

Abstract
Amyotrophic lateral sclerosis (ALS) involves the progressive degeneration of motor neurons in the spinal cord and motor cortex. Mutations to Cu,Zn superoxide dismutase (SOD) linked with familial ALS are reported to increase hydroxyl radical adduct formation from hydrogen peroxide as measured by spin trapping with 5,5′-dimethyl-1-pyrrolline N-oxide (DMPO). In the present study, we have used oxygen-17-enriched water and H2O2 to reinvestigate the mechanism of DMPO/⋅OH formation from the SOD and SOD mutants. The relative ratios of DMPO/⋅17OH and DMPO/⋅16OH formed in the Fenton reaction were 90% and 10%, respectively, reflecting the ratios of H217O2 to H216O2. The reaction of the WT SOD with H217O2 in bicarbonate/CO2 buffer yielded 63% DMPO/⋅17OH and 37% DMPO/⋅16OH. Similar results were obtained from the reaction between familial ALS SOD mutants and H217O2: DMPO/⋅17OH (64%); DMPO/⋅16OH (36%) from A4V and DMPO/⋅17OH (62%); and DMPO/⋅16OH (38%) from G93A. These results were confirmed further by using 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide spin trap, a phosphorylated analog of DMPO. Contrary to earlier reports, the present results indicate that a significant fraction of DMPO/⋅OH formed during the reaction of SOD and familial ALS SOD mutants with H2O2 is derived from the incorporation of oxygen from water due to oxidation of DMPO to DMPO/⋅OH presumably via DMPO radical cation. No differences were detected between WT and mutant SODs, neither in the concentration of DMPO/⋅OH or DEPMPO/⋅OH formed nor in the relative incorporation of oxygen from H2O2 or water.
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease or motor neuron disease, is a condition in which there is degeneration of the motor neurons of the spinal cord, brain stem, and cerebral cortex (1–8). Approximately 10% of ALS cases are familial with the remainder being sporadic. The genetic defect in 20% of familial ALS (FALS) cases now has been linked to Sod 1, the gene that encodes the cytosolic Cu,Zn superoxide dismutase (SOD) enzyme (2, 9, 10). To date, the only humans known to exhibit a genetic defect in the coding regions of SOD are those with the FALS disorder.
The mechanisms by which FALS-linked Sod 1 mutants cause selective degeneration of motor neurons remain unclear. Cu, ZnSOD is an antioxidant enzyme that primarily catalyzes the dismutation of O2⨪ to O2 and H2O2 (11, 12). The remarkably high reaction rate of Cu,ZnSOD is thought to be caused by the electrostatic guidance of the charged species (O2⨪) into the active site by conserved charged amino acid residues (13). It has been hypothesized that mutations at various sites in Sod 1 may push open the loops that form the superoxide-binding pocket and expose the active site to reaction with other oxidants (14). The gain-of-function hypotheses in FALS pathogenesis are based on the premise that the mutant SOD has an increased ability to react with H2O2 to generate higher oxidants (⋅OH or peroxidase activity) or an increased ability to react with peroxynitrite to form nitrated tyrosines (14–20). Evidence for increased peroxidase activity or ⋅OH formation from FALS-associated SOD mutants (A4V and G93A) was first obtained through the electron spin resonance (ESR) spin-trapping technique (16–23). The spin-trapping technique involves trapping of a reactive radical (i.e., superoxide anion or hydroxyl radical) by a nitrone spin trap to yield a more persistent nitroxide spin adduct that can be detected by ESR. By using this technique, Stadtman, Yim, and coworkers (18–20) found that FALS mutants generated increased formation of ⋅OH upon reaction with H2O2. The intensity of the ESR signal due to DMPO/⋅OH was much greater with FALS-associated SOD mutants than with the wild-type (WT) Cu,ZnSOD (16–20). Recently, Fridovich criticized these spin-trapping interpretations and suggested that the DMPO/⋅OH adduct was formed from the peroxidase activity of SOD and not from trapping of free hydroxyl radical (24, 25). Bredesen and Valentine and co-investigators also suggested that the peroxidase activity of SOD was responsible for oxidation of DMPO to DMPO/⋅OH and that FALS-associated SOD mutants exhibit considerably greater peroxidase activity (16, 17). However, no evidence for increased hydroxyl radical formation was obtained by using salicylate hydroxylation and lipid peroxidation assays in transgenic mice that develop progressive motor neuron disease from expressing human FALS-linked Sod 1 mutations (26). Marklund et al. also did not find any evidence for increased reactivity of the FALS mutant SOD D90A with hydrogen peroxide (27). Because of these conflicting reports, we decided to reexamine the previous spin-trapping investigations (16, 20). In the present study, we used two spin traps: 5,5′-dimethyl-1-pyrroline N-oxide (DMPO) and its phosphorylated analog 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO) (28–30). To determine the source of the oxygen atom in the hydroxyl radical adducts, we used oxygen-17-enriched hydrogen peroxide ([17O]-H2O2) and water ([17O]-H2O). Spin-trapping experiments were performed in a loop-gap resonator (31, 32), which enabled us to use exceedingly small volumes of expensive spin traps and 17O-labeled compounds. The mutants studied were the following: A4V (Ala4 → Val) and G93A (Gly93 → Ala) (33). A4V causes the most aggressive form of FALS (34), and G93A has been used previously in several transgenic mouse studies (34).
EXPERIMENTAL PROCEDURES
Materials.
[17O]-H2O (45%) and [17O]-H2O2 (89%) were obtained from ICON Isotopes (Summit, NJ). DMPO was obtained from Sigma and double distilled to remove the paramagnetic impurities. DEPMPO was synthesized and purified as described (30). The bicarbonate/CO2 buffer solutions were prepared by treating with Chelex 100 resin (Bio-Rad) to remove trace polyvalent metal ions. The pH of the Chelex-treated buffer was readjusted to pH 7.4 by bubbling with 95% N2/5% CO2 mixture.
Preparation of SOD Mutants.
Both WT and mutant SODs were expressed in a bacterial system and purified by ammonium sulfate fractionation and anion exchange chromatography as described (35). The metal content of the purified SODs was determined by the colorimetric 4-pyridylazaresorcinol assay (36). The SODs were suspended in 10 mM sodium acetate (pH 5.0). Sufficient cupric citrate and zinc sulfate were added to bring the metal content to 110% of the available copper and zinc binding sites on SOD, and the SODs were incubated overnight at 4°C. The SODs were repurified over a high resolution HQ10 anion exchange resin (PerSeptive Biosystems) by using 20 mM Tris⋅HCl with a 0–200 mM NaCl gradient over 10 column volumes. The second protein peak generally contained the highest activity with 95–103% of copper and zinc. The protein was concentrated in water and frozen until use. Protein concentrations were determined by the bicinchoninic acid method (Pierce). A stock of the WT human SOD was used as the protein standard, whose activities were determined by the cytochrome c method (11) and were ≈5,200 units/mg protein. SOD activity in the metal-replete mutant proteins was equivalent to that of the WT protein as reported (36) and was directly proportional to the copper content in partially metal containing fractions. Zinc-deficient SOD was prepared by dialyzing 1 mg/ml Cu,Zn SOD exhaustively against Chelex 100-treated 100 mM potassium phosphate (pH 3.5) until the zinc content was reduced to <10%, as described (36, 37).
ESR Spin-Trapping.
ESR spectra were recorded at room temperature on a Varian E-109 spectrometer operating at 9.5 GHz and with a 100-KHz field modulation equipped with a TE102 cavity or a loop-gap resonator. Reactions were initiated by the addition of H2O2 to the incubation mixtures containing SOD (100 μg/ml), DMPO (100 mM), and diethylenetriaminepentaacetic acid (DTPA) (100 μM). A typical reaction mixture for ESR analysis consisted of 1.5 μl of WT-SOD or SOD mutants (1 mg/ml), 1 μl of DMPO (1 M) or DEPMPO (0.5 M), 1 μl of [16O]-H2O2 or [17O]-H2O2 (15 mM), and 11.5 μl of bicarbonate/CO2 buffer (25 mM, pH 7.4) containing DTPA (100 μM) in a total volume of 15 μl. For experiments with [17O]-H2O, the reaction mixture consisted of 1.5 μl of WT-SOD or SOD mutants (1 mg/ml), 1 μl of DMPO (1 M) or DEPMPO (0.5 M), 7.5 μl of [17O]-H2O (45%), 1 μl of [16O]-H2O2, and 4 μl of bicarbonate/CO2 buffer (100 mM, pH 7.4) containing DTPA (0.4 mM) in a total volume of 15 μl. The sample was transferred to a capillary tube (0.64 mm i.d. × 0.84 mm o.d. and 100 mm long) that was sealed with miniseal (Baxter Scientific Products, McGaw Park, IL) and placed into the loop-gap resonator. Computer-based simulations of ESR spectra were performed by using software written by Duling (38). The correlation coefficient (r) for the spectral simulation was 0.994 ± 0.001. Spin adduct concentrations were obtained by double integration using 3-carbamoyl-2,2,5,5-tetramethyl-3-pyrroline-1-yloxy as a standard. Spectrometer conditions used in spin trapping studies were: microwave power, 2 mW; time constant, 0.128 s; modulation amplitude, 0.5 G; and scan time, 2 min.
RESULTS
Formation of DMPO- and DEPMPO-Hydroxyl Radical Adducts.
Addition of H2O2 (0.5–5 mM) to a solution containing WT-Cu,ZnSOD (100 μg/ml) or the FALS SOD mutant (100 μg/ml) in a bicarbonate buffer (pH 7.4) containing the metal ion chelator DTPA produced a four-line ESR spectrum (aN = aH = 15 G) with an intensity ratio of 1:2:2:1 corresponding to the DMPO-hydroxyl adduct (DMPO/⋅OH) (Fig. 1A). The spectral intensity of DMPO/⋅OH increased to a steady-state concentration (≈50 μM) within 10 min. The spectral intensity of DMPO/⋅OH obtained from the reaction between H2O2 and the FALS mutants G93A or A4V was nearly the same as that obtained with WT-SOD (Fig. 1A). The concentration of DMPO/⋅OH produced was 93 ± 5% for A4V and 88 ± 6% for G93A relative to the WT-SOD and was found to be independent of DTPA concentration (0.1–1 mM). The yields of DMPO/⋅OH from zinc-deficient WT-SOD or FALS mutant protein in the presence of H2O2 were found to be identical (data not shown). The rate of Cu2+ release during the reaction among WT-SOD, A4V, and G93A (100 μg/ml) with H2O2 (1 mM) in PBS at 37°C also was not significantly different (1.5 nM/s for WT and A4V and 1.1 nM/s for G93A) (data not shown).
Figure 1.

Formation of DMPO/⋅OH and DEPMPO/⋅OH adducts during the reaction between H2O2 and WT-SOD or SOD mutants. (A) H2O2 (5 mM), DMPO (50 mM), DTPA (100 μ M), and WT-SOD (100 μg/ml) or A4V or G93A (100 μg/ml) were incubated in bicarbonate/CO2 buffer (25 mM, pH 7.4). (B) H2O2 (5 mM), DEPMPO (50 mM), DTPA (100 μM), and WT-SOD (100 μg/ml) or A4V or G93A (100 μg/ml) were incubated in bicarbonate/CO2 buffer (25 mM, pH 7.4). The spectra were recorded immediately after the reaction was initiated with the enzyme. Spectra are representative scans of three independent experiments.
Next, we verified whether DEPMPO, a structural analog of DMPO, could trap the hydroxyl radical-like oxidant formed during the reaction between H2O2 and SOD or SOD mutants. Addition of WT-SOD or SOD mutant to a solution containing H2O2 (5 mM) and DEPMPO (25 mM) in a bicarbonate buffer (25 mM, pH 7.4) produced an eight-line spectrum with an intensity ratio of 1:2:2:1:1:2:2:1 (Fig. 1B). Based on the literature data (28, 29), this spectrum (aP = 47.3 G, aH = 13.2 G, and aN = 14.0 G) was assigned to the DEPMPO-hydroxyl adduct (DEPMPO/⋅OH). Formation of this spectrum was absolutely dependent on all of the components. The spectral intensity of DEPMPO/⋅OH obtained from the reaction between H2O2 and the FALS mutants G93A or A4V was nearly the same as that obtained with WT-SOD (Fig. 1B). Contrary to previous reports (16–20), the present spin-trapping data obtained using two different spin traps demonstrate that FALS SOD mutants (A4V and G93A) do not generate a significant increase in hydroxyl radical adduct formation compared with the WT-SOD.
Incorporation of 17O Atom from 17O-H2O2 into DMPO/⋅OH Adduct.
To investigate further the mechanism of formation of DMPO/⋅OH, spin-trapping experiments were carried out in a solution containing 89% 17O-labeled hydrogen peroxide (39). The rationale for using the isotopically enriched H2O2 is as follows: The nuclear spin quantum number (I) for the 17O atom is 5/2 in contrast to the 16O, which has I = 0, and, thus, additional couplings from the 17O atom will be observed if there is any incorporation of oxygen atom into the DMPO/⋅OH adduct from hydrogen peroxide.
By using [17O]-H2O2, it was shown that nearly all of the DMPO/⋅OH formed in the Fenton system (Fe2+ + H2O2) came from hydrogen peroxide as reported (39). The ESR spectrum of DMPO/⋅OH obtained from incubations containing [17O]-H2O2, DMPO, and Fe(II)-EDTA in a bicarbonate/CO2 buffer consists of 90% DMPO/⋅17OH and 10% DMPO/⋅16OH (Fig. 2) matching the isotopic distribution of the hydrogen peroxide. In contrast to the Fenton system, the WT-SOD and the mutants (G93A and A4V) yielded considerably lower amounts of oxygen-17-incorporated hydroxyl adducts (Table 1). We estimate that nearly 35% of DMPO/⋅OH arises from the incorporation of oxygen from a source other than H2O2, presumably water, and that 65% of DMPO/⋅OH is derived from incorporation of oxygen from H2O2 in a reaction catalyzed by SOD or SOD mutants.
Figure 2.

The effect of 17O-labeled H2O2 on DMPO/⋅OH formation. Incubation mixtures contained H2O2 (5 mM), DMPO (50 mM), and DTPA (100 μM) in a bicarbonate/CO2 buffer. The reaction was initiated with either Fe(II)-EDTA (100 μM) or 100 μg of SOD or SOD mutants (G93A and A4V) as shown. Line positions from 17O and 16O couplings are shown by • and ○, respectively. The dotted lines show computer simulations of ESR spectra using the parameters aN = 15.0 G and aH = 15.0 G for DMPO/⋅16OH and aN = 15.0 G, aH = 15.0 G, and a17O = 4.6 G for DMPO/⋅17OH.
Table 1.
The relative concentrations of 17O and 16O spin adducts of DMPO/⋅OH formed during the reaction of SOD with 89% [17O]-H2O2
| Conditions | % of DMPO/⋅OH spin adducts from H2O2 | % of DMPO/⋅OH spin adducts from H2O |
|---|---|---|
| Fe(II)-EDTA | 100 | 0 |
| WT-SOD | 63 ± 0.5 | 37 ± 0.5 |
| A4V | 64 ± 1.4 | 36 ± 1.4 |
| G93A | 62 ± 0.8 | 38 ± 0.8 |
Data were normalized with respect to the 17O content in H217O2 by using the formula: %DMPO/⋅OH from water = 100(%H217O2 − %DMPO/•17OH)/%H217O2.
Incorporation of 17O Atom from [17O]-H2O into DMPO/⋅OH Adduct.
To determine the source of the remaining fraction of DMPO/⋅OH, we investigated the effect of H217O. The Fenton reaction (Fe2+ + H216O2) was carried out in H217O in the presence of DMPO (Fig. 3A). The ESR spectrum of DMPO/⋅OH recorded at a higher gain was identical to that obtained in H216O (Fig. 3A, Lower). This result clearly established that there is no exchange between the 17O in water and the 16O present in DMPO/⋅16OH (DMPO/⋅16OH + H217O –X→ DMPO/⋅17OH + H216O) (39). In contrast to the Fenton reaction, incubation mixtures containing WT-SOD or SOD mutants, H2O2, and DMPO in bicarbonate/CO2 buffer prepared in 45% H217O produced additional hyperfine couplings from DMPO/⋅17OH (marked • in Fig. 3B, Upper). Computer simulation of the total signal (dotted line in Fig. 3B) from both the DMPO/⋅16OH and DMPO/⋅17OH adducts showed that ≈35% of DMPO-hydroxyl adduct was formed from the addition of water to DMPO in a reaction catalyzed by SOD and H2O2 (39). Similar ESR spectra were obtained from incubations containing FALS SOD mutants, H2O2, and DMPO in bicarbonate/CO2 buffer prepared in 45% H217O (Fig. 3C).
Figure 3.

The effect of 17O-labeled H2O on DMPO/⋅OH formation. (A, Upper) Incubation mixture contained Fe(II)-EDTA (100 μM), H2O2 (5 mM), and DMPO (50 mM) in a bicarbonate/CO2 buffer made with H217O; (Lower) obtained at gain × 10. (B, Upper) Incubation mixtures contained H2O2 (5 mM), DMPO (50 mM), WT-SOD (100 μg/ml), and DTPA (100 μM) in a bicarbonate/H2O2 buffer made with H217O (pH 7.4); (Lower) same as Upper but obtained at gain × 10. The dotted lines show a computer simulation of ESR spectra using the parameters aN = 15.0 G and aH = 15.0 G for DMPO/⋅16OH and aN = 15.0 G, aH = 15.0 G, and a17O = 4.6 G for DMPO/⋅17OH. (C) Conditions were the same as in B (Lower) using SOD mutants.
Incorporation of 17O Atom from [17O]-H2O2 into DEPMPO/⋅OH Adduct.
The oxidation potentials of DMPO and DEPMPO are ≈1.87 V (vs. NHE) and 2.24 V (vs. NHE), respectively. We surmised that the 370-mV difference in oxidation potential could help differentiate between the cation radical and hydroxyl radical-mediated formation of hydroxyl adducts. However, similar results were also obtained with DEPMPO trap. The ESR spectrum of DEPMPO/⋅OH obtained from incubations containing [17O]-H2O2, DEPMPO, and Fe(II)-EDTA in a bicarbonate/CO2 buffer consists of 90% DEPMPO/⋅17OH and 10% DEPMPO/⋅16OH matching the distribution of the hydrogen peroxide. (Fig. 4). In contrast to the Fenton system, the WT-SOD and the mutants (G93A and A4V) yielded considerably lower amounts of oxygen-17 incorporated in DEPMPO/⋅OH (Fig. 4 and Table 2). As with DMPO, we estimate that ≈35–40% of DEPMPO/⋅OH is derived from incorporation of oxygen from water, and the rest (60–65%) of DEPMPO/⋅OH arises from incorporation of oxygen from H2O2 in a reaction catalyzed by SOD or SOD mutants.
Figure 4.

The effect of 17O-labeled H2O2 on DEPMPO/⋅OH formation. Incubation mixtures contained H2O2 (5 mM), DEPMPO (20 mM), and DTPA (100 μM) in a bicarbonate/CO2 buffer. The reaction was initiated with either Fe(II)-EDTA (100 μM) or 100 μg of SOD or SOD mutants (G93A and A4V) as shown. Line positions from 17O and 16O couplings are shown by • and ○, respectively. The dotted lines show a computer simulation of ESR spectra using the parameters aN = 14.0 G, aH = 13.2 G, aP = 47.0 G for DEPMPO/⋅16OH and aN = 14.0 G, aH = 13.1 G, aP = 47.0 G, and a17O = 4.1 G for DEPMPO/⋅17OH).
Table 2.
The relative concentrations of 17O and 16O spin adducts of DEPMPO/⋅OH formed during the reaction of SOD with 89% [17O]-H2O2
| Conditions | % of DEPMPO/⋅OH spin adducts from H2O2 | % of DEPMPO/⋅OH spin adducts from H2O |
|---|---|---|
| Fe(II)-EDTA | 100 | 0 |
| WT-SOD | 60 ± 2.82 | 40 ± 2.82 |
| A4V | 66 ± 0.47 | 34 ± 0.47 |
| G93A | 63 ± 0.47 | 37 ± 0.82 |
Data were normalized with respect to the 17O content in H217O2 by using the formula: %DEMPMPO/⋅OH from water = 100(%H217O2 − %DEPMPO/⋅17OH)/%H217O2.
DISCUSSION
SOD-Dependent Radical Reactions.
Table 3 shows the antioxidant and prooxidant reactions of SOD. Cu,ZnSOD is an antioxidant enzyme that primarily catalyzes the dismutation of O2⨪ to O2 and H2O2 (11, 12). In addition to this dismutase activity, SOD also exhibits prooxidant activities, which include the peroxidase activity, hydroxyl radical generating activity and nitration of tyrosine (15–20, 40, 41). A novel superoxide-dependent peroxidase activity recently has been reported for the FALS mutant H48Q in which histidine 48 has been replaced by glutamine (42).
Table 3.
Free radical reactions of
| Dismutase activity | |
| 1 | SOD−Cu2+ + O2⨪ → SOD−Cu+ + O2 |
| 2 | SOD−Cu+ + O2⨪ → SOD−Cu2+ + H2O2 |
| 3 | |
| Peroxidase activity | |
| 4 | SOD−Cu2+ + H2O2 ⇌ SOD−Cu+ + O2⨪ + 2H+ |
| 5 | SOD−Cu+ + H2O2 → SOD−Cu2+−⋅OH + OH− |
| 6 | SOD−Cu2+−⋅OH + His-61 → His⋅ + SOD−Cu2+ + H2O |
| 7 | SOD−Cu2+−⋅OH + HCO2− → SOD−Cu2+ + CO2⨪ + H2O |
| 8 | SOD−Cu2+−⋅OH → inactive SOD (Enz−Cu2+ + 2-oxohistidine) |
| Hydroxyl radical generating activity | |
| 9 | Enz−Cu2+ + H2O2 → Enz−Cu+ + O2⨪ + 2H+ |
| 10 | Enz−Cu+ + H2O2 → Enz−Cu2+ + ⋅OH + OH− |
| Nitration of tyrosine | |
| 11 | SOD−Cu2+ + ONOO− → SOD−CuO....NO2+ |
| 12 | SOD−CuO....NO2+ + Tyr → SOD−Cu2+ + OH− + NO2-Tyr |
The peroxidase activity refers to the “bound” hydroxyl radical to the copper atom at the active site (i.e., SOD−Cu2+−⋅OH) and not to the conventional compound I-type oxidant derived from heme proteins. For example, the horseradish peroxidase/H2O2 system, which forms compound I, does not oxidize DMPO to DMPO/⋅OH.
Mechanism of Formation of DMPO/⋅OH.
The mechanism of formation of hydroxyl radical adduct was attributed to the trapping of enzyme-bound oxidant SOD − Cu2+ − ⋅OH by DMPO (SOD − Cu2+ − ⋅OH + DMPO → SOD − Cu2+ + DMPO/⋅OH) (15). However, the present data show only a partial transfer of oxygen-17 from H217O2. We interpret the present spin-trapping data by a mechanism involving both oxidation and hydroxylation of DMPO by Cu2+ − ⋅OH (43–45).
The incorporation of 17O from water into DMPO/⋅OH can occur by at least two known mechanisms. In the first, a Cu2+-catalyzed nucleophilic addition of H217O to DMPO results in the formation of the corresponding hydroxylamine of DMPO/⋅17OH with subsequent air oxidation forming DMPO/⋅17OH (45). This mechanism is not favored in the SOD/H2O2 system because DTPA did not prevent the incorporation of 17O from water into DMPO/⋅OH. In the second, a strong oxidant oxidizes DMPO to its cation radical DMPO⋅+, which reacts with H217O to form DMPO/⋅17OH directly (44). The significant difference in these two mechanisms is the need for a very strong oxidant to form DMPO⋅+, which has an oxidation potential greater than 1.8 V. The oxidation of the corresponding hydroxylamine of radical adducts occurs even in air and is rapid with ferricyanide (46). Direct incorporation of 17O-atom into DMPO/⋅OH in the presence of H217O or H217O2 may be explained by the following reaction mechanism:
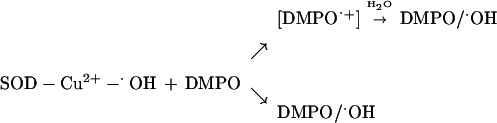 |
The significance of this reaction, however, remains unclear, because DMPO (≅200 mM) did not prevent the inactivation of SOD by H2O2 (data not shown).
In the presence of formate, the spectrum due to the DMPO–carbon dioxide anion radical adduct (DMPO/⋅CO2−) or DEPMPO/⋅CO2− was obtained (29, 30). This result is attributed to trapping of the carbon dioxide radical anion (⋅CO2−) produced from oxidation of formate by copper-bound hydroxyl radical (reaction 7 in Table 3) (47–49). The ESR spectra of DMPO/⋅CO2− (aN = 15.7 G, aH = 18.8 G) and DEPMPO/⋅CO2− (aN = 14.5 G, aH = 17.3 G and aP = 51.6 G) were observed in incubations containing WT-SOD (100 μg/ml), H2O2 (5 mM), and formate (200 mM) in a bicarbonate/CO2 buffer (25 mM, pH 7.4) (data not shown).
Oxidative Mechanisms in FALS Pathogenesis.
Many mechanisms have been suggested to explain how mutations to SOD cause the selective degeneration of motor neurons. SOD has been implicated in increasing oxidative injury by mechanisms involving the increased generation of hydroxyl radical, increased peroxidase activity, or increased nitration of tyrosine by peroxynitrite. Based on the present data, we can rule out the formation of free hydroxyl radical from loosely bound copper (reactions 9 and 10 in Table 3). The present data also do not favor the previous hypothesis that FALS mutants exhibit an increased peroxidase activity relative to WT-SOD. Furthermore, the mechanism of generating DMPO/⋅OH adduct in the SOD/H2O2 system is more complex than previously reported.
Previously, the gain in function of FALS mutants was attributed to increased toxicity caused by released copper from the active site of FALS mutants. The ALS mutant SODs have reduced affinity for zinc (36). Furthermore, neurofilament subunits bind zinc with sufficient affinity that they can remove zinc from SOD (36). The increased loss of zinc from SOD will diminish the scavenging of superoxide and increase the catalysis of tyrosine nitration by peroxynitrite (36). The accumulation of nitrotyrosine has been demonstrated in both familial and sporadic ALS patients as well as in ALS–SOD transgenic mice (50). These results suggest that the loss of zinc from SOD may enhance peroxynitrite formation and motor neuron death in ALS.
In summary, the 17O results demonstrate that approximately two-thirds of the DMPO/⋅OH arises from reaction with an oxidant from SOD, with the remainder being derived from the oxidation of the spin trap with subsequent addition of water. No differences were detected between WT and mutant SODs, neither in the concentration of DMPO/⋅OH or DEPMPO/⋅OH formed nor in the relative incorporation of oxygen from H2O2 or water.
Acknowledgments
Both B.K. and R.J.S. express thanks to Drs. T. Siddique and M. S. Ahmed for stimulating their interest in ALS research and Drs. J. Sampson, J. Crow, and Y. Zhuang for their assistance in preparing SODs with defined metal content. This work has been supported by grants from the National Institutes of Health RR01008 and GM22923 and from the Amyotrophic Lateral Sclerosis Association.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: ALS, amyotrophic lateral sclerosis; FALS, familial amyotrophic lateral sclerosis; SOD, superoxide dismutase; A4V, SOD mutant with substitution of alanine to valine at position 4; DEPMPO, 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide; DMPO, 5,5′-dimethyl-1-pyrroline N-oxide; DMPO/⋅OH; DMPO-hydroxyl radical spin adduct; DMPO/⋅CO2−, DMPO-carbon dioxide radical anion spin adduct; DEPMPO/⋅CO2−, DEPMPO-carbon dioxide radical anion spin adduct; G93A, SOD mutant with substitution of glycine to alanine at position 93; WT, wild-type.
References
- 1.Price D L, Cleveland D W, Koliatsos V E. Neurobiol Dis. 1994;1:3–11. doi: 10.1006/nbdi.1994.0002. [DOI] [PubMed] [Google Scholar]
- 2.Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan J P, Deng H X, et al. Nature (London) 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Bruijn L I, Cleveland D W. Neuropath Appl Neurobiol. 1996;22:373–387. doi: 10.1111/j.1365-2990.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 4.Cudkowicz M E, McKenna-Yasek D, Sapp P E, Chin W, Geller B, Hayden D L, Schoenfeld D A, Hosler B A, Horvitz H R, Brown R H. Ann Neurol. 1997;41:210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- 5.Deng H X, Hentati A, Tainer J A, Iqbal Z, Cayabyab A, Hung W Y, Getzoff E D, Hu P, Herzfeldt B, Roos R P, et al. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 6.Siddique T. Cold Spring Harbor Symp Quant Biol. 1996;61:699–708. [PubMed] [Google Scholar]
- 7.Smith R G, Appel S H. Annu Rev Med. 1995;46:133–145. doi: 10.1146/annurev.med.46.1.133. [DOI] [PubMed] [Google Scholar]
- 8.Rowland L P. Proc Natl Acad Sci USA. 1995;92:1251–1253. doi: 10.1073/pnas.92.5.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siddique T, Figlewicz D A, Pericak-Vance M A, Haines J L, Rouleau G, Jeffers A J, Sapp P, Hung W Y, Bebout J, McKenna-Yasek D, et al. N Engl J Med. 1991;324:1381–1384. doi: 10.1056/NEJM199105163242001. [DOI] [PubMed] [Google Scholar]
- 10.Wong P C, Pardo C A, Borchelt D R, Lee M K, Copeland N G, Jenkins N A, Sisodia S S, Cleveland D W, Price D L. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 11.McCord J M, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 12.Fridovich I. Annu Rev Biochem. 1975;44:147–159. doi: 10.1146/annurev.bi.44.070175.001051. [DOI] [PubMed] [Google Scholar]
- 13.Sampson J B, Crow J, Strong P M, Beckman J S. Mineral and Metal Neurotoxicity. Boca Raton, FL: CRC; 1996. pp. 395–405. [Google Scholar]
- 14.Beckman J S, Carson M, Smith C D, Koppenol W H. Nature (London) 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 15.Parge H E, Hallewell R A, Tainer J A. Proc Natl Acad Sci USA. 1992;89:6109–6113. doi: 10.1073/pnas.89.13.6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiedau-Pazos M, Goto J J, Rabizadeh S, Gralla E B, Roe J A, Lee M K, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 17.Bredesen, D. E., Wiedau-Pazos M., Goto, J. J., Rabizadeh, S., Roe, J. A., Gralla, E. B., Ellerby, L. M. & Valentine, J. S. (1996) Neurology 47, Suppl. 2, S36–S38. [DOI] [PubMed]
- 18.Yim M B, Kang J H, Yim H S, Kwak H S, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yim M B, Chock P B, Stadtman E R. J Biol Chem. 1993;268:4099–4105. [PubMed] [Google Scholar]
- 20.Yim H S, Kang J H, Chock P B, Stadtman E R, Yim M B. J Biol Chem. 1997;272:8861–8863. doi: 10.1074/jbc.272.14.8861. [DOI] [PubMed] [Google Scholar]
- 21.Janzen E G. Acc Chem Res. 1971;3:31–40. [Google Scholar]
- 22.Buettner G R. Free Radical Biol Med. 1987;3:259–303. doi: 10.1016/s0891-5849(87)80033-3. [DOI] [PubMed] [Google Scholar]
- 23.Buettner G R, Mason R P. Methods Enzymol. 1990;186:127–133. doi: 10.1016/0076-6879(90)86101-z. [DOI] [PubMed] [Google Scholar]
- 24.Fridovich I. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 25.Fridovich I. Annu Rev Biochem. 1995;64:97–112. doi: 10.1146/annurev.bi.64.070195.000525. [DOI] [PubMed] [Google Scholar]
- 26.Bruijn L I, Beal M F, Becher M W, Schulz J B, Wong P C, Price D L, Cleveland D W. Proc Natl Acad Sci USA. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marklund S L, Andersen P M, Forsgren L, Nilsson P, Ohlsson P I, Wikander G, Oberg A. J Neurochem. 1997;69:675–681. doi: 10.1046/j.1471-4159.1997.69020675.x. [DOI] [PubMed] [Google Scholar]
- 28.Fréjaville C, Karoui H, Le Moigne F, Culcasi M, Pietri S, Lauricella R, Tuccio B, Tordo P. J Chem Soc Chem Commun. 1994;15:1793–1794. doi: 10.1021/jm00002a007. [DOI] [PubMed] [Google Scholar]
- 29.Karoui H, Hogg N, Fréjaville C, Tordo P, Kalyanaraman B. J Biol Chem. 1996;271:6000–6009. doi: 10.1074/jbc.271.11.6000. [DOI] [PubMed] [Google Scholar]
- 30.Vásquez-Vivar J, Hogg N, Pritchard K A, Jr, Martasek P, Kalyanaraman B. FEBS Lett. 1997;403:127–130. doi: 10.1016/s0014-5793(97)00036-7. [DOI] [PubMed] [Google Scholar]
- 31.Froncisz W, Hyde J S. J Magn Reson. 1982;47:515–521. [Google Scholar]
- 32.Altenbach C, Marti T, Khorana H G, Hubbell W L. Science. 1990;248:1088. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- 33.Rosen D R, Sapp P, O’Regan J, McKenna-Yasek D, Schlumpf K S, Haines J L, Gusella J F, Horvitz H R, Brown R H., Jr Am J Med Gen. 1994;51:61–69. doi: 10.1002/ajmg.1320510114. [DOI] [PubMed] [Google Scholar]
- 34.Ripps M E, Huntley G W, Hof P R, Morrison J H, Gordon J W. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rabizadeh S, Gralla E B, Borchelt D R, Gwinn R, Valentine J S, Sisodia S, Wong P, Lee M, Hahn H, Bredesen D E. Proc Natl Acad Sci USA. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crow J P, Sampson J B, Zhuang Y, Thompson J A, Beckman J S. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 37.Pantoliano M W, McDonnel P J, Valentine J S. J Am Chem Soc. 1979;101:6454–6456. [Google Scholar]
- 38.Duling D R. J Magn Res. 1994;B 104:105–110. doi: 10.1006/jmrb.1994.1062. [DOI] [PubMed] [Google Scholar]
- 39.Lloyd R V, Hanna P M, Mason R P. Free Radical Biol Med. 1997;22:885–888. doi: 10.1016/s0891-5849(96)00432-7. [DOI] [PubMed] [Google Scholar]
- 40.Beckman J S. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 41.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin J C, Smith C D, Beckman J S. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 42.Liochev S I, Chen L L, Hallewell R A, Fridovich I. Arch Biochem Biophys. 1997;346:263–268. doi: 10.1006/abbi.1997.0298. [DOI] [PubMed] [Google Scholar]
- 43.Gunther M R, Hanna P M, Mason R P, Cohen M S. Arch Biochem Biophys. 1995;316:515–522. doi: 10.1006/abbi.1995.1068. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharjee S, Khan Md N, Chandra H, Symons M C R. J Chem Soc Perkin Trans. 1996;2:2631–2634. [Google Scholar]
- 45.Hanna P M, Chamulitrat W, Mason R P. Arch Biochem Biophys. 1992;296:640–644. doi: 10.1016/0003-9861(92)90620-c. [DOI] [PubMed] [Google Scholar]
- 46.Sentjurc M, Mason R P. Free Radical Biol Med. 1992;13:151–160. doi: 10.1016/0891-5849(92)90077-t. [DOI] [PubMed] [Google Scholar]
- 47.Hodgson E K, Fridovich I. Biochemistry. 1975;14:5294–5303. doi: 10.1021/bi00695a010. [DOI] [PubMed] [Google Scholar]
- 48.Sinet P M, Garber P. Arch Biochem Biophys. 1981;212:411–416. doi: 10.1016/0003-9861(81)90382-9. [DOI] [PubMed] [Google Scholar]
- 49.Cabelli D E, Allen D, Bielski B H, Holcman J. J Biol Chem. 1989;264:9967–9971. [PubMed] [Google Scholar]
- 50.Beal M F, Ferrante R J, Browne S E, Matthews R T, Kowall N W, Brown R H. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]