and normal tissues using the bDNA QuantiGene 2.0 amplification system. Values for each gene were normalized to RPL19 as a housekeeping gene. The ratio between cancerous and normal tissues is calculated and shown in a five-tiered categorical scale.
A gene expression panel of 14 prostate cancer genes was selected based on expression patterns in published DNA array experiments19,20,21 that assessed gene expression differences between normal and cancerous tissues. In these array studies and confirmed by PCR and protein detection, the most consistently overexpressed gene in prostate cancer compared to normal prostate tissue is α-methylacyl-CoA racemase (AMACR).22,23,24 Therefore as expected, in our measurements with the bDNAFFPE assay (QuantiGene 2.0), AMACR was overexpressed in all four cases of prostate cancer compared to normal prostate tissues. Three additional genes that are up-regulated in published array data sets (MYO6, TPD52, and EZH2)25,26,27 were also noticeably increased in measurements of our prostate cancer samples with the bDNAFFPE assay. For the remaining genes in agreement with expression levels in published array data sets, the increase in expression was more variable with the bDNAFFPE assay across the four specimens. For example in sample 55, only three genes were over expressed in cancer compared to normal tissue. However overall, measurements with the bDNAFFPE assay provided the expected overexpression compared to normal tissue of a panel of 14 prostate cancer genes.
Discussion
The primary goal of this study is to determine whether the bDNA assay using the QuantiGene Regent System provides an alternative method to qPCR for measuring gene expression in FFPE tissues. Major advantages of the QuantiGene Reagent System are its simple assay format, resembling that of an enzyme-linked immunosorbent assay, and the avoidance of RNA isolation. By comparing the bDNA assay QuantiGene Reagent System to qPCR for quantification of gene expression in FFPE tissues we observed that the bDNA assay is more reliable and sensitive that qPCR. The validity of the bDNA QuantiGene Reagent System appears to be comparable to that of qPCR in FFPE tissues. By comparing the expression levels of 14 cancer genes in macrodissected normal and cancerous prostate tissues, we observed an expected amount of overexpression in cancer.
The initial application of the bDNA assay with the QuantiGene 1.0 Reagent System for gene expression measurements in FFPE tissues provided promising results8 and motivated us to undertake a more complete characterization and preliminary validation of the assay for a set of five genes. The results of our study clearly demonstrate that the most significant benefit of the bDNA assay with the QuantiGene 1.0 Reagent System lies in its reproducibility. A similar result was previously noted with the QuantiGene 1.0 Reagent System when measuring RNA in frozen tissues.28 In a Food and Drug Administration-sponsored study, the QuantiGene 1.0 Reagent System demonstrated excellent precision and accuracy in comparison to TaqMan or StartRT-PCR.29 When comparing QuantiGene 1.0 Reagent System measurements in frozen and FFPE tissues from the same xenografts, we observed similar excellent reliability coefficients, suggesting that formalin treatment of RNA does not affect the reproducibility of the bDNA QuantiGene 1.0 assay.
However, we also note limitations in the study design and in the QuantiGene 1.0 Reagent System. The limitation in the study design is the relatively small number of samples. To evaluate the performance of the QuantiGene 1.0 Reagent System in frozen and FFPE tissues, we measured five genes in 20 samples from 10 xenografts. While this number of samples sufficed to determine the assay reliability, it was not enough to for the intermethod comparison (Table 3). The correlation coefficients comparing bDNA and qPCR measurements in frozen and fixed sample preparations are not significantly different based on the large confidence intervals. While it might appear that the bDNAFFPE assay provides measurements that correlate better than the bDNAFROZEN assay with the reference QPCRFROZEN assay, a definite result requires a much larger sample size.
The lowest correlation coefficients are observed for KRT18. The components of sample and assay variability are large for this gene, and as a result the component of xenograft variability is small (see Supplementary Figure 1 at http://jmd.amjpathol.org). A possible explanation for the inconsistent measurements of KRT18 is the presence of 138 pseudogenes related to KRT18 and KRT8 in the human genome in addition to multiple splice variants.30 This problem cannot be solved by improving the primer design, and it might be necessary to exclude genes of this kind from gene expression panels that are used to measure clinical samples. If the assay variability is due to suboptimal primer design, RNA amplification by random priming could provide an improvement. In this case, the primer design would not be limited to within 300 bases of the gene 5′ to the polyA tail, as it was after reverse transcription using oligo(dT). The approach that we are putting forward in this study can be implemented to select genes that have adequate measurement characteristics to provide reliable prognostic or treatment-associated information in FFPE tissues.
A major advantage of the bDNA assay is its simple and rapid workflow. Whole sections from tissue blocks that are directly solubilized in tissue homogenization solution, added to wells with the detection probe set, and signals are measured by a plate reader. The ideal sample for analysis in bDNAFFPE assay therefore consists of >80% purity of the cell type of interest. However, a potential limitation of the bDNA assay (QuantiGene 1.0) is the absence of enrichment of the analyte of interest through lack of RNA preamplification. While preamplification is a major source of gene-specific measurement errors, it vastly increases the assay sensitivity, which is particularly important for low abundance transcripts. Extrapolation from our results to smaller amounts of sample input suggests that whole transcriptome preamplification is required for measurements of analyte concentrations that are obtained from laser-captured samples. The new QuantiGene 2.0 Reagent System increases sensitivity over the QuantiGene 1.0 Reagent System by to 14-fold (see Supplementary Figure 2 at http://jmd.amjpathol.org). To facilitate quantification of low abundance transcripts in macrodissected tissue preparations, we therefore used the 2.0 system instead of the 1.0 system (Figure 2). A comparison of the two systems demonstrated a high correlation in measurements of purified RNA. The recent development of a multiplex bead-based QuantiGene Plex 2.0 assay using the 2.0 amplification system will further increase the number of genes that can be measured in limited amounts of FFPE tissues to up to 30.
Our study highlights the reliability of the bDNA assay for measurement of RNA in FFPE tissues. The simple workflow of the QuantiGene Reagent System facilitates high-throughput gene expression measurements. In addition, the superb reproducibility of the bDNA assay with the QuantiGene Reagent System makes this platform ideally suited to obtain reliable measurements of gene expression panels in clinical samples. The QuantiGene Reagent System will greatly improve the use of gene expression measurements as molecular biomarkers in patient treatment and management.
Acknowledgements
We thank Kim Adolphson at Fred Hutchinson Cancer Research Center Histopathology Shared Resources for the tissue sections.
Footnotes
Supported by Fred Hutchinson Cancer Research Center grant P30 CA015704 and Prostate SPORE grant P50 CA097186 and Ovarian SPORE grant P50 CA083636 (to B.K.).
Supplemental material for this article can be found on http://jmd.amjpathol.org.
J.E.D., B.M., and G.K.M. are employed by Panomics, Inc., the manufacturer of the QuantiGene Reagent System, and declare competing financial interests.
Supplementary data
References
- 1.Finke J, Fritzen R, Ternes P, Lange W, Dolken G. An improved strategy and a useful housekeeping gene for RNA analysis from formalin-fixed, paraffin-embedded tissues by PCR. Biotechniques. 1993;14:448–453. [PubMed] [Google Scholar]
- 2.Park YN, Abe K, Li H, Hsuih T, Thung SN, Zhang DY. Detection of hepatitis C virus RNA using ligation-dependent polymerase chain reaction in formalin-fixed, paraffin-embedded liver tissues. Am J Pathol. 1996;149:1485–1491. [PMC free article] [PubMed] [Google Scholar]
- 3.Macabeo-Ong M, Ginzinger DG, Dekker N, McMillan A, Regezi JA, Wong DT, Jordan RC. Effect of duration of fixation on quantitative reverse transcription polymerase chain reaction analyses. Mod Pathol. 2002;15:979–987. doi: 10.1097/01.MP.0000026054.62220.FC. [DOI] [PubMed] [Google Scholar]
- 4.Cronin M, Pho M, Dutta D, Stephans JC, Shak S, Kiefer MC, Esteban JM, Baker JB. Measurement of gene expression in archival paraffin-embedded tissues: development and performance of a 92-gene reverse transcriptase-polymerase chain reaction assay. Am J Pathol. 2004;164:35–42. doi: 10.1016/S0002-9440(10)63093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrahamsen HN, Steiniche T, Nexo E, Hamilton-Dutoit SJ, Sorensen BS. Towards quantitative mRNA analysis in paraffin-embedded tissues using real-time reverse transcriptase-polymerase chain reaction: a methodological study on lymph nodes from melanoma patients. J Mol Diagn. 2003;5:34–41. doi: 10.1016/S1525-1578(10)60449-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Godfrey TE, Kim SH, Chavira M, Ruff DW, Warren RS, Gray JW, Jensen RH. Quantitative mRNA expression analysis from formalin-fixed, paraffin-embedded tissues using 5′ nuclease quantitative reverse transcription-polymerase chain reaction. J Mol Diagn. 2000;2:84–91. doi: 10.1016/S1525-1578(10)60621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999;27:4436–4443. doi: 10.1093/nar/27.22.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang W, Maqsodi B, Ma Y, Bui S, Crawford KL, McMaster GK, Witney F, Luo Y. Direct quantification of gene expression in homogenates of formalin-fixed, paraffin-embedded tissues. Biotechniques. 2006;40:481–486. doi: 10.2144/000112133. [DOI] [PubMed] [Google Scholar]
- 9.Specht K, Haralambieva E, Bink K, Kremer M, Mandl-Weber S, Koch I, Tomer R, Hofler H, Schuuring E, Kluin PM, Fend F, Quintanilla-Martinez L. Different mechanisms of cyclin D1 overexpression in multiple myeloma revealed by fluorescence in situ hybridization and quantitative analysis of mRNA levels. Blood. 2004;104:1120–1126. doi: 10.1182/blood-2003-11-3837. [DOI] [PubMed] [Google Scholar]
- 10.Bibikova M, Talantov D, Chudin E, Yeakley JM, Chen J, Doucet D, Wickham E, Atkins D, Barker D, Chee M, Wang Y, Fan JB. Quantitative gene expression profiling in formalin-fixed, paraffin-embedded tissues using universal bead arrays. Am J Pathol. 2004;165:1799–1807. doi: 10.1016/S0002-9440(10)63435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomlins SA, Mehra R, Rhodes DR, Shah RB, Rubin MA, Bruening E, Makarov V, Chinnaiyan AM. Whole transcriptome amplification for gene expression profiling and development of molecular archives. Neoplasia. 2006;8:153–162. doi: 10.1593/neo.05754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horn PA, Thomasson BM, Wood BL, Andrews RG, Morris JC, Kiem HP. Distinct hematopoietic stem/progenitor cell populations are responsible for repopulating NOD/SCID mice compared with nonhuman primates. Blood. 2003;102:4329–4335. doi: 10.1182/blood-2003-01-0082. [DOI] [PubMed] [Google Scholar]
- 13.Urdea MS, Horn T, Fultz TJ, Anderson M, Running JA, Hamren S, Ahle D, Chang CA. Branched DNA amplification multimers for the sensitive, direct detection of human hepatitis viruses. Nucleic Acids Symp Ser. 1991:197–200. [PubMed] [Google Scholar]
- 14.Gleaves CA, Welle J, Campbell M, Elbeik T, Ng V, Taylor PE, Kuramoto K, Aceituno S, Lewalski E, Joppa B, Sawyer L, Schaper C, McNairn D, Quinn T. Multicenter evaluation of the Bayer VERSANT HIV-1 RNA 3.0 assay: analytical and clinical performance. J Clin Virol. 2002;25:205–216. doi: 10.1016/s1386-6532(02)00011-2. [DOI] [PubMed] [Google Scholar]
- 15.Bramlett KS, Yao S, Burris TP. Correlation of farnesoid X receptor coactivator recruitment and cholesterol 7alpha-hydroxylase gene repression by bile acids. Mol Genet Metab. 2000;71:609–615. doi: 10.1006/mgme.2000.3106. [DOI] [PubMed] [Google Scholar]
- 16.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 17.Zheng Z, Luo Y, McMaster GK. Sensitive and quantitative measurement of gene expression directly from a small amount of whole blood. Clin Chem. 2006;52:1294–1302. doi: 10.1373/clinchem.2005.065078. [DOI] [PubMed] [Google Scholar]
- 18.Flagella M, Bui S, Zheng Z, Nguyen CT, Zhang A, Pastor L, Ma Y, Yang W, Crawford KL, McMaster GK, Witney F, Luo Y. A multiplex branched DNA assay for parallel quantitative gene expression profiling. Anal Biochem. 2006;352:50–60. doi: 10.1016/j.ab.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Choo KW, Kong W. Identification of differentially expressed genes in multiple microarray experiments using discrete fourier transform. Front Biosci. 2007;12:1845–1851. doi: 10.2741/2192. [DOI] [PubMed] [Google Scholar]
- 20.Jung YY, Oh MS, Shin DW, Kang SH, Oh HS. Identifying differentially expressed genes in meta-analysis via Bayesian model-based clustering. Biometrics J. 2006;48:435–450. doi: 10.1002/bimj.200410230. [DOI] [PubMed] [Google Scholar]
- 21.Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427–4433. [PubMed] [Google Scholar]
- 22.Jiang Z, Wu CL, Woda BA, Iczkowski KA, Chu PG, Tretiakova MS, Young RH, Weiss LM, Blute RD, Jr, Brendler CB, Krausz T, Xu JC, Rock KL, Amin MB, Yang XJ. Alpha-methylacyl-CoA racemase: a multi-institutional study of a new prostate cancer marker. Histopathology. 2004;45:218–225. doi: 10.1111/j.1365-2559.2004.01930.x. [DOI] [PubMed] [Google Scholar]
- 23.Ananthanarayanan V, Deaton RJ, Yang XJ, Pins MR, Gann PH. Alpha-methylacyl-CoA racemase (AMACR) expression in normal prostatic glands and high-grade prostatic intraepithelial neoplasia (HGPIN): association with diagnosis of prostate cancer. Prostate. 2005;63:341–346. doi: 10.1002/pros.20196. [DOI] [PubMed] [Google Scholar]
- 24.Luo J, Dunn TA, Ewing CM, Walsh PC, Isaacs WB. Decreased gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: implications for finasteride therapy of prostate carcinoma. Prostate. 2003;57:134–139. doi: 10.1002/pros.10284. [DOI] [PubMed] [Google Scholar]
- 25.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 26.Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, Paris PL, Hofer MD, Storz-Schweizer M, Kuefer R, Fletcher JA, Hsi BL, Byrne JA, Pienta KJ, Collins C, Sellers WR, Chinnaiyan AM. Overexpression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer Res. 2004;64:3814–3822. doi: 10.1158/0008-5472.CAN-03-3881. [DOI] [PubMed] [Google Scholar]
- 27.Dunn TA, Chen S, Faith DA, Hicks JL, Platz EA, Chen Y, Ewing CM, Sauvageot J, Isaacs WB, De Marzo AM, Luo J. A novel role of myosin VI in human prostate cancer. Am J Pathol. 2006;169:1843–1854. doi: 10.2353/ajpath.2006.060316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canales RD, Luo Y, Willey JC, Austermiller B, Barbacioru CC, Boysen C, Hunkapiller K, Jensen RV, Knight CR, Lee KY, Ma Y, Maqsodi B, Papallo A, Peters EH, Poulter K, Ruppel PL, Samaha RR, Shi L, Yang W, Zhang L, Goodsaid FM. Evaluation of DNA microarray results with quantitative gene expression platforms. Nature Biotechnol. 2006;24:1115–1122. doi: 10.1038/nbt1236. [DOI] [PubMed] [Google Scholar]
- 29.Willey JC, Crawford EL, Knight CR, Warner KA, Motten CA, Herness EA, Zahorchak RJ, Graves TG. Standardized RT-PCR and the standardized expression measurement center. Methods Mol Biol. 2004;258:13–41. doi: 10.1385/1-59259-751-3:13. [DOI] [PubMed] [Google Scholar]
- 30.Hesse M, Zimek A, Weber K, Magin TM. Comprehensive analysis of keratin gene clusters in humans and rodents. Eur J Cell Biol. 2004;83:19–26. doi: 10.1078/0171-9335-00354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.